

Биологические мембраны: Журнал мембранной и клеточной биологии, 2019, T. 36, № 1, стр. 44-52
Влияние микробных метаболитов на функции митохондрий в условиях ацидоза и дефицита субстратов окисления
Н. И. Федотчева a, b, *, В. В. Теплова a, Н. В. Белобородова b
a Институт теоретической и экспериментальной биофизики РАН
142290 Московской обл., Пущино, ул. Институтская, 3, Россия
b Федеральный научно-клинический центр реаниматологии и реабилитологии
107031 Москва, ул. Петровка, 25, Россия
* E-mail: nfedotcheva@mail.ru
Поступила в редакцию 03.08.2018
После доработки 22.08.2018
Принята к публикации 21.08.2018
Аннотация
Дисфункция митохондрий является основным фактором в развитии полиорганной недостаточности при сепсисе. В данной работе исследовали роль микробных метаболитов в индукции митохондриальных нарушений. Изучали влияние фенольных кислот микробного происхождения на функции митохондрий в условиях ацидоза и дефицита субстрата окисления, сопутствующих развитию сепсиса. Эти условия значительно усиливали действие бензойной, фенилпропеновой, фенилпропионовой и фенилуксусной кислот на индукцию митохондриальной поры, окисление NADH и окислительное фосфорилирование в изолированных митохондриях печени. Наиболее эффективным фактором, усиливающим действие фенольных кислот, оказался дефицит субстрата окисления. Инкубация митохондрий с фенольными кислотами в отсутствие субстрата окисления вызывала значительное ингибирование дегидрогеназ цикла трикарбоновых кислот. Полученные данные свидетельствуют о потенциальной роли фенольных кислот микробного происхождения в развитии митохондриальной дисфункции при воспалительном процессе и сепсисе.
ВВЕДЕНИЕ
Многочисленные данные свидетельствует о нарушениях функций митохондрий при воспалительных процессах и сепсисе [1–3]. В настоящее время общепризнано, что дисфункция митохондрий является одним из главных механизмов появления и развития полиорганной недостаточности при сепсисе. К повреждающим факторам, действующим на митохондрии, относят усиленную продукцию цитокинов и активных форм кислорода, общую и локальную гипоксию, истощение антиоксидантной защиты и ряд других факторов [4–6]. Было обнаружено снижение активности отдельных участков дыхательной цепи в биоптатах скелетной мышцы и сердечной мышцы, ингибирование пируватдегидрогеназы в скелетной мышце [7–9] и в клетках крови [10, 11], цитохромоксидазы в тромбоцитах [12]. Эти изменения проявляются уже на ранних стадиях сепсиса. Данные о роли микробных метаболитов в этих процессах отсутствуют или появляются только в последнее время. Хотя микробные метаболиты необходимы для нормального метаболизма и гомеостаза организма, однако в избыточных концентрациях они могут оказывать токсичное действие на отдельные функции и организм в целом, в том числе влиять на проницаемость мембран и гематоэнцефалический барьер [13, 14]. Показано участие микробных метаболитов (короткоцепочечных жирных кислот и фенольных кислот) в эпигенетическом контроле экспрессии генов, регулирующих продукцию провоспалительных и антивоспалительных цитокинов [15, 16].
Данные о влиянии микробных метаболитов на митохондриальные функции к настоящему времени немногочисленны и относятся преимущественно к их роли в развитии почечной и сердечной недостаточности. Микробные метаболиты в основном рассматриваются как уремические токсины. Было показано, что в митохондриях почки индолацетат, индоксилсульфат и фенилацетат ингибируют сукцинатдегидрогеназу и дыхание митохондрий почки более чем на 20% при концентрации этих токсинов в диапазоне от 1 до 2 мМ [17]. Кроме того, фенилацетат увеличивал продукцию активных форм кислорода в митохондриях почки [18]. Таким образом, микробные метаболиты активно участвуют в регуляции биохимических и физиологических процессов в организме. Актуальность исследований, направленных на изучение влияния микробных метаболитов на функции митохондрий, обусловлена также ключевой ролью митохондриальных дегидрогеназ в формировании фенотипа иммунных клеток крови под влиянием бактериальных липополисахаридов [19]. Наибольший интерес представляют те микробные метаболиты, гиперпродукция которых наблюдается при бактериемии и сепсисе. К ним относятся низкомолекулярные ароматические кислоты – продукты деградации фенилаланина, тирозина и полифенолов различными видами бактерий [20, 21].
Ранее нами было показано, что микробные метаболиты фенольной природы, к которым относятся бензойная, фенилпропеновая (фенилакриловая), фенилпропионовая, фенилуксусная, фенилмолочная кислоты, а также гидроксифенилуксусная и гидроксифенилмолочная кислоты влияют на функции митохондрий [20, 21]. При нормальных физиологических условиях (рН 7.4 и при отсутствии дополнительных воздействий) эти микробные метаболиты в высоких концентрациях ингибировали дыхание, снижали мембранный потенциал и активировали продукцию активных форм кислорода в митохондриях печени крыс [22]. В данной работе исследовано влияние этих микробных метаболитов на индукцию митохондриальной поры, окислительное фосфорилирование и активность дегидрогеназ в условиях ацидоза и дефицита субстратов окисления, сопутствующих развитию сепсиса. Обнаружено, что действие фенольных кислот резко усиливается при закислении среды инкубации и после предварительной инкубации с митохондриями в отсутствие субстратов окисления. В этих условиях действующие концентрации фенольных кислот могут снижаться на порядок. Полученные данные указывают на потенциальную роль микробных метаболитов в развитии митохондриальной дисфункции при воспалительном процессе и сепсисе.
МАТЕРИАЛЫ И МЕТОДЫ
Выделение митохондрий печени крыс. Митохондрии выделяли из печени взрослых крыс линии Wistar (самцы, вес 200–250 г) стандартным методом дифференциального центрифугирования [23]. Печень гомогенизировали в холодной среде, содержащей 300 мМ сахарозы, 1 мМ EGTA, 10 мМ трис-HCl буфера (рН 7.4). Гомогенат центрифугировали при 4°С 10 мин при 600 g, полученный супернатант центрифугировали 10 мин при 9000 g для осаждения митохондрий. Осажденные митохондрии промывали средой выделения без EGTA, ресуспендировали в среде того же состава до концентрации белка 60 мг/мл и хранили на льду.
Измерение мембранного потенциала и кальциевой емкости митохондрий. Pазноcть электpичеcких потенциалов на внутpенней мембpане митоxондpий опpеделяли по pаcпpеделению липофильного катиона тетpафенилфоcфония (ТФФ+), концентpацию котоpого во внешней cpеде определяли c помощью ТФФ+-cелективного электpода. Транспорт ионов Са2+ в митохондрии изучали с помощью компьютеризированной установки “Record 4” (Россия) и Са2+-селективного электрода, регистрирующего изменения концентрации кальция во внешней среде в ответ на последовательные добавки СаСl2 в конечной концентрации 20 мкМ. Кальциевую емкость митохондрий определяли по способности митохондрий аккумулировать и удерживать последовательные добавки ионов кальция до пороговой концентрации, необходимой для открытия неспецифической митохондриальной поры [24]. При измерениях митохондрии находились в среде, содержащей 125 мМ КCl, 1.5 мМ KH2PO4 и 15 мМ HEPES–трис (рН 7.25), а также глутамат (4 мМ) и малат (2 мМ) в качестве субстратов окисления.
Оценка редокс-состояния пиридиновых нуклеотидов и окислительного фосфорилирования митохондрий. Редокс-состояние пиридиновых нуклеотидов и окислительное фосфорилирование в суспензии митохондрий определяли по флуоресценции пиридиновых нуклеотидов (возбуждение при 340 нм, эмиссия при 460 нм), регистрируемой на флуориметре Hitachi-F700 (Japan), как описано нами ранее [25]. Митохондрии (0.6 мг белка в мл) добавляли в среду, содержащую 125 мМ КCl, 1.5 мМ KH2PO4 и 15 мМ HEPES–трис (рН 7.25), а также глутамат (4 мМ) и малат (2 мМ) в качестве субстратов окисления. Для оценки окислительного фосфорилирования добавляли ADP (100 мкМ). Полное окисление пиридиновых нуклеотидов было индуцировано добавлением разобщителя FCCP (0.5 мкМ).
Определение активности дегидрогеназ по восстановлению метилтиазолилтетразолия (МТТ). МТТ-тест основан на анализе изменений оптической плотности раствора формазана, образующегося при восстановлении МТТ дегидрогеназами [26]. В 2 мл среды инкубации, содержащей 125 мМ KCl, 15 мМ HEPES, pH 7.4, 150 мкМ МТТ и 5 мМ субстрата, добавляли митохондрии (0.5 мг белка/мл) и инкубировали в течение 5 мин. Предварительную инкубацию митохондрий с тестируемыми соединениями проводили в отсутствие субстрата в течение 10 мин, затем добавляли субстрат и МТТ и инкубировали еще 5 мин. После инкубации в каждую пробу добавляли 10 мкл 10% Тритона Х-100 для полного лизиса митохондрий и измеряли оптическую плотность суспензии при 580 нм на спектрофотометре Ocean Optics USB4000. Другие экспериментальные условия указаны в подписях к рисункам.
Статистический анализ. В работе представлены данные 4–5 независимых экспериментов, проведенных на разных препаратах митохондрий. Статистическую достоверность различий между группами определяли с помощью критерия Стьюдента, различие считалось достоверным при p < 0.05.
Все использованные в работе реактивы были от фирмы Sigma (CША). Тестируемые соединения растворяли в воде; рН полученного раствора доводили до 7.25.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Влияние фенольных кислот на Са2+-индуцированную митохондриальную пору. Действие фенольных кислот на функции митохондрий оценивалось по их влиянию на аккумуляцию ионов кальция и индукцию митохондриальной поры (mitochondrial permeability transition pore, MPTP). Рисунок 1 иллюстрирует влияние рН среды инкубации на кальциевую емкость митохондрий в контроле и в присутствии фенольных кислот. Как видно на рис. 1а и 1б, закисление среды инкубации приводило к почти 2-кратному снижению пороговых концентраций ионов кальция, индуцирующих открытие поры в изолированных интактных митохондриях, в то время как защелачивание, напротив, способствовало повышению этих концентраций по сравнению с их значениями при физиологическом рН. Эти результаты согласуются с данными о стимулирующем влиянии ацидоза на индукцию митохондриальной поры. Как было показано, закисление среды инкубации активировало открытие поры не только в изолированных митохондриях, но и в клетках [27, 28].
Рис. 1.
Влияние фенольных кислот на индукцию МРТ поры ионами кальция при ацидозе. а, б – Влияние рН среды инкубации на индуцированное последовательными добавками СаСl2 (20 мкМ) открытие митохондриальной поры; в – действие фенилакриловой кислоты (ФАК, 100 мкМ) и циклоспорина (CsA, 2 мкМ) на индукцию МРТР при рН 7.4 и 6.8; г – влияние фенольных кислот (все по 100 мкМ) на кальциевую емкость митохондрий при рН 7.4 и рН 6.8. Все измерения проводились кальций-селективным электродом. Митохондрии (1 мг белка/мл) были добавлены в среду измерения: 120 мМ KCl, 15 мМ HEPES, 1.5 мМ фосфата, 5 мМ сукцината, 2 мкМ ротенона (рН 7.25). На панелях (а, в) приведены данные типичных экспериментов, на панелях (б, г) – средние значения ± SEM, полученные из 4–5 независимых экспериментов. Звездочками (*) показаны величины, отличающиеся достоверно от контрольных значений (p < 0.05).
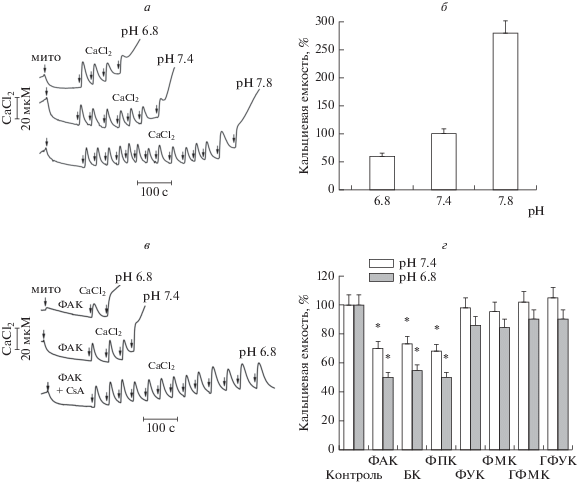
В присутствии 100 мкМ фенилакриловой кислоты кальциевая емкость снижалась в 1.5 и 2 раза при рН 7.4 и рН 6.8 соответственно (рис. 1в). Циклоспорин А (CsA) восстанавливал эти значения до исходных как в контроле, так и в присутствии фенилакриловой кислоты. На рис. 1г показано влияние каждой из исследованных фенольных кислот на кальциевую емкость в норме и при закислении среды инкубации. Результаты показывают, что бензойная (БК), фенилакриловая (ФАК) и фенилпропионовая (ФПК) кислоты снижают кальциевую емкость, в то время как другие фенольные кислоты – фенилмолочная (ФМК), гидроксифенилмолочная (ГФМК) и гидроксифенилуксусная (ГФУК) практически не влияют на этот показатель как в норме, так и при ацидозе. При закислении среды эффект БК, ФАК и ФПК почти в 2 раза выше, чем при рН 7.4 при одних и тех же концентрациях фенольных кислот.
Влияние фенольных кислот на редокс-состояние пиридиновых нуклеотидов и окислительное фосфорилирование в изолированных митохондриях. На рис. 2 показано влияние фенольных кислот на окислительное фосфорилирование, которое оценивали по изменению флуоресценции пиридиновых нуклеотидов в ответ на добавку ADP при физиологическом значении (рН 7.4) и при закислении среды инкубации (рН 6.8). При добавлении ФАК, БК или ФПК наблюдалось уменьшение флуоресценции пиридиновых нуклеотидов, соответствующее снижению их редокс-состояния, и неполное восстановление исходной флуоресценции после фосфорилирования ADP (рис. 2, а2). Добавление разобщителя (FCCP) приводило к полному окислению пиридиновых нуклеотидов. Время фосфорилирования в присутствии этих соединений также увеличивалось по сравнению с контролем и особенно сильно, в несколько раз, при рН 6.8. Скорости окисления пиридиновых нуклеотидов после добавления фенольных кислот (рис. 2, б2) и скорости восстановления пиридиновых нуклеотидов, окисленных после фосфорилирования ADP (рис. 2, в2), также значительно снижались. Еще более выраженный эффект фенольные кислоты оказывали на восстановление пиридиновых нуклеотидов, окисленных после фосфорилирования ADP, – при рН 6.8 оно не достигало 20% от исходного уровня. Кроме того, при закислении среды инкубации эти кислоты действовали уже при низких концентрациях, от 10–20 мкМ, как это показано на примере ФАК на рис. 2г и 2д. Другие фенольные кислоты – фенилмолочная, гидроксифенилмолочная и гидроксифенилуксусная – не оказывали заметного влияния на окислительное фосфорилирование и редокс-состояние пиридиновых нуклеотидов.
Рис. 2.
Влияние фенольных кислот на окислительно-восстановительный статус пиридиновых нуклеотидов и окислительное фосфорилирование в митохондриях. Влияние фенольных кислот (100 мкМ) на флуоресценцию пиридиновых нуклеотидов (ПН) в контроле (рН 7.4, левые панели) и при подкислении (рН 6.8, правые панели) в процессе окислительного фосфорилирования. Панели а1 и а2 – оригинальные кривые, где 1 – контроль, 2 – бензойная кислота (БК), 3 – фенилакриловая кислота (ФАК), 4 – фенилпропионовая кислота (ФПК). Панели б1 и б2 – окисление ПН до добавления ADP в % от контроля; панели в1 и в2 – степень восстановления ПН после фосфорилирования ADP (100 мкМ) в % от контроля; панели г и д концентрационная зависимость действия ФАК на окисление/восстановление ПН. Фенольные кислоты (100 мкМ) добавляли к среде, содержащей 120 мМ KCl, 1.5 мМ KH2PO4, 10 мМ HEPES–трис, 4 мМ глутамата и 2 мМ малата в качестве субстратов окисления и 0.6 мг/мл митохондриального белка печени крысы. Приведены данные типичных экспериментов, проведенных не менее чем в четырех повторах на разных препаратах митохондрий. Значения, представленные столбиками, являются средними значениями ± SEM из 4–5 независимых экспериментов. Звездочками (*) показаны величины, отличающиеся достоверно от контрольных значений (p < 0.05).
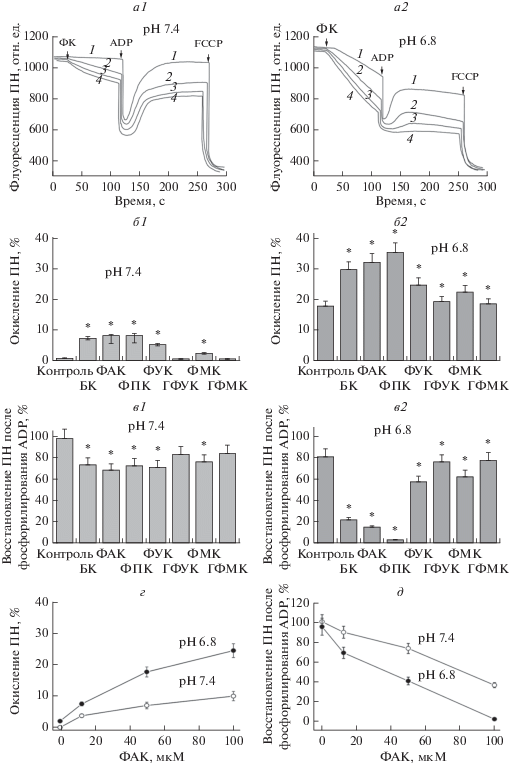
Полученные данные показывают, что в присутствии ФАК, БК и ФПК митохондрии не способны к поддержанию уровня редокс-состояния пиридиновых нуклеотидов и восстановлению пиридиновых нуклеотидов, окисленных после фосфорилирования ADP в процессе окислительного фосфорилирования. Редокс-состояние пиридиновых нуклеотидов в ответ на добавку ADP является интегральным показателем, включающим ряд факторов, в том числе возможное влияние фенольных кислот на активности ATP-азы и NAD-зависимых дегидрогеназ. Представленные данные показывают, что эти соединения снижают уровень восстановленности пиридиновых нуклеотидов и в отсутствие ADP. Можно предположить, что эти нарушения связаны с влиянием фенольных кислот на активность дегидрогеназ. В связи с этим в следующих экспериментах исследовалось влияние фенольных кислот на активность митохондриальных дегидрогеназ.
Влияние фенольных кислот на активность митохондриальных дегидрогеназ. Влияние фенольных кислот на активность митохондриальных дегидрогеназ оценивалось спектрофотометрически по восстановлению акцептора электронов МТТ. На рис. 3а показаны значения оптической плотности восстановленного МТТ после 5-мин инкубации митохондрий с каждой фенольной кислотой при окислении NAD-зависимых субстратов и сукцината. В концентрации 100 мкМ БК, ФАК и ФПК ингибировали восстановление МТТ на 15–20%. Другие фенольные кислоты в такой же концентрации не влияли или слабо активировали восстановление акцептора. Фенольные кислоты в меньшей степени влияли на окисление сукцината, при этом специфичность восстановления МТТ ферментом подтверждалась полным ингибированием восстановления акцептора в присутствии малоната, специфического ингибитора сукцинатдегидрогеназы (СДГ).
Рис. 3.
Влияние фенольных кислот на активность митохондриальных дегидрогеназ. Влияние фенольных кислот (100 мкМ) на восстановление метилтиазолилтетразолия (МТТ) при инкубации с субстратом окисления (5 мМ) – пируватом, глутаматом, α-кетоглутаратом (КГЛ) или сукцинатом (а); после предварительной инкубации без субстрата (б) и с ингибиторами (в). После 5-мин инкубации добавляли Тритон X-100 (10 мкл) и измеряли оптическую плотность препарата. Фенольные кислоты добавляли к среде, содержащей 120 мМ KCl, 1.5 мМ KH2PO4, 10 мМ HEPES, 5 мМ субстрата, 150 мкМ МТТ и 0.5 мг/мл митохондриального белка печени крысы. Ингибиторы: 2 мкМ ротенона (rot), 250 мкМ N-этилмалеимид (NEM), 5 мМ малонатата. Показаны средние значения (столбики) ± SEM из 5–10 независимых экспериментов. Звездочками (*) показаны величины, отличающиеся достоверно от контрольных значений (p < 0.05).
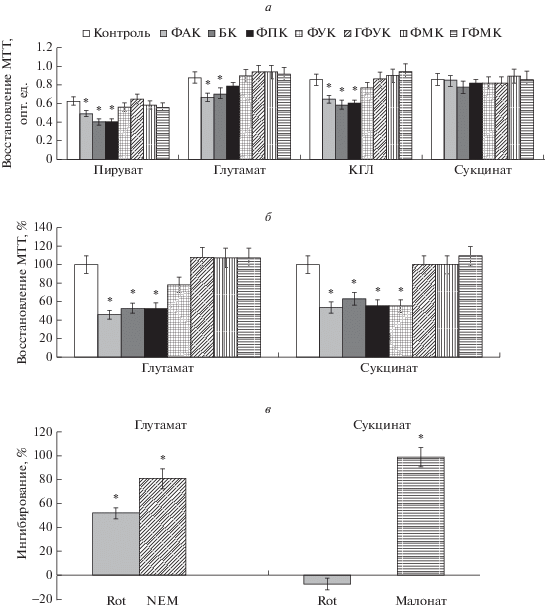
Поскольку субстрат может защищать фермент от ингибирования, как это показано для ряда дегидрогеназ, было исследовано влияние фенольных кислот на восстановление МТТ после предварительной инкубации митохондрий с фенольными кислотами в отсутствие субстрата. На рис. 3б показано влияние предварительной инкубации с фенольными кислотами на восстановление МТТ при последующем добавлении субстрата. Видно, что ингибирование, вызванное инкубацией с ФАК, БК или ФПК, увеличивается более чем в 2 раза по сравнению с ингибированием при стандартной инкубации в присутствии субстрата окисления. Ингибирующий эффект в этих условиях оказывала также ФУК, в то время как другие фенольные кислоты по-прежнему либо не влияли, либо слабо активировали восстановление акцептора. Инкубация митохондрий с фенольными кислотами в отсутствие субстрата сопровождалась также ингибированием СДГ. На рис. 3в показано, что ингибитор первого комплекса дыхательной цепи ротенон снижал восстановление МТТ более чем на 50%; ингибитор NAD-зависимого окисления N-этилмалеимид снижал восстановление МТТ на 80% при окислении глутамата, а ингибитор СДГ малонат ингибировал восстановление МТТ на 100% при окислении сукцината. Эти данные подтверждают зависимость восстановления акцептора от субстрата окисления.
Полученные данные позволяют разделить фенольные кислоты микробного происхождения на две группы: одна с прооксидантным, другая с антиоксидантным действием. К первой группе относятся БК, ФАК, ФПК, ФУК, ко второй группе – ФМК, ГФМК, ГФУК. Прооксидантные фенольные кислоты снижали пороговую концентрацию кальция, индуцирующую открытие MPTP, снижали степень восстановления NAD в процессе окислительного фосфорилирования и оказывали ингибирующий эффект на активность митохондриальных дегидрогеназ. Эффекты этих соединений резко усиливались при закислении среды инкубации, а также после предварительной инкубации в отсутствие субстрата окисления. Антиоксидантные фенольные кислоты либо не влияли на эти параметры, либо восстанавливали их до контрольных значений.
Действие фенольных кислот на открытие поры ионами кальция хорошо согласуются с данными об активации МРТР при индуцированном сепсисе на животных. Индуцированный сепсис, как и фенольные кислоты, снижал пороговые концентрации ионов кальция, требуемые для открытия MPTP [29]. Кроме того, фенольные кислоты снижали эффективность окислительного фосфорилирования и активность митохондриальных дегидрогеназ, что также характерно для митохондриальной дисфункции при сепсисе. Среди факторов, индуцирующих нарушения функций митохондрий при сепсисе, выделяют гипоксию, избыточную продукцию активных форм кислорода, дефицит метаболических субстратов и другие факторы [10]. Наши данные показывают, что митохондриальная дисфункция может быть также связана с гиперпродукцией микробных метаболитов. Известно, что проявление таких признаков дисфункции митохондрий, как снижение активности митохондриальных ферментов и комплексов дыхательной цепи, синтеза ADP, активации продукции активных форм кислорода зависит от стадии воспалительного процесса. В экспериментах на животных было обнаружено, что изменения активности дыхательной цепи происходят в течение первых 24 ч после индукции сепсиса, резервы антиоксидантной защиты истощаются в течение 48 ч, продукция активных форм кислорода митохондриями активируется на начальных стадиях сепсиса [30, 31]. Как следует из полученных нами данных, микробные метаболиты могут вносить существенный вклад в эти процессы, регулируя активность митохондриальных дегидрогеназ и индукцию митохондриальной поры. Этот эффект может усиливаться начиная с ранней стадии, связанной с их гиперпродукцией, и на последующих стадиях, сопровождаемых развитием ацидоза, окислительного стресса и истощением субстратов окисления. В условиях ацидоза действующие концентрации фенольных кислот могут снижаться на порядок, приближаясь к обнаруженным в крови септических больных [20, 21]. Еще больший эффект в усилении их действия оказывал дефицит субстрата окисления. В этом случае ингибированию подвергались не только NAD-зависимые дегидрогеназы, но и сукцинатдегидрогеназа.
Одним из механизмов действия прооксидантных фенольных кислот на митохондрии может быть окисление или связывание тиоловых групп, показанное нами ранее для ФАК [32]. Другой механизм может быть связан с тем, что фенольные кислоты являются субстратами пероксидазы и при окислительном стрессе могут вступать в реакцию с перекисью водорода. При этом образуются такие продукты окисления как феноксил-радикалы, которые могут оказывать токсичное влияние на белки и липиды [33]. Антиоксидантное действие, характерное для фенольных кислот, содержащих гидроксигруппу в бензойном кольце, хорошо согласуется с аналогичным действием 3,4-дигидроксифенилпропионовой и 3,4-дигидроксифенилуксусной кислот, которые снижали окислительный стресс в нейрональных клетках [34]. С другой стороны, антиоксидантное действие этих фенольных кислот по отношению к активированным нейтрофилам, показанное нами ранее [21], может приводить к дисбалансу в иммунном ответе организма, особенно при наличии бактериемии на ранних стадиях воспалительного процесса. Таким образом, микробные метаболиты участвуют в развитии воспалительного процесса и митохондриальной дисфункции, характерной для сепсиса. Как следует из представленных данных, их действие на митохондрии резко усиливается при ацидозе и дефиците субстратов окисления, сопутствующих появлению синдрома полиорганной недостаточности.
Работа выполнена при финансовой поддержке РНФ (проект № 15-15-00110).
Список литературы
Arulkumaran N., Deutschman C.S., Pinsky M.R., Zuckerbraun B., Schumacker P.T., Gomez H., Gomez A., Murray P., Kellum J.A. 2016. Mitochondrial function in sepsis. ADQI XIV Workgroup. Shock. 45, 271–281.
Crouse E.D. 2004. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 4, 729–741.
Lee I., Hüttemann M. 2014. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta. 1842 (9), 1579–1586.
Bhatia M., Moochhala S. 2004. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. 202, 145–156.
Brown K.A., Brain S.D., Pearson J.D., Edgeworth J.D., Lewis S.M., Treacher D.F. 2006. Neutrophils in development of multiple organ failure in sepsis. Lancet. 368, 157–169.
Singer M. 2014. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. 5 (1), 66–72.
Patil N.K., Parajuli N., MacMillan-Crow L.A., Mayeux P.R. 2014. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Renal. Physiol. 306 (7), F734–F743.
Vary T.C., Hazen S. 1999. Sepsis alters pyruvate dehydrogenase kinase activity in skeletal muscle. Mol. Cell Biochem. 198 (1–2), 113–118.
Levy R.J., Deutschman C.S. 2007. Cytochrome c oxidase dysfunction in sepsis. Crit. Care Med. 35, S468–S475.
Puskarich M.A., Kline J.A., Watts J.A., Shirey K., Hosler J., Jones A.E. 2016. Early alterations in platelet mitochondrial function are associated with survival and organ failure in patients with septic shock. J. Crit.Care. 31, 63–67.
Nuzzo E., Berg K.M., Andersen L.W., Balkema J., Montissol S., Cocchi M.N., Liu X., Donnino M.W. 2015. Pyruvate dehydrogenase activity is decreased in the peripheral blood mononuclear cells of patients with sepsis. A prospective observational trial. Ann. Am. Thorac. Soc. 11, 1662–1666. doi 10.1513/AnnalsATS.201505-267BC
Lorente L., Martín M.M., López-Gallardo E., Blanquer J., Solé-Violán J., Labarta L., Díaz C., Jiménez A., Montoya J., Ruiz-Pesini E. 2015. Decrease of oxidative phosphorylation system function in severe septic patients. J. Crit. Care. 30 (5), 935–939.
Wang H.X., Wang Y.P. 2016. Gut microbiota–brain axis. Chin. Med. J. (Engl.). 129 (19), 2373–2380. doi 10.4103/0366-6999.190667
Zhang L.S., Davies S.S. 2016. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 8 (1), 46. doi 10.1186/s13073-016-0296-x
Zhang H., Du M., Yang Q., Zhu M.J. 2015. Butyrate suppresses murine mast cell proliferation and cytokine production through inhibiting histone deacetylase. J. Nutr. Biochem. 27, 299–306. doi 10.1016/j.jnutbio. 2015.09.020
Waldecker M., Kautenburger T., Daumann H., Busch C., Schrenk D. 2008. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 19 (9), 587–593.
Mutsaers H.A., Wilmer M.J., Reijnders D., Jansen J., van den Broek P.H., Forkink M., Schepers E., Glorieux G., Vanholder R., van den Heuvel L.P., Hoenderop J.G., Masereeuw R. 2013. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Biophys. Acta. 1832 (1), 142–150. doi 10.1016/j.bbadis.2012.09.006
Schmidt S., Westhoff T.H., Krauser P., Zidek W., van der Giet M. 2008. The uraemic toxin phenylacetic acid increases the formation of reactive oxygen species in vascular smooth muscle cells. Neprol. Dial. Transplant. 23, 65–71.
O’Neill L.A. 2016. A metabolic road block in inflammatory macrophages. Cell Rep. 17 (3), 625–626.
Белобородова Н.В., Байрамов И.Т., Оленин А.Ю., Федотчева Н.И. 2011. Экзометаболиты некоторых анаэробных микроорганизмов микрофлоры человека. Биомед. химия. 57 (1), 95–105.
Beloborodova N.V., Bairamov I.T., Olenin A.Yu., Shubina V., Teplova V.V., Fedotcheva N.I. 2012. Effect of phenolic acids of microbial origin on production of reactive oxygen species in mitochondria and neutrophils. J. Biomed. Sci. 19, 89. doi 10.1186/1423-0127-19-89
Федотчева Н.И., Теплова В.В., Белобородова Н.В. 2010. Участие фенольных кислот микробного происхождения в дисфункции митохондрий при сепсисе. Биол. мембраны. 27 (1), 60–66.
Fedotcheva N.I., Teplova V.V., Fedotcheva T.A., Rzheznikov V.M., Shimanovskii N.L. 2009. Effect of progesterone and its synthetic analogues on the activity of mitochondrial permeability transition pore in isolated rat liver mitochondria. Biochem. Pharmacol. 78, 1060–1068.
Федотчева Т.А., Теплова В.В., Федотчева Н.И. 2018. Активация кальций-зависимой циклоспорин-чувствительной митохондриальной поры доксорубицином в комплексе с ионами железа. Биол. мембраны. 35 (1), 79–84.
Solomatin A.S., Yakovlev R.Y., Teplova V.V., Nadezhda I., Fedotcheva N.I., Kondrachova M.N., Kulakova I.I., Nikolay B. Leonidov N.B. 2018. Effect of detonation nanodiamond surface composition on physiological indicators of mitochondrial functions. J. Nanopart. Res. 20, 201. https://doi.org/10.1007/s11051-018-4297-0.
Федотчева Н.И., Литвинова Е.Г., Захарченко М.В., Хундерякова Н.В., Фадеев Р.С., Теплова В.В., Федотчева Т.А., Белобородова Н.В., Кондрашова М.Н. 2017. Субстрат-специфичное восстановление солей тетразолия в изолированных митохондриях, тканях и лейкоцитах. Биохимия. 82 (2), 309–322.
Kristian T., Bernardi P., Siesjö B.K. 2001. Acidosis promotes the permeability transition in energized mitochondria: Implications for reperfusion injury. J. Neurotrauma. 18 (10), 1059–1074.
Teixeira J., Basit F., Swarts H.G., Forkink M., Oli-veira P.J., Willems P.H., Koopman W.J. 2018. Extracellular acidification induces ROS- and mPTP-mediated death in HEK293 cells. Redox Biol. 15, 394–404.
Larche J., Lancel S., Hassoun S.M., Favory R., Decoster B., Marchetti P., Chopin C., Neviere R. 2006. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J. Am. Coll. Cardiol. 48, 377–385.
Brealey D., Karyampudi S., Jacques T.S., Novelli M., Stidwill R., Taylor V., Smolenski R.T., Singer M. 2004. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R491–R497.
Eyenga P., Roussel D., Morel J., Rey B., Romestaing C., Teulier L., Sheu S.S., Goudable J., Négrier C., Viale J.P. 2014. Early septic shock induces loss of oxidative phosphorylation yield plasticity in liver mitochondria. J. Physiol. Biochem. 70 (2), 285–296.
Федотчева Н.И., Теплова В.В., Белобоpодова Н.В. 2012. Роль тиоловых антиоксидантов в восстановлении функций митохондрий, модифицированных микробными метаболитами. Биофизика. 57 (5), 820–826.
Kagan V.E., Tyurina Y.Y. 1998. Recycling and redox cycling of phenolic antioxidants. Ann. N.Y. Acad. Sci. 854, 425–434.
González-Sarrías A., Núñez-Sánchez M.Á., Tomás-Barberán F.A., Espín J.C. 2017. Neuroprotective effects of bioavailable polyphenol-derived metabolites against oxidative stress-induced cytotoxicity in human neuroblastoma SH-SY5Y Cells. J. Agric. Food Chem. 65 (4), 752–758.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии