

Биологические мембраны: Журнал мембранной и клеточной биологии, 2019, T. 36, № 6, стр. 400-408
Оценка влияния нокаута гена β-аррестина-2 у мышей на выживаемость культивируемых астроцитов при токсическом действии тромбина и последствия тромбоза головного мозга
М. Д. Галков a, b, *, А. Е. Иванова b, М. В. Гуляев a, Е. В. Киселева c, И. Г. Савинкова b, Л. Р. Горбачева a, b
a Московский государственный университет им. М.В. Ломоносова
119234 Москва, Ленинские горы, 1, Россия
b Российский национальный исследовательский медицинский университет
им. Н.И. Пирогова
117997 Москва, ул. Островитянова, 1, Россия
c Институт биологии развития им. Н.К. Кольцова РАН
119334 Москва, ул. Вавилова, 26, Россия
* E-mail: galkovm@yandex.ru
Поступила в редакцию 17.04.2019
После доработки 04.07.2019
Принята к публикации 05.07.2019
Аннотация
Полифункциональная сериновая протеаза тромбин привлекает внимание многих исследователей, в том числе в связи с широким спектром ее эффектов, реализующихся в нервной ткани. Основным рецептором тромбина является рецептор, активируемый протеазами, типа 1, функциональная активность которого сопряжена с адаптерным белком β-аррестином-2. С использованием мышей с нокаутом гена β-аррестина-2 нами оценено возможное участие данного белка в токсическом действии тромбина in vitro и in vivo. Установлено, что тромбин вызывает дозозависимую гибель культивируемых астроцитов, полученных из мозга мышей с нокаутом гена β-аррестина-2, через 48 ч после воздействия. Вместе с тем не обнаружено влияния тромбина в используемых концентрациях на выживаемость астроцитов животных дикого типа. Согласно результатам экспериментов in vivo нокаут целевого гена не влиял на тяжесть последствий фокальной фотоиндуцированной ишемии головного мозга, что может быть связано с вовлечением в повреждение нервной ткани дополнительных типов клеток и молекулярных механизмов. Таким образом, нами впервые показана необходимость β-аррестина-2 для выживания культивируемых астроцитов мыши при токсическом действии тромбина. В то же время требуются дальнейшие исследования на уровне ЦНС в целом для уточнения ключевых мишеней тромбина в каждом из клеточных компонентов нервной ткани и роли сигнализации β-аррестина.
ВВЕДЕНИЕ
Тромбин – важнейшая сериновая протеаза системы гемостаза, локальное повышение концентрации которой в нервной ткани наблюдается при тромбозах различной этиологии вследствие повышения проницаемости гематоэнцефалического барьера (ГЭБ) [1, 2]. Эта протеаза не только участвует в тромбообразовании, но и вовлечена в регуляцию ряда сопровождающих нарушения мозгового кровообращения физиологических процессов таких, как воспалительные реакции, изменение сосудистой проницаемости и гибель нейронов [1–3]. Эффекты тромбина реализуются через рецепторы, активируемые протеазами (ПАР), прежде всего, ПАР1 [3–6]. При этом ПАР, будучи рецепторами, сопряженными с G-белками (GPCRs), являются объектом регуляции многофункциональных адаптерных белков – β-аррестинов [3, 5–8].
Тромбин активирует ПАР1 посредством расщепления его N-конца в специфическом сайте (Арг41-Сер42), что приводит к формированию “привязанного” лиганда, взаимодействующего со второй внеклеточной петлей рецептора, с последующим запуском Gq- или G12/13-белокзависимых каскадов. В результате этого наблюдается опосредованное RhoA нарушение барьерных свойств эндотелия, экспонирование адгезивных молекул на поверхности эндотелиоцитов, усиление пролиферативной и секреторной активности глиальных клеток, гибель нейронов [3–8]. В этом случае β-аррестины, связываясь с ПАР1, чьи специфические сайты внутриклеточных петель фосфорилированы киназами GPCRs (GRKs), препятствуют дальнейшей активации G-белков, инициируя десенситизацию и интернализацию рецептора (рис. 1а) [5, 6].
Рис. 1.
Различные внутриклеточные сигнальные пути тромбина, запускаемые при активации ПАР1, и участие в них β‑аррестинов. а – Классический сигнальный путь, запускаемый тромбином через G-белки ([3] с изменениями); б – сигнальный путь, запускаемый тромбином опосредованно через активированный протеин С (АПС) ([3] с изменениями); в – непосредственная активация β-аррестиновой сигнализации тромбином ([7] с изменениями) (GRKs – киназы рецепторов, сопряженных с G-белками, ПКС – протеинкиназа С, ТМ – тромбомодулин, ПС – протеин С, ЭРПС – эндотелиальный рецептор протеина С, КАВ1 – кавеолин типа 1, Dvl-2 – регуляторный фосфопротеин Dishevelled-2, RhoA и Rac1 – GTP-азы).
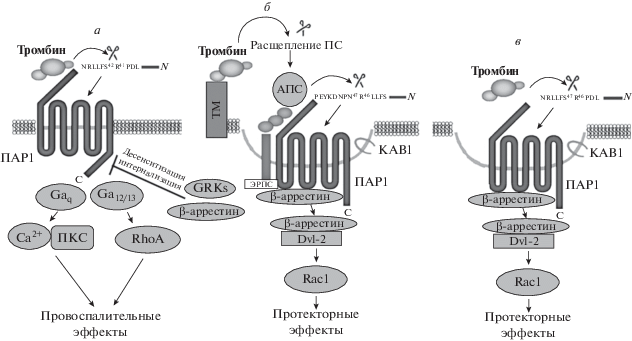
Помимо этого, тромбин обладает и цитопротекторными свойствами, которые не зависят от G-белков и связаны, прежде всего, со способностью протеазы расщеплять протеин С (ПС) с образованием его активированной формы – АПС. АПС, в свою очередь, в отличие от тромбина протеолитически расщепляет ПАР1 между Арг46 и Асн47, что приводит к связыванию с рецептором β-аррестинов, рекрутирующих ряд эффекторных белков. В результате наблюдается стабилизация эндотелиальных барьеров с вовлечением Rac1, антиапоптотическое действие, подавление нейровоспаления и т.д. В этом случае для АПС-зависимой активации ПАР1 необходима колокализация рецептора с эндотелиальным рецептором ПС и тромбомодулином в кавеоле (рис. 1б) [3, 8]. Кроме того, на основе результатов экспериментов, проведенных на культуре эндотелиоцитов, выдвинута гипотеза о том, что и сам тромбин, непосредственно активируя ПАР1, расположенный в кавеоле, может запускать схожие цитопротекторные сигнальные пути с вовлечением β-аррестинов (рис. 1в) [7].
Следовательно, возможно инициирование разнонаправленных внутриклеточных сигналов посредством активации ПАР1 как через G-белки, так и через β-аррестины. Подобный сигнальный дуализм, названный “смещенным агонизмом”, определяется характером “молекулярного ансамбля”, в котором пребывает рецептор, и паттерном его фосфорилирования GRKs [9–12]. Считается, что большинство GPCRs регулируются двумя представителями семейства β-аррестинов – β-аррестином-1 и -2 [11].
Вместе с тем роль β-аррестина-2 в развитии тромбин-индуцированных повреждений нервной ткани не выяснена. В связи с этим актуально исследование данного вопроса как в условиях in vitro с использованием первичных культур астроцитов – важнейшего компонента нервной ткани [13], так и in vivo в модели фотоиндуцированной ишемии головного мозга.
МАТЕРИАЛЫ И МЕТОДЫ
Животные. В экспериментах использовали линию мышей с нокаутом гена β-аррестина-2 (группа β-аррестин-2–/–) [14], любезно предоставленных Р.Р. Гайнетдиновым. В качестве контрольной группы использовали животных генетически родственной линии С57BL/6 с полноценной экспрессией данного белка (дикий тип). Все эксперименты выполнены согласно Директиве 2010/63/EU Европейского Парламента и Совета Европейского Союза.
Генотипирование животных. Нокаут гена β-аррестина-2 подтверждали методом полимеразной цепной реакции (ПЦР). Геномную ДНК выделяли из лизатов фрагментов хвоста мышей обеих групп с использованием набора Extract DNA (Евроген, Россия). ПЦР проводили с использованием Taq-полимераз, стандартного набора реагентов (Thermo Scientific, США) и соответствующих праймеров. Далее продукты ПЦР разделяли в 2% агарозном геле с добавлением бромистого этидия, в качестве маркеров использовали DNA ladder NL002 (Евроген, Россия), визуализацию проводили с помощью УФ-света в трансиллюминаторе.
У животных дикого типа в ходе ПЦР амплифицировался фрагмент, ограниченный двумя праймерами (прямым: 5'-GATCAAAGCCCTCGATGATC-3' и обратным: 5'-ACAGGGTCCACTTTGTCCA-3'), фланкирующими потенциальное место вставки генетической конструкции. Модификация целевого гена у животных с нокаутом, осуществленная методом гомологичной рекомбинации [14], привела к появлению в гене β-аррестина-2 последовательности донорской ДНК с фрагментом, комплементарным прямому праймеру 5'-GCTAAAGCGCATGCTCCAGA-3'. Таким образом, у мышей дикого типа и животных с нокаутом были амплифицированы фрагменты ДНК, ограниченные одним обратным и отличающимися прямыми праймерами, что привело к различиям в размерах ПЦР-продуктов. Фрагмент длиной 350 п.н. указывал на присутствие аллеля гена с нокаутом, фрагмент длиной 300 п.н. – на аллель дикого типа. В эксперимент отбирали животных, гомозиготных по аллелю гена с нокаутом.
Проведение вестерн-блотинга. Культуры астроцитов, полученных от мышей с нокаутом целевого гена и животных дикого типа, лизировали в буфере RIPA с добавлением ингибиторов протеаз (Sigma Aldrich, США). Вестерн-блотинг проводили по ранее описанной методике [15] (в одну лунку геля наносили 20 мкг белка). В качестве первичных антител использовали неконъюгированные антитела против β-аррестина-2 (1 : 1000; Abcam, Великобритания) и β-актина (1 : 1000; Abcam), в качестве вторичных – антитела против IgG мыши, конъюгированные с пероксидазой хрена (1 : 3000; Santa Cruz Biotechnology, США). Присутствие целевых белков регистрировали посредством хемилюминесценции с помощью набора ECL (Thermo Scientific, США).
Культивирование астроцитов. В экспериментах in vitro использовали первичные культуры астроцитов, полученные из мозга новорожденных мышей возрастом от 0 до 3 дней по ранее описанному протоколу [15]. Далее клетки инкубировали в среде DMEM/F12 (Gibco, США), содержащей пируват, глутамин, пенициллин/стрептомицин и 10% эмбриональную телячью сыворотку (Invitrogen, США), в течение 10–12 дней при 37°С и 5% CO2 в культуральных флаконах. За 4 дня до экспериментов астроциты пересевали на 48-луночные планшеты с плотностью 5 × 104 клеток/лунка. Однородность культуры составляла более 90%, что подтверждено окрашиванием антителами к глиальному фибриллярному кислому белку (Abcam, Великобритания) – маркеру астроцитов. За 24 ч до эксперимента клетки переводили в среду с пониженной концентрацией сыворотки (до 2%), далее астроциты опытной группы подвергали воздействию тромбина в концентрациях 10, 50 и 100 нМ в течение 48 ч.
Оценка выживаемости клеток. Выживаемость клеток оценивали с помощью реагента WST-1 (Sigma Aldrich, США) – тетразолиевой соли, которая восстанавливается дегидрогеназами живых клеток до формазана, значительно поглощающего свет в видимой части спектра [15]. К культуре клеток добавляли WST-1 в соотношении 10 : 1 и инкубировали в течение 1 ч. Затем среду переносили в 96-луночный планшет, в котором производили измерение оптической плотности при длине волны 450 нм. Результаты нормировали на соответствующие контрольные значения, принятые за 100%.
Фотоиндуцированная ишемия. В экспериментах использовали самцов мышей массой 25–28 г и возрастом 4–5 мес. Животных содержали в стандартных условиях (22°С, световой день – 12 ч) без ограничения доступа к воде и корму.
Ишемию индуцировали посредством фототромбоза [16] сенсомоторной области коры левого полушария (лазерный луч фокусировали на 2.2 мм латеральнее точки bregma [17]). Для анестезии использовали смесь коммерческих препаратов “Золетил100” (Virbac Sante Animale, Франция) и “Ксила” (Interchemie werken “De Adelaar” B.V., Нидерланды) в дозах 35 и 3.5 мг/кг (в/б) соответственно. В качестве фотосенсибилизатора использовали бенгальский розовый (Sigma Aldrich, США) в дозе 10 мг/кг, вводимый в/в посредством вживленного в яремную вену животного катетера. Тромбоз индуцировали с помощью лазерной установки (λ = 532 нм, диаметр светового пучка – 3 мм, Viacho, Китай) путем освещения поверхности черепа (освещенность – 50 лк) в течение 5 мин.
Оценка неврологического статуса. Сенсомоторный дефицит определяли с помощью теста “Решетка” [18]. До и через 24 ч после индукции фототромбоза животное помещали на поверхность решетки и с помощью видеорегистратора в течение 5 мин фиксировали число случаев свободного проскальзывания передней и задней контралатеральных поврежденному полушарию конечностей в ячейки (моторные ошибки). Для учета общей двигательной активности животного этот показатель для каждой из анализируемых конечностей нормировали на 100 шагов, сделанных задней ипсилатеральной лапой.
МРТ-метод. Прижизненную визуализацию ишемического очага выполняли методом магнитно-резонансной томографии (МРТ) с использованием 7 Tл научно-исследовательского томографа BioSpec 70/30 USR (Bruker, Германия) путем получения Т2-взвешенных изображений.
Метод окрашивания хлоридом 2,3,5-трифенилтетразолия (ТТС). Оценку объема поражения головного мозга через 24 ч после вызванной ишемии проводили с помощью окрашивания секций мозга толщиной 1 мм 1% раствором ТТС (Sigma Aldrich, США) на фосфатно-солевом буфере по стандартной методике [19]. Площади ишемического очага, а также площади каждого полушария для дальнейшего вычисления объема поражения [20] измеряли с помощью программы ImageJ (NIH, США).
Статистическая обработка. Данные анализировали с помощью программы GraphPad Prism 6 (GraphPad Software Inc., США). Результаты представляли как среднее арифметическое ± стандартная ошибка, различия считали статистически значимыми при р < 0.05. Количество культур астроцитов для каждого воздействия составило не менее семи, число животных в экспериментах in vivo – восемь особей в группе. Используемые статистические критерии указаны отдельно под соответствующими рисунками. Нормальное распределение данных подтверждено критерием Шапиро–Уилка.
РЕЗУЛЬТАТЫ
В настоящем исследовании для изучения роли белка β‑аррестина-2 в развитии цитотоксического действия тромбина мы использовали линию мышей с нокаутом гена β-аррестина-2. Согласно данным, полученным в ходе генотипирования животных методом ПЦР, у всех мышей группы β-аррестин-2–/– имелся нокаут обоих аллелей целевого гена в отличие от животных дикого типа (рис. 2а). Результаты вестерн-блотинга полностью подтвердили отсутствие экспрессии β-аррестина-2 у животных с нокаутом (рис. 2б).
Рис. 2.
Подтверждение нокаута гена β-аррестина-2. а – Результаты ПЦР-анализа; б – результаты вестерн-блотинга.

Влияние нокаута гена β-аррестина-2 на выживаемость астроцитов в условиях действия тромбина. Для оценки влияния тромбина на выживаемость одного из основных компонентов глии – астроцитов в условиях in vitro мы выбрали подход, предполагающий использование первичных культур клеток мозга. Проведенные эксперименты показали, что тромбин во всех использованных концентрациях (10, 50 и 100 нМ) не влиял на относительное количество живых астроцитов животных дикого типа через 48 ч после добавления протеазы (рис. 3).
Рис. 3.
Влияние тромбина на выживаемость астроцитов, полученных от мышей дикого типа и с нокаутом гена β-аррестина-2 (двухфакторный дисперсионный анализ с поправкой Бонферрони на множественное сравнение; * – р < 0.05, *** – р < 0.001 по сравнению с соответствующей контрольной группой).
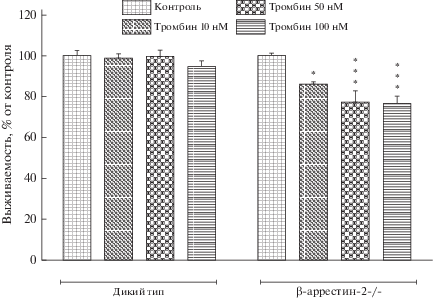
В то же время мы выявили дозозависимый токсический эффект тромбина на астроциты, полученные из мозга мышей с нокаутом гена β-аррестина-2: протеаза вызывала значимое снижение относительного содержания живых клеток в культуре (рис. 3). Так, выживаемость астроцитов животных данной группы при действии на них тромбина в концентрации 10 нМ составила 86.1 ± ± 1.2% от контрольного значения, в концентрации 50 нМ – 77.2 ± 5.8%, а в концентрации 100 нМ – 76.7 ± 3.8%. Таким образом, нами показано, что токсическое действие тромбина зависит от наличия β-аррестина-2 в культивируемых астроцитах мыши.
Влияние нокаута гена β-аррестина-2 на выраженность вызванного фототромбозом поражения у мышей. Появление тромбина в ткани мозга in vivo может наблюдаться при повреждении сосудистой стенки, что сопровождается не только формированием тромба, но и повышением проницаемости ГЭБ [1, 2]. Эти события в полной мере наблюдаются в ходе поражения мозга в результате ишемического инсульта. По этой причине в следующей серии экспериментов для выяснения роли β-аррестина-2 в реализации повреждений нервной ткани, сопряженных с повышенными концентрациями тромбина, мы использовали модель фокальной фотоиндуцированной ишемии [16].
Последствия фототромбоза регистрировали через 24 ч после его индукции, так как известно, что к этому моменту очаг поражения достигает максимальных размеров на фоне существенного повреждения сосудистой стенки [21], значительно повышается проницаемость ГЭБ [22], активируются глиальные клетки [23], а также возрастает концентрация ряда провоспалительных цитокинов в нервной ткани [24]. Следовательно, именно в указанный период времени реализуется широкий спектр негативных эффектов ишемии, в развитие которых существенный вклад вносит тромбин [1, 2].
С помощью метода МРТ показано, что выбранная концентрация фотосенсибилизатора и координаты индукции очага ишемии позволяют локализовать повреждение исключительно в коре больших полушарий, преимущественно в ее сенсомоторной области передних и задних конечностей (рис. 4а).
Рис. 4.
Индуцированное фототромбозом повреждение головного мозга. а – Очаг поражения на МРТ-изображении (срез на уровне точки bregma: 1 – первичная моторная кора, 2 и 3 – соматосенсорная кора задних и передних конечностей соответственно [17]); б – объем поражения головного мозга у мышей дикого типа и с нокаутом по гену β-аррестина-2 через 24 ч после фототромбоза (t-тест для несвязанных выборок); в – изменение площади поражения мозга относительно точки bregma (тест Манна–Уитни для сравнения площади повреждения между двумя группами на каждом расстоянии); г – серия репрезентативных фотографий секций мозга, окрашенных ТТС (треугольником отмечен очаг поражения).
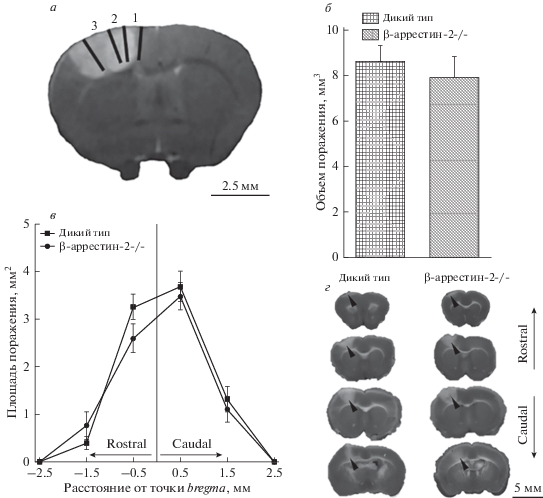
Средний объем поражения, измеренный методом ТТС-окрашивания через 24 ч после индукции ишемии, т.е. в острый период реализации ее негативных последствий, в группе β-аррестин-2–/– составил 7.9 ± 0.9 мм3. Данный показатель статистически значимо не отличался от такового, подсчитанного у животных дикого типа и составившего 8.6 ± 0.7 мм3 (рис. 4б). В конфигурации очага поражения также не выявлено никаких изменений: средняя площадь повреждения была максимальной в области точки bregma и снижалась сходным образом в ростральном и каудальном направлениях у животных обеих групп (рис. 4в).
Повреждение мозга в результате ишемии характеризуется изменениями на уровне не только нервной ткани, но и всего организма, что приводит к появлению сенсомоторного дефицита у животных [18]. Так, с помощью сенсомоторного теста “Решетка” нами выявлен статистически значимый моторный дефицит и передних, и задних контралатеральных поврежденному полушарию конечностей. Данное нарушение развивалось в обеих экспериментальных группах через 24 ч после индукции ишемии (рис. 5а, 5б).
Рис. 5.
Результаты сенсомоторного теста “Решетка” до и через 24 ч после индукции фототромбоза у животных дикого типа и с нокаутом гена β-аррестина-2: а – Передняя контралатеральная конечность; б – задняя контралатеральная конечность (двухфакторный дисперсионный анализ с поправкой Бонферрони на множественное сравнение; * – р < 0.05 по сравнению со значениями до тромбоза).
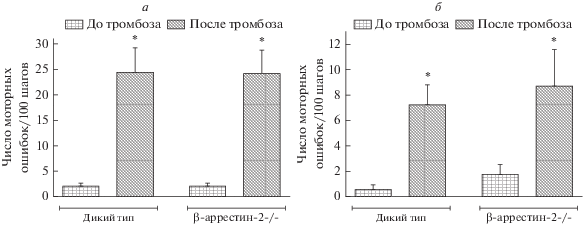
У животных дикого типа количество моторных ошибок на 100 шагов для передней и задней контралатеральных конечностей до фототромбоза составило 2.1 ± 0.6 и 0.5 ± 0.4 соответственно, после фототромбоза – 24.4 ± 4.9 и 7.2 ± 1.6. В группе β-аррестин-2–/– аналогичные показатели составляли 1.9 ± 0.7 и 1.7 ± 0.8 до индукции ишемии и 24.2 ± 4.7 и 8.7 ± 2.9 – после. Однако не отмечено статистически значимых различий в проявлении сенсомоторного дефицита между двумя группами животных. Из представленных данных следует, что в экспериментах in vivo не выявлено значимого влияния нокаута гена β-аррестина-2 на выраженность вызванных фототромбозом нарушений у мышей.
ОБСУЖДЕНИЕ
Сериновая протеаза тромбин – полифункциональный белок, участвующий не только в свертывании крови, но и в регуляции широкого спектра функций клеток. Результаты исследований последних лет свидетельствуют о зависимости эффектов тромбина как от его концентрации, так и от длительности воздействия и состояния клеток [1–3, 7, 8, 15]. Показано, что тромбин в высокой концентрации (нМ) повышает пролиферацию культивируемых астроцитов, активирует индуцибельную NO-синтазу, усиливает секрецию провоспалительных цитокинов и вызывает клеточную гибель [1, 15]. В то же время в низкой концентрации (пМ) тромбин способствует усилению синтеза белков теплового шока и запускает цитопротекторные каскады, активируя ПАР1 [1], экспонированный, как и другие типы ПАР, на мембранах астроцитов [25].
Ранее с использованием культур астроцитов крысы показано дозозависимое усиление клеточной пролиферации через 3 и 24 ч после добавления в культуральную среду тромбина [15, 25]. Однако в настоящем исследовании он не вызывал изменения относительного числа живых астроцитов мышей дикого типа даже в максимальной из использованных концентраций (100 нМ) через 48 ч после воздействия. Возможно, запуск пролиферации может быть краткосрочным ответом на тромбин, тогда как в дальнейшем запускается процесс клеточной гибели за счет индуцированной протеазой провоспалительной активации клеток. Это приводит к компенсации краткосрочного ответа и, как следствие, к отсутствию изменений количества клеток через 48 ч.
Статистически значимое снижение выживаемости астроцитов, полученных от животных группы β-аррестин-2–/–, может свидетельствовать о нарушении процесса десенситизации и интернализации ПАР1 из-за отсутствия β-аррестина-2. Следовательно, в результате невозможности ограничения ПАР1-опосредованного сигнала усиливается развитие токсического действия тромбина.
Помимо этого, тромбин в комплексе с тромбомодулином может способствовать генерации АПС. В наших экспериментах нельзя исключить возможность нивелирования токсичности тромбина в культуре астроцитов животных дикого типа за счет АПС, который реализует свои защитные эффекты по механизму, зависимому от β-аррестина-2 [3, 8]. В то же время реализацию цитопротекторных эффектов АПС в культуре астроцитов мышей с нокаутом целевого гена затрудняет отсутствие β-аррестина-2, что способствует усилению некомпенсированной токсичности тромбина.
Развитие ишемии головного мозга приводит к росту концентрации тромбина в нервной ткани за счет запуска системы гемостаза на фоне сниженных барьерных свойств ГЭБ [1, 26, 27]. Ранее было показано, что ингибиторы тромбина улучшают исход фотоиндуцированной ишемии [27], что подтверждает важную роль данной сериновой протеазы в повреждении мозга при тромбообразовании.
Настоящее исследование не выявило вклада адаптерного белка β-аррестина-2 в развитие нарушений, индуцированных фототромбозом: не обнаружено различий между двумя группами животных ни на уровне сенсомоторных реакций, ни в объеме ишемического поражения мозга. Данный факт может быть связан с вовлечением в ответ на ишемическое повреждение не только астроцитов, но и других компонентов нейроваскулярной единицы, где, возможно, ключевое значение имеет β-аррестин-1.
Более того, in vivo возможна реализация всех эффектов тромбина: и провоспалительных посредством активации G-белков, и защитных, связанных с активацией системы АПС. Также возможен показанный на культуре эндотелиоцитов непосредственный запуск протеазой β-аррестиновых сигнальных путей. В связи с этим направленность действия тромбина в высоких концентрациях на уровне всего организма будет определяться соотношением его отдельных эффектов, а также особенностями β-аррестиновой сигнализации в каждом из типов клеток мозга.
Таким образом, в настоящей работе впервые показана необходимость β‑аррестина-2 для выживания культивируемых астроцитов мыши при токсическом действии тромбина. В то же время требуются дальнейшие исследования, проводимые на уровне ЦНС в целом, для уточнения ключевых мишеней данной протеазы в каждом из клеточных компонентов нервной ткани и роли β-аррестиновой сигнализации.
Коллектив авторов выражает благодарность Д.А. Горбачеву (Институт биоорганической химии имени академиков М.М. Шемякина и Ю.А. Овчинникова РАН) и В.С. Вьюшкову (МГУ имени М.В. Ломоносова) за помощь в генотипировании животных и интерпретации полученных данных.
Работа поддержана грантом РФФИ № 18-34-00977.
Список литературы
Suo Z., Citron B.A., Festoff B.W. 2004. Thrombin: A potential proinflammatory mediator in neurotrauma and neurodegenerative disorders. Curr. Drug Targets – Inflamm. Allergy. 3, 105–114.
Gingrich M.B., Traynelis S.F. 2000. Serine proteases and brain damage – is there a link? Trends Neurosci. 23, 399–407.
Gorbacheva L.R., Kiseleva E.V., Savinkova I.G., Strukova S.M. 2017. New conception of the influence of hemostatic proteases on inflammation, neurotoxicity, and tissue regeneration. Biochem. 82, 1018–1032.
Ramachandran R., Hollenberg M. 2008. Proteinases and signalling: Pathophysiological and therapeutic implications via PARs and more. Br. J. Pharmacol. 153, 263–282.
Traynelis S.F., Trejo J. 2007. Protease-activated receptor signaling: New roles and regulatory mechanisms. Curr. Opin. Hematol. 14, 230–235.
Ossovskaya V.S., Bunnett N.W. 2004. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 84, 579–621.
Roy R.V., Ardeshirylajimi A., Dinarvand P., Yang L., Rezaie A.R. 2016. Occupancy of human EPCR by protein C induces β-arrestin-2 biased PAR1 signaling by both APC and thrombin. Blood. 128, 1884–1893.
Griffin J.H., Zlokovic B. V, Mosnier L.O. 2015. Activated protein C: Biased for translation. Blood. 125, 2898–2908.
Reiter E., Ahn S., Shukla A. K., Lefkowitz R. J. 2012. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–199.
Whalen E.J., Rajagopal S., Lefkowitz R.J. 2011. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. 17, 126–139.
Premont R.T., Gainetdinov R.R. 2007. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 69, 511–534.
Violin J.D., Lefkowitz R.J. 2007. β-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 28, 416–422.
Sofroniew M.V., Vinters H.V. 2010. Astrocytes: Biology and pathology. Acta Neuropathol. 119, 7–35.
Bohn L.M., Lefkowitz R.J., Gainetdinov R.R., Peppel K., Caron M.G., Lin F.-T. 1999. Enhanced morphine analgesia in mice lacking β-arrestin-2. Science. 286, 2495–2498.
Ivanova A.E., Gorbacheva L.R., Strukova S.M., Pinelis V.G., Reiser G. 2014. Activated protein C and thrombin participate in the regulation of astrocyte functions. Biochem. 8, 50–59.
Lee J.-K., Park M.-S., Kim Y.-S., Moon K.-S., Joo S.-P., Kim T.-S., Kim J.-H., Kim S.-H. 2007. Photochemically induced cerebral ischemia in a mouse model. Surg. Neurol. 67, 620–625.
Franklin K.B., Paxinos G. 2001. Mouse brain in stereotaxic coordinates. 2nd ed. London: Acad. Press, 350 p.
Baskin Y.K., Dietrich W.D., Green E.J. 2003. Two effective behavioral tasks for evaluating sensorimotor dysfunction following traumatic brain injury in mice. J. Neurosci. Methods. 129, 87–93.
Benedek A., Móricz K., Jurányi Z., Gigler G., Lévay G., Hársing L., Mátyus P., Szénási G., Albert M. 2006. Use of TTC staining for the evaluation of tissue injury in the early phases of reperfusion after focal cerebral ischemia in rats. Brain Res. 6, 159–164.
Pialat J.-B., Cho T.-H., Beuf O., Joye E., Moucharaffie S., Langlois J.-B., Nemoz C., Janier M., Berthe-zene Y., Nighoghossian N., Desvergne B., Wiart M. 2007. MRI monitoring of focal cerebral ischemia in peroxisome proliferator-activated receptor (PPAR)-deficient mice. NMR Biomed. 6, 335–342.
Kang H.-M., Sohn I., Park C. 2015. Use of indocyanine green for optical analysis of cortical infarcts in photothrombotic ischemic brains. J. Neurosci. Methods. 248, 46–50.
Kim G.W., Lewen A., Copin J.-C., Watson B.D., Chan P.H. 2001. The cytosolic antioxidant, copper/zinc superoxide dismutase, attenuates blood-brain barrier disruption and oxidative cellular injury after photothrombotic cortical ischemia in mice. Neuroscience. 105, 1007–1018.
Schroeter M., Jander S., Stoll G. 2002. Non-invasive induction of focal cerebral ischemia in mice by photothrombosis of cortical microvessels: Characterization of inflammatory responses. J. Neurosci. Methods. 117, 43–49.
Cotrina M.L., Lou N., Tome-Garcia J., Goldman J., Nedergaard M. 2017. Direct comparison of microglial dynamics and inflammatory profile in photothrombotic and arterial occlusion evoked stroke. Neuroscience. 343, 483–494.
Wang H., Ubl J.J., Reiser G. 2002. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia. 37, 53–63.
Watson B.D., Dietrich W.D., Busto R., Wachtel M.S., Ginsberg M.D. 1985. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 17, 497–504.
Xi G., Reiser G., Keep R.F. 2003. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective? J. Neurochem. 84, 3–9.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии