

Биологические мембраны: Журнал мембранной и клеточной биологии, 2019, T. 36, № 2, стр. 109-114
2-APB стимулирует выброс Са2+ из Са2+-депо
Д. С. Ивашин a, О. А. Рогачевская a, М. Ф. Быстрова a, С. С. Колесников a, *
a Институт биофизики клетки РАН,
142290 Московская обл., Пущино, ул. Институтская, 3, Россия
* E-mail: staskolesnikov@yahoo.com
Поступила в редакцию 26.10.2018
После доработки 20.11.2018
Принята к публикации 30.11.2018
Аннотация
С помощью микрофотометрии (Сa2+ imaging), генетически кодируемого Са2+-сенсора G-CEPIA1er и Са2+-зонда Fura-2 анализировались Cа2+-сигналы, инициируемые различными стимулами в цитоплазме и эндоплазматическом ретикулуме (ЭР) клеток HEK-293. Показано, что 2-APB, являющийся блокатором SOC-каналов и антагонистом IP3-рецепторов, инициировал выброс Са2+ из ЭР. В присутствии тапсигаргина, ингибировавшего ретикулярную Са2+-ATP-азу SERCA и частично опустошавшего Са2+-депо, 2-APB не вызывал выброса депонированного Са2+. Мы предполагаем, что эти эффекты можно объяснить способностью 2-APB стимулировать ретикулярные Са2+-каналы CLAC, которые активируются при высокой концентрации Са2+ в люмене ЭР и предотвращают перегрузку Са2+-депо.
ВВЕДЕНИЕ
Клеточные функции регулируются внутриклеточным Са2+, уровень и динамика которого контролируются рядом внутриклеточных транспортных и сигнальных систем. Одним из важнейших является фосфоинозитидный путь, который сопрягает многие гептаспиральные (G-protein-coupled receptor, GPCR) и некоторые тирозинкиназные рецепторы с мобилизацией внутриклеточного Са2+ [1]. Это сопряжение протекает в несколько стадий, включая агонист-зависимую активацию фосфолипазы С (PLC), катализирующей гидролиз фосфолипида PIP2 (phosphatidylinositol 4,5-bisphosphate), в результате чего продуцируются два вторичных медиатора: водорастворимый инозитолтрисфосфат (IP3, inositol 1,4,5-trisphosphate) и липидный диацилглицерин (DAG, diacylglycerol). В свою очередь, IP3 стимулирует IP3-рецепторы, являющиеся внутриклеточными IP3-активируемыми Са2+-каналами, функционирующими в эндоплазматическом ретикулуме и ответственными за высвобождение депонированного Са2+ [1, 2]. Этот первоначальный агонист-зависимый Са2+-сигнал может инициировать так называемый Са2+-индуцированный выброс Са2+ (Са2+-induced Са2+ release, CICR), который играет ключевую роль в превращении локальных Са2+-сигналов в глобальные, а также в распространении Са2+-волн в цитоплазме клеток [3–5]. Регенеративный процесс CICR происходит за счет того, что в определенной области концентраций внутриклеточный Са2+ стимулирует IP3-рецепторы [2] и/или рианодиновые рецепторы, которые также являются внутриклеточными Са2+-каналами, обеспечивающими выброс Са2+ из Са2+-депо [6].
Существенный прогресс в исследовании внутриклеточных сигнальных процессов был достигнут в значительной степени благодаря ингибиторному анализу, который опирается на широкий спектр соединений, способных проникать в клетки и избирательно модулировать активность сигнальных белков. Например, рианодиновые рецепторы активируются кофеином, но ингибируются рианодином, что позволяет оценивать их вклад в генерацию внутриклеточных Са2+-сигналов [6, 7]. Фармакологический инструментарий для функциональных исследований IP3-рецепторов менее эффективен. Среди применяемых агонистов заслуживает внимания лишь проникающий предшественник IP3 (Bt3-Ins(145)P3/AM), хотя индуцируемый им подъем уровня внутриклеточного IP3 и его динамика сильно зависят от скорости гидролиза эфирной группы внутриклеточными эстеразами и от интенсивности метаболизма IP3 в данной клетке. Имеется несколько соединений, которые считаются антагонистами IP3-рецепторов: гепарин, ксестоспонгины (xestospongin С, xestospongin D) и 2-APB (2-amino-ethoxydiphenyl borate) [8–10]. Их использование в качестве ингибиторов IP3-зависимого выброса Са2+ связано с рядом проблем. Так, гепарин не проникает через мембрану, и его быстрая доставка в цитоплазму клетки может быть осуществлена либо с помощью инъекции, либо путем пермеабилизации (permeabilization) с использованием детергентов или токсинов, формирующих поры в плазматической мембране [11]. Существенный недостаток этого подхода состоит в том, что условия, при которых поры образуются преимущественно в плазматической мембране, но не во внутриклеточных органеллах, подобрать трудно.
Хотя в ранних работах утверждалось, что ксестоспонгины ингибируют IP3-зависимый выброс Са2+ [9], в дальнейшем прямое ингибирование IP3-рецепторов ксестоспонгинами было поставлено под сомнение (например, [12]). Более того, в недавней работе с использованием пермеабилизованных клеток было показано, что ксестоспонгины С и D не являются ингибиторами IP3-рецепторов всех трех типов [11]. В пионерской работе Мариямы и соавторов [10] было показано, что 2-APB является проникающим через мембрану соединением, способным ингибировать IP3-индуцируемый выброс Са2+. Оказалось, что в этих эффектах основной мишенью 2-APB являлся IP3-рецептор типа 1 [11]. Между тем ряд работ указывает на то, что действие 2-APB на внутриклеточную Са2+-сигнализацию может быть опосредовано и иными механизмами. К ним относятся блокирование входа наружного Са2+ через потенциал-независимые Сa2+-проницаемые каналы, включая депо-управляемые каналы (SOC, store-operated channel) [13–15], ингибирование ретикулярной Са2+-ATP-азы SERCA [16] и активация Са2+-проницаемых каналов TRPV1, TRPV2 и TRPV3 [17].
Обычно анализ роли различных внутриклеточных сигнальных и транспортных систем в генерации Са2+-сигналов основан на мониторинге цитозольного Са2+ с использованием высокоаффинных флуоресцентных Са2+-зондов (Fura-2, Fluo-4) в сочетании с ингибиторным анализом. В силу недостаточной специфичности многих ингибиторов, отмеченной выше, трудно однозначно интерпретировать результаты ингибиторного анализа, основываясь лишь на одном измеряемом параметре – концентрации свободного Са2+ в цитозоле клетки. Это ставит задачу одновременного мониторинга Са2+ и в других компартментах, вовлеченных в Са2+-обмен, таких как эндоплазматический/саркоплазматический ретикулум, митохондрии и лизосомы. Наиболее эффективно эта задача решается с использованием генетически кодируемых Са2+-зондов на основе флуоресцентных белков, содержащих в своей последовательности сигнал локализации в том или ином компартменте [18]. В настоящей работе мы использовали генетически кодируемый низкоаффинный (Kd = 672 мкМ) Са2+-индикатор G-CEPIA1er [19] для мониторинга Са2+ в эндоплазматическом ретикулуме (ЭР) клеток HEK-293.
МАТЕРИАЛЫ И МЕТОДЫ
В работе использовались клетки линии HEK-293, которые культивировались во влажной атмосфере с 5% СО2 при 37°С в планшете с 12 лунками с 1 мл среды DMEM с добавлением 10% эмбриональной бычьей сыворотки в каждой. Клетки трансфицировались плазмидным вектором pCMV-G-CEPIA1er (Addgene). Для временной трансфекции в лунку добавляли 1 мкг pCMV-G-CEPIA1er и 2 мкл липофектамина 3000 (Thermo-Fisher). После инкубации клеток в течение 5–6 ч при 37°С трансфекционная среда замещалась на обычную культуральную среду. Регистрации проводились через 24–72 ч после трансфекции. Качество трансфекции оценивалось по интенсивности и распределению эмиссии флуоресцентного белка GFP, являющегося индикаторной частью сенсора G-CEPIA1er.
Для фотометрических экспериментов клетки инкубировали при комнатной температуре (23–25°C) в присутствии проникающего агента Fura-2 AM (4 мкМ) и детергента Pluronic (0.02%) (оба из Molecular Probes) в течение 20–30 мин, что обеспечивало загрузку клеток Са2+-зондом Fura-2. Внеклеточный раствор содержал (мМ): NaCl – 140, KCl – 5, CaCl2 – 2, MgSO4 – 1, HEPES – 10, глюкоза – 10. В случае необходимости 2 мМ CaCl2 заменялся на 1 мМ EGTA + 0.8 мМ CaCl2, чтобы снизить концентрацию свободного Са2+ во внеклеточном растворе до 250 нМ. Фотометрические эксперименты проводили с использованием инвертированного флуоресцентного микроскопа Axiovert 200 (Carl Zeiss), оборудованного объективом Plan NeoFluar 20×/0.75 и цифровой EMCCD камерой iXon 888 (Andor Technology). Флуоресценцию Са2+-индикаторов возбуждали при длинах волн 340 ± 5 и 380 ± 5 нм в случае Fura-2 и 480 ± 5 нм в случае G-CEPIA1er. Эмиссию обоих зондов регистрировали в области 525 ± 20 нм. Изменение свободного Са2+ в цитозоле и в ЭР индивидуальных клеток оценивали по отношению F340/F380 и ΔF/F0 соответственно, где F340 и F380 – интенсивности эмиссии Fura-2 при возбуждении при 340 и 380 нм соответственно, а ΔF = F0 – F, где F и F0 – текущая интенсивность эмиссии G-CEPIA1er и его эмиссия в начале регистрации, соответственно. Количественный фотометрический анализ изображений проводили с использованием программы Imaging Workbench 6 (INDEC).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Микрофотометрия клеток HEK-293, экспрессирующих Са2+-сенсор G-CEPIA1er и нагруженных Са2+-зондом Fura-2, позволила сопоставить динамику свободного Са2+ в цитозоле и ЭР индивидуальных клеток в различных условиях. Одна из типичных регистраций представлена на рис. 1. Поскольку в клетках HEK-293 функционируют GPCR-рецепторы нуклеотидов P2Y-типа, сопряженные с фосфоинозитидным каскадом, кратковременная аппликация 2–5 мкМ АТР инициировала импульс Са2+ в цитозоле клеток (рис. 1, верхняя кривая) и синхронное падение и восстановление уровня ретикулярного Са2+ (рис. 1, нижняя кривая). Такое согласованное поведение Са2+ в цитоплазме и ЭР было вполне ожидаемо, поскольку многие агонисты GPCR-рецепторов, включая АТР, стимулируют выброс Са2+, депонированного в ЭР [1].
Рис. 1.
Репрезентативная регистрация цитоплазматического и ретикулярного Са2+ в одиночной клетке линии HEK-293 (n = 51). Верхняя панель – мониторинг цитоплазматического Са2+ по флуоресценции Са2+-зонда Fura-2. Данные представлены как отношение F340/F380, где F340 и F380 – интенсивности эмиссии Fura-2 при возбуждении на 340 и 380 нм. Нижняя панель – одновременный мониторинг Са2+ в ЭР по флуоресценции Са2+-сенсора G-CEPIA1er. Данные представлены в виде ΔF/F0, где ΔF = F0 – F, F и F0 – текущая интенсивность эмиссии G-CEPIA1er и эмиссия в начале регистрации, соответственно. Моменты аппликации 2 мкМ ATP, 2 мкМ тапсигаргина и 50 мкМ 2-APB обозначены на верхней панели линиями над экспериментальной кривой.
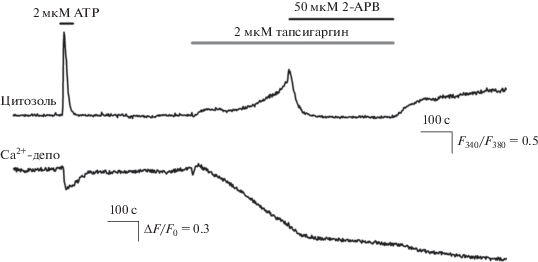
Выброс депонированного Са2+ приводит к истощению Са2+-депо, которое пополняется Са2+-ATP-азой SERCA (sarco-endoplasmic reticulum Ca2+ ATPase), перекачивающей Са2+ из цитоплазмы в ЭР, а также за счет входа наружного Са2+ через SOC-каналы (store-operated channels), активируемые при опустошении Са2+-депо [20, 21]. Сообразно с этой универсальной закономерностью, тапсигаргин (thapsigargin) (2 мкМ), эффективный игибитор ретикулярной Са2+-ATP-азы [20], вызывал постепенное падение Са2+ в ЭР (рис. 1, нижняя кривая) за счет утечки депонированного Са2+, которая обеспечивается специализированными ионными каналами [22] и, возможно, фоновой активностью IP3-рецепторов. Опустошение Са2+-депо ожидаемо сопровождалось активацией SOC-каналов и увеличением входа наружного Са2+, что приводило к росту концентрации Са2+ в цитоплазме (рис. 1, верхняя кривая). Аппликация 2-APB (50 мкМ), блокирующего SOC-каналы, возвращала цитозольный Са2+ практически к уровню покоя, очевидно, за счет активности Са2+-насосов плазматической мембраны. Последующее удаление 2-APB приводило к деблокированию SOC-каналов, увеличению входа наружного Са2+ и, соответственно, росту цитозольного Са2+ (рис. 1, верхняя кривая). Следует отметить, что концентрация полуингибирования различных изоформ SERCA тапсигаргином лежит в области низких наномолярных концентраций [23]. Учитывая это обстоятельство, можно было ожидать, что тапсигаргин при внеклеточной концентрации 2 мкМ должен был полностью подавить активность ретикулярной Са2+-ATP-азы. Поэтому существенное уменьшение скорости падения депонированного Са2+, инициированное 2-APB на фоне тапсигаргина (рис. 1, нижняя кривая), можно было объяснить тем, что это соединение блокировало Са2+-каналы утечки и ингибировало фоновую активность IP3-рецепторов.
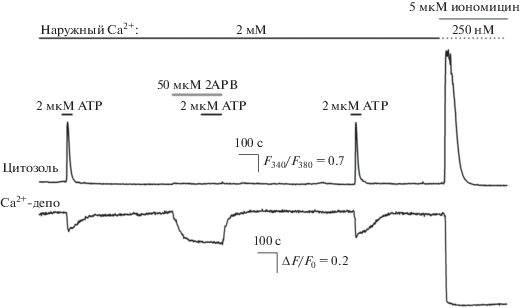
Это предположение, однако, противоречило данным экспериментов, в которых анализировалось действие 2-APB (50 мкМ) на Са2+-ответы клеток, инициированные ATP (2 мкМ). Оказалось, что клетки, в которых ATP индуцировал импульсы цитозольного Са2+, обратимо теряли чувствительность к АТР в присутствии 2-APB (50 мкМ) (рис. 2, верхняя кривая). Этот эффект 2-APB обычно интерпретируется как свидетельство блокады IP3-рецепторов. Между тем, мониторинг депонированного Са2+ указывал на то, что 2-APB также вызывал обратимое падение депонированного Са2+ (рис. 2, нижняя кривая). 2-APB опустошал Са2+-депо на 25–30%, если принять за ноль уровень Са2+, который достигался в ЭР после аппликации Са2+-ионофора иономицина (5 мкМ). Последний быстро проникал в клетку и опустошал ЭР, уровень Са2+ в котором был существенно выше, чем в цитоплазме (рис. 2, нижняя кривая). Поэтому подавление клеточных ответов на ATP в присутствии 2-APB не могло быть связано со слишком низким уровнем депонированного Са2+ и несомненно было обусловлено вызванным 2-APB блокированием IP3-рецепторов. В то же время быстрое падение Са2+ в ЭР (рис. 2, нижняя кривая) свидетельствовало о том, что 2-APB активировал утечку Са2+, что трудно согласовать с действием, оказываемым этим ингибитором на фоне тапсигаргина (рис. 1, нижняя кривая).
Рис. 2.
2-APB стимулирует выброс депонированного Са2+ в клетках HEK-293. Верхняя панель – репрезентативная регистрация (44 клетки) цитоплазматического Са2+ при стимуляции клетки ATP (2 мкМ) и 2-APB (50 мкМ) при 2 мМ Са2+ в экстраклеточном растворе. Перед аппликацией иономицина наружный Са2+ снижался до 250 нМ. Нижняя панель – одновременный мониторинг Са2+ в ЭР.
Уровень Са2+ в ЭР определяется балансом между поступлением Са2+, обеспечиваемым Са2+ насосом SERCA, и выходом Са2+ в цитозоль через различные Са2+-проницаемые каналы, функционирующие в ЭР. Среди таковых IP3- и рианодиновый рецепторы, которые обеспечивают быстрый выброс Са2+ и генерацию Са2+-сигналов [1, 3]. Другие Са2+-проницаемые каналы выполняют функцию регулируемой Ca2+-утечки [22]. Среди них следует отметить ретикулярные Са2+-каналы с функциональным названием CLAC (Ca2+ load-activated Ca2+ channel), которые формируются канальными субъединицами TMCO1 (transmembrane and coiled-coil domains 1). CLAC-каналы активируются за счет тетрамеризации субъединиц TMCO1, которая иницируется при высокой концентрации Са2+ в люмене ЭР. Это рассматривается как механизм, предотвращающий перегрузку Са2+-депо [24]. Если предположить, что 2-APB способен активировать CLAC-каналы, понижая аффинность TMCO1 к свободному Са2+ в люмене ЭР, то можно объяснить различие в эффектах 2-APB, наблюдавшееся нами в различных условиях (рис. 1 и 2). Действительно, при длительной аппликации тапсигаргина ЭР опустошался (рис. 1, нижняя кривая), и 2-APB был не способен стимулировать CLAC-каналы из-за низкой концентрации Са2+ в люмене ЭР. В условиях, при которых уровень Са2+ в ЭР был высок, 2-APB инициировал выброс ретикулярного Са2+, повышая активность CLAC-каналов (рис. 2, нижняя кривая). Этот эффект 2-APB был обратим за счет восполнения Са2+ в ЭР активной SERCA. Доказательство функциональности предполагаемого механизма требует отдельных экспериментов непосредственно с CLAC-каналами.
Работа поддержана Российским научным фондом (грант 18-14-00347).
Список литературы
Berridge M.J. 2016. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296.
Mikoshiba K. 2015. Role of IP3 receptor signaling in cell functions and diseases. Adv. Biolog. Reg. 57, 217–227.
Berridge M.J., Bootman M.D., Roderick H.L. 2003. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529.
Zhang S., Fritz N., Ibarra C., Uhlen P. 2011. Inositol 1,4,5-trisphosphate receptor subtype-specific regulation of calcium oscillations. Neurochem. Res. 36, 1175–1185.
Iino M. 2010. Spatiotemporal dynamics of Ca2+ signaling and its physiological roles. Proc. Japan Acad. Ser. B. Phys. Biol. Sci. 86, 244–256
Thomas N.L., Williams A.J. 2012. Pharmacology of ryanodine receptors and Ca2+-induced Ca2+ release. Wiley Interdiscipl. Rev. Membr. Transp. Signal. 1, 383–397.
Kong H., Jones P.P. Koop A., Zhang L., Duff H.J., Chen W.S.R. 2008. Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem J. 414, 441–452.
Ghosh T.K., Eis P.S., Mullaney J.M., Ebert C.L., Gill D.L. 1988. Competitive, reversible, and potent antagonism of inositol 1,4,5-trisphosphate-activated calcium release by heparin. J. Biol. Chem. 263, 11 075–11 079.
Gafni J., Munsch J.A., Lam T.H., Catlin M.C., Costa L.G., Molinski T.F., Pessah I.N. 1997. Xestospongins: Potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 19, 723–733.
Maruyama T., Kanaji T., Nakade S., Kanno T., Mikoshiba K. 1997. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. Jpn. J. Biochem. 122, 498–505.
Saleem H., Tovey S.C., Molinski T.F., Taylor C.W. 2014. Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br. J. Pharm. 171, 3298–3312.
Solovyova N., Fernyhough P., Glazner G., Verkhratsky A. 2002. Xestospongin C empties the ER calcium store but does not inhibit InsP3-induced Ca2+ release in cultured dorsal root ganglia neurones. Cell Calcium 32, 49–52.
Ma H.-T., Venkatachalam K., Li H.-S., Montell C., Kurosaki T., Patterson R.L., Gill D.L. 2001. Assessment of the role of the inositol 1,4,5-trisphosphate receptor in the activation of transient receptor potential channels a and store-operated Ca2+ entry. J. Biol. Chem. 276, 18888–18896.
Gregory R.B., Rychkov G., Barritt G.J. 2001. Evidence that 2-aminoethoxydiphenyl borate is a novel inhibitor of store operated Ca2+ channels in liver cells, and acts through a mechanism which does not involve inositol trisphosphate receptors. Biochem. J. 354, 285–290.
Xu S.-Z., Zeng F., Boulay G., Grimm C., Harteneck C., Beech D.J. 2005. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: A differential, extracellular and voltage-dependent effect. Br. J. Pharm. 145, 405–414.
Missiaen L., Callewaert G., De Smedt H., Parys J.B. 2001. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific leak from the non-mitochondrial Ca2+ stores in permeabilised A7r5 cells. Cell Calcium. 29, 111–116.
Hu H.-Z., Gu Q., Wang C., Colton C.K., Tang J., Kinoshita-Kawada M., Lee L.Y., Wood J.D., Zhu M.X. 2004. 2-Aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 279, 35741–35748.
Kwon S.-K., Hirabayashi Y., Polleux F. 2016. Organelle-specific sensors for monitoring Ca2+ dynamics in neurons. Front. Syn. Neurosci. 8, 29.
Suzuki J., Kanemaru K., Ishii K., Ohkura M., Okubo Y., Iino M. 2014. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 5, 4153.
Tadini-Buoninsegni F., Smeazzetto S., Gualdani R., Moncelli M.R. 2018. Drug interactions with the Ca2+-ATPase from sarco(endo)plasmic reticulum (SERCA). Front. Mol. Biosci. 5, 36.
Lopez J.J., Albarran L., Gómez L.J., Smani T., Salido G.M., Rosado J.A. 2016. Molecular modulators of store-operated calcium entry. Biochim. Biophys. Acta. 1863, 2037–2043.
Carreras-Sureda A., Pihán P., Hetz C. 2018. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium. 70, 24–31.
Treiman M., Caspersen C., Christensen S.B. 1998. A tool coming of age: Thapsigargin as an inhibitor of sarcoendoplasmic reticulum Ca2+-ATPases. Trends Pharm. Sci. 19, 131–135.
Wang Q.C., Zheng Q., Tan H., Zhang B., Li X., Yang Y., Yu J., Liu Y., Chai H., Wang X., Sun Z., Wang J.-Q., Zhu S., Wang F.i, Yang M., Guo C., Wang H., Zheng Q., Li Y., Chen Q., Zhou A., Tang T.-S. 2016. TMCO1 is an ER Ca2+ load-activated Ca2+ channel. Cell. 165, 1454–1466.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии