

Биологические мембраны: Журнал мембранной и клеточной биологии, 2020, T. 37, № 3, стр. 175-187
Участие Са2+-проницаемых AMPA-рецепторов в синаптической пластичности
Л. П. Долгачева a, *, С. Т. Тулеуханов b, В. П. Зинченко a, **
a Федеральный исследовательский центр “Пущинский научный центр биологических исследований
Российской академии наук”
142290 Московская обл, Пущино, Россия
b Казахский национальный университет им. аль-Фараби
050040 Алматы, Казахстан
* E-mail: dolgacheva@mail.ru
** E-mail: vpz@mail.ru
Поступила в редакцию 26.04.2019
После доработки 17.12.2019
Принята к публикации 23.12.2019
Аннотация
АМРА-рецепторы являются ключевыми молекулами синаптической передачи и участвуют в синаптической пластичности. При нарушении процессов, контролирующих сборку AMPA-рецепторов, миграцию и синапс-специфическую экспрессию, снижаются такие когнитивные функции головного мозга, как восприятие, обработка и анализ сигналов, запоминание, хранение и обмен информацией между нейронами. Содержание рецепторов в синапсе регулируется экзоцитозом, эндоцитозом и рециркуляцией. Несколько вспомогательных субъединиц и белков-партнеров участвуют в этих процессах и модулируют активность AMPA-рецепторов. Несмотря на то, что Ca2+-проводящие AMPA-рецепторы (CP-AMPARs), не содержащие субъединицу GluA2, экспрессированы далеко не во всех нейронах, они участвуют в синаптической пластичности, включая долговременное потенцирование и депрессию, и обеспечивают баланс возбуждения и торможения в мозге. Активация CP-AMPARs в нейронах, вызывает быстрый постсинаптический вход Ca2+, который индуцирует синаптическую пластичность через взаимодействие с другими Са2+-зависимыми сигнальными системами. Цель данного обзора состоит в том, чтобы обратить внимание исследователей на последние достижения в области участия СР-АМРА-рецепторов в синаптической пластичности.
ВВЕДЕНИЕ
Глутаматные АМРА-рецепторы классически рассматриваются как рецепторы-каналы, деполяризующие мембрану за счет проводимости по Na+, что обеспечивает прохождение возбуждающего сигнала через нейрональные синапсы, активацию потенциал-зависимых кальциевых каналов и снятие магниевого блока с NMDA-рецепторов. Однако отдельные популяции АМРА-рецепторов, лишенные субъединицы GluA2, обладают кальциевой проводимостью, которая не зависит от мембранного потенциала [1], что дает возможность запускать процесс секреции нейротрансмиттеров без деполяризации и участия потенциал-зависимых кальциевых и NMDA-каналов. Са2+-проницаемость этих рецепторов имеет решающее значение при некоторых формах синаптической пластичности [1, 2] и клеточной гибели, происходящей при неврологических заболеваниях и расстройствах [3]. СP-АМРА-рецепторы вовлечены во множество нормально протекающих физиологических процессов, таких как процессы памяти и забывания [4]. Значительные изменения экспрессии GluA2-субъединицы происходят при различных патологических состояниях. Список нейродегенеративных заболеваний и нарушений развития мозга, в патогенезе которых СР-АМРА-рецепторы играют заметную роль, постоянно расширяется [3, 5, 6]. К ним относятся разные формы эпилепсии, болезни Альцгеймера и Паркинсона, шизофрения, амиотрофический боковой склероз, синдром аутизма, инсульт, глиобластома, наркомания и др. В настоящем обзоре представлены данные о роли СР-АМРА глутаматных рецепторов в синаптической функции и пластичности, гибели и выживании нейронов [5, 7].
1. ИОНОТРОПНЫЕ РЕЦЕПТОРЫ ГЛУТАМАТА
L-Глутамат является основным возбуждающим медиатором в синапсах центральной нервной системы позвоночных. Глутаматные ионотропные рецепторы подразделяются на три основных типа в соответствие с лигандом, селективно активирующим данный тип рецепторов: рецепторы N-метил-D-аспартата (NMDA-рецептор), α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (AMPA-рецептор) и каиновой кислоты (KA-рецептор). NMDA-рецепторы являются лиганд- и потенциал-управляемыми каналами, и их активация зависит не только от связывания глутамата, но также от сопутствующей деполяризации постсинаптической мембраны, которая снимает блокирование этого канала ионами магния. АMPA-рецепторы являются только лиганд-управляемыми ионными каналами, участвующими в быстрой передаче возбуждения. Семейство KA глутаматных рецепторов отличается от других ионотропных глутаматных рецепторов разнообразием субъединиц, функций и локализацией, и поэтому представляет значительный интерес для модуляции синаптической передачи [8, 9]. KA-рецепторы могут быть тормозящими, поскольку часто локализованы в пресинаптической мембране ГАМК-ергичеcких нейронов [10, 11].
2. AMPA-РЕЦЕПТОРЫ
AMPARs представляют собой тетрамерные ионные каналы, которые вместе с другими ионотропными глутаматными рецепторами (NMDA- и KA-рецепторами) опосредуют большую часть возбуждающей нейротрансмиссии в центральной нервной системе [12]. При связывании глутамата канал АМРА-рецептора открывается, что приводит к преимущественному входу натрия (или натрия и кальция), и вызывает деполяризацию постсинаптического нейрона [13, 14]. AMPAR-зависимая быстрая деполяризация постсинаптической мембраны необходима для открытия Na+ каналов, а также для снятия Mg2+ блока с NMDA-рецепторов [15]. AMPARs принимают участие в процессах долговременной потенциации (LTP) и долговременной депрессии (LTD) [14]. Когнитивные функции головного мозга, такие, как восприятие, обработка и анализ сигналов, запоминание, хранение и обмен информацией, снижаются при нарушении процессов, контролирующих активность AMPARs [16, 17]. Изменения активности AMPARs описаны при развитии многих заболеваний, таких как болезнь Альцгеймера, инсульт и эпилепсия.
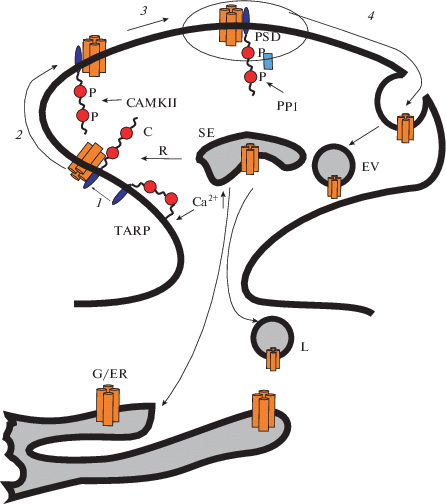
Постсинаптические AMPARs конститутивно обновляются с большой скоростью [18]. Молекулярные и клеточные механизмы, контролирующие транспортировку АМРА-рецепторов в синапс, стабилизацию и время жизни в синапсе, являются предметом интенсивного и широкого исследования. АМРАRs транспортируются в синапс путем латеральной диффузии в плазматической мембране или экзоцитозом [19]. Удаление AMPARs из плазмалеммы осуществляется в результате рецептор-зависимого эндоцитоза [20]. При этом небольшие окаймленные структуры сливаются между собой, образуя эндосомы. В ранних эндосомах рецепторы сортируются по различным путям транспортировки [21]: путь возвращения в плазмалемму, путь посттрансляционной модификации, и путь лизосомальной деградации (рис. 1).
Рис. 1.
Участие TARP в транспорте АМРАRs. Увеличение концентрации Са2+ в постсинапсе вызывает диссоциацию С-конца вспомогательного белка ТАRP ( ) от цитоплазматической мембраны (1), инициирует взаимодействие TARP с AMPAR (
) от цитоплазматической мембраны (1), инициирует взаимодействие TARP с AMPAR ( ) и активирует протеинкиназу CAMKII. Фосфорилирование CAMKII киназой С-концевых участков
белка TARP (старгазин) (2) ускоряет движение AMPAR к постсинаптическому уплотнению PSD, (3) вызывает связывание TARP со скаффолд-белком PSD-95 и способствует удерживанию AMPAR
в PSD (3). Дефосфорилирование этих же остатков фосфатазой (PP1) нарушает взаимодействие TARP-PSD-95
и освобождает AMPAR из постсинаптического уплотнения (4). Удаление AMPAR из плазмалеммы происходит в результате рецептор-зависимого эндоцитоза.
Небольшие окаймленные структуры сливаются между собой и образуют ранние эндосомы (EV).
В более зрелых, сортирующих эндосомах (SE), рецепторы распределяются по различным
путям: путь возвращения в плазмалемму или рециклирование (R), путь посттрансляционной
модификации через аппарат Гольджи (G/ER), путь лизосомальной (L) деградации.
) и активирует протеинкиназу CAMKII. Фосфорилирование CAMKII киназой С-концевых участков
белка TARP (старгазин) (2) ускоряет движение AMPAR к постсинаптическому уплотнению PSD, (3) вызывает связывание TARP со скаффолд-белком PSD-95 и способствует удерживанию AMPAR
в PSD (3). Дефосфорилирование этих же остатков фосфатазой (PP1) нарушает взаимодействие TARP-PSD-95
и освобождает AMPAR из постсинаптического уплотнения (4). Удаление AMPAR из плазмалеммы происходит в результате рецептор-зависимого эндоцитоза.
Небольшие окаймленные структуры сливаются между собой и образуют ранние эндосомы (EV).
В более зрелых, сортирующих эндосомах (SE), рецепторы распределяются по различным
путям: путь возвращения в плазмалемму или рециклирование (R), путь посттрансляционной
модификации через аппарат Гольджи (G/ER), путь лизосомальной (L) деградации.
AMPARs функционируют как тетрамеры, построенные из димерных комбинаций четырех основных субъединиц, GluA1–4 [22]. Тетрамеризация субъединиц происходит в результате взаимодействия между лиганд-связывающими, трансмембранными и N-концевыми сегментами [23, 24]. Функциональные свойства каналов AMPARs в значительной степени определяются составом субъединиц и регулируются редактированием посттранскрипционной РНК, посттрансляционной модификацией и вспомогательными белками. Процессы редактирования и посттрансляционной модификацией приводят к изменению числа рецепторов и появлению в синапсе рецепторов с другими свойствами.
При посттрансляционной модификации субъединицы рецепторных белков могут быть модифицированы путем ацетилирования, метилирования, фосфорилирования и др. Важную роль в моделировании функций глутаматных рецепторов играет фосфорилирование серина, треонина и тирозина С-концевого внутриклеточного домена субъединиц. Фосфорилирование/дефосфорилирование АМРА-рецептора может приводить к синаптической пластичности в норме и при патологии [25–28].
2.1. Редактирование субъединицы GluA2 на уровне мРНК
Субъединица GluA2, содержание которой определяет проводимость рецептора, проходит редактирование на уровне мРНК. Группа Хайнемана была первой, кто показал, что отсутствие редактированной субъединицы GluA2 определяет Ca2+-проводимость AMPA-рецепторов [29, 30]. Процесс редактирования мРНК гена данной субъединицы состоит в том, что перед началом трансляции мРНК взаимодействует с белком ADAR (аденозиндезаминаза РНК), который заменяет триплет CAG на CIG (аденозин заменяется на инозин). Соответствующие изменения происходит и в структуре белка – глутамин в 607-й позиции заменяется на аргинин. Также в 764 позиции аргинин заменяется на глицин [31]. Изменение заряда в поре канала приводит к потере кальциевой проницаемости, сильно снижает проводимость одиночного канала рецептора и предотвращает блокирование канала внутриклеточными полиаминами [32]. Высокая проводимость для Са2+-рецепторов, не содержащих субъединицу GluA2, предполагает их значительную роль в индукции синаптической LTP, как в норме, так и при повреждении нейронов в патологических условиях.
2.2. Вспомогательные белки AMPA-рецепторов
Функции AMPARs в синапсе зависят не только от субъединичного состава (GluA1–4) [33], но также от взаимодействия рецептора с вспомогательными белками [34, 35]. Участие различных вспомогательных белков в транспорте AMPA-рецепторов к синапсу интенсивно исследуется в последнее время [36]. Большинство субъединиц AMPAR находятся в комплексе с вспомогательными субъединицами, которые участвуют в экспрессии рецепторов, рециркуляции, пластичности, а также определяют их биофизические свойства [37]. В настоящее время известны такие вспомогательные белки AMPARs, как TARP, CNIH, GSG1L и CKAMP [38–40].
2.2.1. Вспомогательные трансмембранные регуляторные белки семейства TARP. Трансмембранные регуляторные белки (TARP) [41] направляют AMPARs в синапсы, участвуют в удержании/стабилизации AMPA-рецепторов в синапсе и модулируют активность рецептора, регулируя воротный механизм канала [42, 43]. На рис. 1, 2 показаны схемы взаимодействия субъединицы АМРА-рецептора и белка TARP при транспорте АМРА-рецепторов к синапсу.
Рис. 2.
Взаимодействие субъединицы АМРА-рецептора и белка TARP. Две экстраклеточные петли EX1 и EX2 белка TARP (серый овал) взаимодействуют с лиганд-связывающим доменом LBD – (серый овал) субъединицы AMPA-рецептора, управляя воротным механизмом канала. Проксимальная область С-концевого участка TARP взаимодействует с участком поры AMPA-рецептора (остаток Q/R, желтый круг), оказывая влияние на проводимость канала и полиаминный блок. Красные круги – участки фосфорилирования. Синий квадрат – PDZ –связывающие домены GluAR и TARP, обеспечивающие взаимодействие с PSD-95. Glu – глутамат.
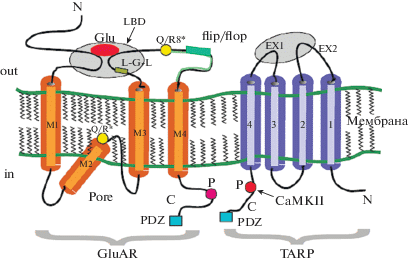
Семейство TARP включает шесть изоформ, которые по-разному изменяют свойства AMPA-рецепторов. TARP γ2, γ5, и γ7 участвуют в регуляции биофизических свойств Са2+-проводящих рецепторов (CP-AMPARs). TARP γ2, γ5 уменьшают чувствительность этих рецепторов к полиаминам [44, 45], а TARP γ7 избирательно усиливает синаптическую экспрессию CP-AMPAR и подавляет экспрессию кальций-непроницаемых AMPARs [46].
TARP γ2, γ3, γ4 и γ8 удерживают синаптические AMPARs посредством связывания со скаффолд-белком постсинаптического уплотнения PSD-95, и эта стабилизация весьма важна для развития LTP [34]. Образованный комплекс TARP-PSD-95 уменьшает латеральную подвижность AMPARs в синапсе, а нарушение этого взаимодействия позволяет AMPARs в комплексе с TARP мигрировать из синапса [42]. Комплекс TARP-PSD-95 регулируется фосфорилированием ряда сериновых остатков во внутриклеточном С-концевом домене TARP. Фосфорилирование С-концевого домена TARP кальций/кальмодулин-зависимой протеинкиназой CaMKII ингибирует его связывание с отрицательно заряженными фосфолипидами в липидном бислое и способствует связыванию с PSD-95 и удержанию рецептора в синапсе [47]. Дефосфорилирование этих же остатков фосфатазой PP1 [34], способствует ассоциации внутриклеточного домена TARP с фосфолипидами, нарушая взаимодействие TARP-PSD-95, и, следовательно, освобождает AMPARs из постсинаптического уплотнения [47]. Интернализация субъединиц GluA1 и GluA2 инициируется различными фосфатазами. Интернализация субъединицы GluA2 селективно блокируется ингибиторами PP2A (окадаевая кислота и фостриецин), тогда как интернализация GluA1 предотвращается ингибиторами фосфатаз PP2A, PP1 и PP2B [48].
При высокочастотной стимуляции связывание глутамата с AMPARs уменьшает взаимодействие с TARP γ2 [49], инициируя удаление рецепторов из синапсов [50, 51], чтобы позже, для развития LTP, пополнить синапс новыми или модифицированными рецепторами из внесинаптических компартментов. TARP γ2 (старгазин) функционирует как положительный аллостерический модулятор AMPARs: замедляет деактивацию рецептора, уменьшает десенситизацию, ускоряет восстановление после десенситизации, увеличивает сродство к агонистам, проницаемость для кальция и проводимость одиночного канала и уменьшает блокирование канала внутриклеточными полиаминами [41, 44, 52]. Продолжительность процесса удержания старгазином AMPARs в синапсе увеличивается при длительном потенцировании за счет CaMKII-зависимого фосфорилирования остатков серина в C-концевом домене старгазина [35, 46]. Таким образом, взаимодействие между старгазином и PSD-95 регулирует обмен AMPARs между экстрасинаптическим и синаптическим компартментами.
2.2.2. Вспомогательные трансмембранные регуляторные белки семейства CKAMP. Белки, содержащие цистиновый узел и модулирующие AMPARs (Cystine-knot AMPA receptor-modulating proteins CKAMPs), представляют собой семейство, состоящее из четырех белков, которые влияют на транспорт, субклеточную локализацию и функцию AMPARs. Наиболее изученными членами CKAMP семейства являются белки CKAMP39, CKAMP44, CKAMP52 и CKAMP59. Все члены семейства CKAMP являются рецепторными трансмембранными белками типа 1. Внеклеточные домены этих белков обогащены цистеином. Дисульфидные связи между цистеинами способствуют стабилизации глобулярной структуры, которая важна для взаимодействия с AMPARs и модуляции их активности. CKAMP44 и CKAMP52 были обнаружены в AMPA-рецепторных комплексах, которые также содержали TARP [54], что свидетельствовало о связывании CKAMP и TARP с различными областями субъединиц AMPARs. Показано, что белки TARP γ8 и CKAMP44, которые высоко экспрессированы в гранулярных клетках зубчатой фасции гиппокампа, снижают скорость деактивации рецептора. Для эффективного транспорта AMPARs в определенную область на клеточной мембране необходима коэкспрессия обеих этих вспомогательных субъединиц [54]. Следует отметить, что все четыре члена семейства CKAMP различаются по профилю экспрессии и модулирующему влиянию на функцию AMPARs [55]. Последние исследования о роли семейства CKAMP белков приведены в обзоре [56].
2.2.3. Трансмембранный вспомогательный белок GSG1L. GSG1L (Germline-specific gene 1-like) был идентифицирован как вспомогательная субъединица AMPARs и имеет некоторое структурное сходство с TARPs [39]. Этот трансмембранный вспомогательный белок, в отличие от старгазина, снижает проводимость одиночного канала и кальциевую проницаемость рекомбинантных CP-AMPARs, тем самым расширяет функциональные свойства этих рецепторов [57].
2.2.4. Трансмембранный вспомогательный белок CNIH. Белки CNIH (Cornichon homolog protein) были идентифицированы протеомным анализом как белки, взаимодействующие с AMPARs [38]. Изменение уровня CNIH-2 в гиппокампе изменяло кинетику синаптических AMPARs, что указывало на регулирующую роль CNIH-2 в передаче, опосредованной AMPARs [58]. Совместная экспрессия рекомбинантных AMPARs с белками CNIH увеличивала экспрессию этих рецепторов и замедляла их деактивацию и десенситизацию [59, 60]. В нейронах гиппокампа и мозжечка CNIH-2 оказывал влияние на взаимодействие AMPARs и белков TARP (γ7), регулируя количество TARPs в AMPA-рецепторном комплексе и изменяя параметры воротного механизма рецептора [61]. Нокаут CNIH-2/3 у мышей уменьшал количество AMPARs, содержащих субъединицу GluA1 в синапсе, что приводило к снижению, как AMPAR-опосредованной передачи, так и LTP [62].
Таким образом, взаимодействие АМРА-рецепторов с различными вспомогательными белками усиливает синаптическую экспрессию CP-AMPARs, рециркуляцию, пластичность, а также определяет биофизические свойства CP-AMPARs, уменьшает чувствительность этих рецепторов к полиаминам, меняет проводимость одиночного канала и кальциевую проницаемость рецептора. Необходимо заметить, что регуляция активности AMPARs вспомогательными белками усложняется тем, что влияние каждой из вспомогательных субъединиц зависит от состава субъединиц рецептора и модулируется другими вспомогательными субъединицами [54].
3. СИНАПТИЧЕСКАЯ ПЛАСТИЧНОСТЬ
Синаптическая пластичность представляет собой способность синапсов менять силу передачи в зависимости от нейронной активности в ответ на внешние раздражители. Термин “синаптическая пластичность” для описания усиления синаптической передачи в 1948 году ввел Конорский [63]. Синаптическую пластичность также можно определить как структурную и функциональную адаптацию нейронных сетей к изменениям, связанным с обучением и памятью, влиянием окружающей среды и повреждением головного мозга [64].
Феномен увеличения силы синапса между двумя нейронами при передаче информации в головном мозге может сохраняться длительное время и известен как LTP. LTP является хорошо охарактеризованным видом синаптической пластичности, которая коррелирует с обучением и памятью [65]. Во многих работах показано, что LTP, вызываемая даже короткой высокочастотной стимуляцией, сохраняется в течение нескольких дней или даже недель in vivo. В зависимости от продолжительности эффекта синаптическая пластичность разделена на несколько видов: (1) – кратковременная пластичность, когда изменения происходят в диапазоне от миллисекунд до минут и позволяют синапсам выполнять кратковременные вычислительные функции в нейронных цепях [66], (2) – долговременная пластичность, при которой изменения могут длиться от нескольких часов до дней, недель или даже месяцев [67, 68], (3) – гомеостатическая пластичность, которая может происходить как в синапсах, так и в нейронах, и позволяет стабилизировать уровни возбудимости и межнейронные связи, несмотря на изменения в окружающей среде, вызванные метаболизмом и пластичностью [69]. В LTP также можно выделить “раннюю фазу”, которая длится приблизительно 60 мин, требует активации NMDA-рецепторов, последующего входа Са2+ и активации CaMKII [70, 71]. В это время также происходит транспорт новых AMPARs. И “позднюю” фазу LTP, которая продолжается дни и недели и требует экспрессии генов и синтеза белка [72, 73].
Длительная депрессия (LTD), это ослабление синаптической передачи в результате определенного режима высокочастотной активности синапсов. Известно, что эффект LTP или LTD зависит от того, какова частота стимуляции путей к пресинаптическому нейрону и каков уровень поляризации мембраны постсинаптического нейрона. Основной механизм долговременной синаптической потенциации или депрессии обусловлен увеличением или уменьшением количества AMPA-рецепторов в синапсе [16, 74]. AMPA-рецепторы собираются в эндоплазматическом ретикулуме и затем транспортируются к плазматической мембране. Их содержание в синапсе находится в динамическом равновесии между увеличением посредством экзоцитоза и удалением посредством эндоцитоза [75].
4. УЧАСТИЕ CP-AMPARs В СИНАПТИЧЕСКОЙ ПЕРЕДАЧЕ И В ЕЕ ПЛАСТИЧНОСТИ
CP-AMPARs осуществляют альтернативный NMDA-рецептор-зависимому путь повышения Са2+ в клетках и играют важную роль в синаптической передаче и в синаптической пластичности, вызывая LTP [2, 76–78]. CP-AMPARs имеют большую проводимость одиночного канала 7–8 pS, тогда как канал с GluA2 имеет проводимость около 300 fS [79]. Последние исследования дают убедительные доказательства участия CP-AMPARs в синаптической передаче и в ее пластичности [1, 3, 5, 80–82] как при нормальном функционировании мозга в процессах запоминания и обучении [2, 83], так и при патогенезе нервных заболеваний [84]. Также как NMDA-рецепторы, CP-AMPARs могут вызывать Ca2+-зависимую эксайтотоксическую гибель клеток при различных патологиях [5, 7]. При ишемии происходит увеличение CP-AMPARs в синапсе, что способствуют гибели нейронов [85]. А селективные блокаторы CP-AMPARs обладают нейропротекторным действием и препятствуют гибели нейронов при ишемии [86]. Повышенная экспрессия СР-AMPARs у мышей приводит к судорогам и преждевременной смерти в возрасте нескольких недель [87]. Кроме того, оказалось, что содержание CP-AMPARs увеличивается в вентральной области покрышки мозга мышей после однократной инъекции кокаина, что, как полагают, способствует пластичности, вызванной этим соединением [88].
При развитии LTP транспорт CP-AMPARs в синаптическую мембрану и удаление из нее усиливается. При этом CP-AMPARs транспортируются в синапсы гиппокампа из внесинаптических и/или внутриклеточных источников. Встраивание и удаление CP-AMPARs в синапс регулируется процессами фосфорилирования и дефосфорилирования C-концевого остатка S845 субъединицы GluA1 cAMP-зависимой протеинкиназой (PKA) и Ca2+-кальмодулин-зависимой протеинфосфатазой 2B (PP2B фосфатаза), соответственно. В синапсах области CA1 гиппокампа показано, что для индукции LTP необходима CP-AMPARs-зависимая активация PKA-зависимого синтеза белка [89]. Фосфорилирование приводит к накоплению GluA1 в постсинапсе, тогда как убиквитинирование субъединицы GluA1 E3-лигазой Nedd4-1 ведет к деградации AMPARs. Дефосфорилирование C-концевого домена GluA1 по серину 845 фосфатазой PP2B инициирует удаление CP-AMPARs из зоны постсинаптического уплотнения. Оптимизация этих процессов обеспечивается белком AKAP 150 (A-kinase anchoring protein), который заякоривает киназу и фосфатазу. Транспортировка самого белка AKAP150 активируется пальмитоилированием двух цистеиновых остатков.
В ряде работ показано, что после периода повышенной активности количество CP-AMPARs в синапсе возрастает [90, 91]. Последние исследования показали, что соотношение транспортируемых Са2+-проводящих и Са2+-непроводящих AMPA-рецепторов зависит от активности синапса. При слабой стимуляции в синапсе активируется транспорт CP-AMPARs, а при сильной стимуляции активируется транспорт AMPARs, содержащих GluA2 [92]. Показано, что для потенциации LTP при слабой стимуляции, которая направляет CP-AMPARs в синапсы, необходимо пальмитоилирование AKAP150, но при сильной стимуляции, которая рекрутирует AMPARs, содержащие GluA2, этого не происходит. Таким образом, пальмитоилирование AKAP150 регулирует субъединичный состав AMPARs в зависимости от степени активности синапса [92].
При развитии LTP CP-AMPARs включаются в состав синапса на определенное время (<25 мин), а затем заменяются AMPARs, содержащими GluA2 [90, 93, 94]. Эксперименты с использованием филантотоксина-433, избирательно блокирующего CP-AMPARs [95], показали, что его аппликация во время и сразу после индукции LTP предотвращает LTP, но после полного развития LTP филантотоксин был уже не эффективен [90, 93], что также говорит о быстром удалении CP-AMPARs из синапса в этот период.
5. РОЛЬ ПОЛИАМИН-ЗАВИСИМОЙ РЕГУЛЯЦИИ СP-AMPARs В ПЛАСТИЧНОСТИ
В закрытом состоянии СР-АМРАRs блокируются потенциал-зависимо полиаминами [96, 97], такими как спермин и спермидин. Однако повторные активации постепенно приводят к снятию этого блока и усилению потока ионов через канал, что может быть причиной кратковременной пластичности синапсов, экспрессирующих СP-AMPARs [97, 98]. Известно, что блокирование ионных каналов цитоплазматическими полиаминами в зависимости от потенциала является регуляторным механизмом для многих семейств катионных каналов [99]. Вспомогательный белок старгазин ослабляет полиаминовый блок и усиливает активность CP-AMPARs, увеличивая проводимость одиночного канала и проницаемость для кальция. Принимая во внимание тот факт, что экспрессия СР-АМРА-рецепторов меняется при сетевой активности и увеличивается в мозге при развитии [100] и при различных заболеваниях [101, 102], полиамин-зависимая регуляция СP-AMPARs является важным, постсинаптическим механизмом регуляции усиления синаптической передачи [98].
6. ИЗМЕНЕНИЯ ЭКСПРЕССИИ CP-AMPARs ПРИ ПАТОЛОГИИ
Изменения экспрессии CP-AMPARs или их активности наблюдаются при ряде серьезных неврологических заболеваниях, включая инсульт, эпилепсию, черепно-мозговые травмы и нейродегенеративные нарушения [101–104]. В работе [98] показано, что в нейронах гиппокампа области СА1 постишемическая пластичность АМРАR проявляется в увеличении содержания CP-AMPARs и связана с возникающим при ишемии ацидозом. Нарушение транспорта CP-AMPARs отмечено при болезни Альцгеймера. Амилоидные β-олигомеры индуцируют дефосфорилирование GluA1 по Ser-845 и подавляют механизм вовлечения и доставки CP-AMPARs в синапсы на ранних стадиях заболевания [105, 106], что приводит к удалению AMPARs из плазматической мембраны, потере дендритных шипиков и синаптической депрессии [107].
7. УЧАСТИЕ CP-AMPARs В ПЛАСТИЧНОСТИ ИНТЕРНЕЙРОНОВ
ГАМК-еpгичеcкие тормозные интернейроны играют решающую роль в развитии и созревании нейронных сетей мозга, регуляции синаптической пластичности и в ритмогенезе [53, 108, 109]. Ослабление ГАМК-еpгичеcкой пеpедачи вызывает гипеpвозбуждение и гипеpcинxpонизацию нейpонов в cети [110]. Наpушение pаботы тормозных нейpонов пpиводит к pазвитию нейpодегенеpативныx заболеваний, таких как cиндpом Туpетта, болезнь Паpкинcона, шизофpения, эпилепcия и аутизм [111–114]. Тормозные нейроны не являются однородной популяцией и различаются по морфологическим, электрофизиологическим характеристикам и набору экспрессируемых белков [115]. Несколько подтипов ГАМК-еpгичеcкиx нейpонов, cодеpжат Ca2+-связывающие белки в буфеpных концентрациях, что может ослаблять кратковременную пластичность и задерживать освобождение трансмиттера [116]. Последние исследования показали, что CP-AMPARs коэкспрессированы с некоторыми Ca2+-связывающими белками в нейронах определенного подтипа. Показано, что быстроразряжающиеся (fast spiking) интернейроны неокортекса [117, 118], гиппокампа [119, 120] и мозжечка [121], содержащие парвальбумин, также содержат CP-AMPARs. При этом во время базальной синаптической активности CP-AMPARs взаимодействуют с парвальбумином и Na+/Ca2+-обменником плазмалеммы [117, 121]. Таким образом, активация CP-AMPARs в быстроразряжающихся интернейронах, содержащих парвальбумин, обеспечивает быстрый постсинаптический вход Ca2+, который индуцирует процесс синаптической пластичности [120–124].
Учитывая тот факт, что в мозге взрослых особей CP-AMPARs локализованы в основном в ГАМК-ергических нейронах, возбуждающее действие агонистов этих рецепторов и нейропротекторное действие антагонистов можно объяснить иннервацией этими нейронами других тормозных нейронов, контролирующих возбуждающие нейроны. Недавно было показано [125], что в гиппокампе крысы определенная субпопуляция ГАМК-ергических нейронов, содержащих CP-AMPARs, может иннервировать ГАМК-ергические нейроны, содержащие кальций-проводящие каинатные рецепторы.
В заключение можно сказать, что CP-AMPARs, наряду с NMDA-рецепторами, благодаря высокой кальциевой проводимости, являются активными участниками процессов синаптической пластичности в норме и в патологических условиях. Повышая базальный уровень Са2+ в клетках по NMDA-рецептор-независимому пути, CP-AMPARs играют важную роль в синаптической пластичности, вызывая LTP. Активация CP-AMPARs, локализованных в ГАМК-ергических нейронах, может усиливать Са2+-зависимую секрецию ГАМК и, таким образом, участвовать в подавлении возбуждения иннервируемых нейронов [126, 127]. В отличие от NMDA-рецепторов, вклад которых в синаптические процессы изменяется в основном за счет кальций-зависимой десенситизации [128], активность СР-АМРАRs меняется за счет быстрого транспорта рецепторов в синапс после PKA-зависимого фосфорилирования и удаления из синапса после РР2В-зависимого дефосфорилирования. Также как NMDA-рецепторы, AMPARs могут вызывать Ca2+-зависимую эксайтотоксическую гибель клеток при различных патологиях. Экспрессия СР-AMPA-рецепторов зависит от сетевой активности и увеличивается при ишемии, эпилепсии. При этом селективные ингибиторы CP-AMPARs обладают нейропротекторным действием. Таким образом, изученные свойства, локализация и функции CP-AMPARs позволяют рассматривать их в качестве потенциальной мишени селективного фармакологического воздействия.
Работа выполнена при финансовой поддержке Комитета науки МОН Реcпублики Казахcтан (гpант № AP05133528) и РФФИ (грант № 19-04-00138).
Список литературы
Isaac J.T., Ashby M.C., Mcbain C.J. 2007. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 54, 859–871.
Wiltgen B.J., Royle G.A., Gray E.E., Abdipranoto A., Thangthaeng N., Jacobs N., Saab F., Tonegawa S., Heinemann S.F., O’Dell T.J., Fanselow M.S., Vissel B. 2010. A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. PLoS ONE. 5, e12818.
Liu S.J., Zukin R.S. 2007. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Clem R.L., Huganir R.L. 2010. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 330 (6007), 1108–1112.
Pellegrini-Giampietro D.E., Gorter J.A., Bennet M.V., Zukin R.S. 1997. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 20, 464–470.
Bowie D. 2008. Ionotropic glutamate receptors & CNS disorders. CNS Neurol Disord Drug Targets. 7(2), 129–143.
Kwak S., Weiss J.H. 2006. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr. Opin. Neurobiol. 16, 281–287.
Contractor A., Swanson G.T., Sailer A., O’Gorman S., Heinemann S.F. 2000. Identification of the kainite receptor subunits underlying modulation of excitatory synaptic transmission in the CA3 region of the hippocampus. J. Neurosci. 20 (22), 8269–8278.
Schmitz D., Frerking M., Nicoll R.A. 2000. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 27 (2), 327–338.
Rodríguez-Moreno A., Lerma J. 1998. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 20 (6), 1211–1218.
Кононов А.В., Баль Н.В., Зинченко В.П. 2012. Регуляция спонтанных синхронных осцилляций Сa2+ в нейронах гиппокампа ГАМКергическими нейронами, содержащими каинатные рецепторы без десенситизации. Биол. мембраны. 29 (1), 133–138.
Traynelis S.F., Wollmuth L.P., McBain C.J., Menniti F.S., Vance K.M., Ogden K.K., Hansen K.B., Yuan H., Myers S.J., Dingledine R. 2010. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 62, 405–496.
Scannevin R.H., Huganir R.L. 2000. Postsynaptic organization and regulation of excitatory synapses. Nat. Rev. Neurosci. 1 (2), 133–141.
Henley J.M., Wilkinson K.A. 2016. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 17, 337–350.
Mayer M.L., Westbrook G.L., Guthrie P.B. 1984. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 309, 261–263.
Henley J.M., Wilkinson K.A. 2013. AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin. Neurosci. 15 (1), 11–27.
Cheng G.R., Li X.Y., Xiang Y.D., Liu D., McClintock S.M., Zeng Y. 2017. The implication of AMPA receptor in synaptic plasticity impairment and intellectual disability in fragile X syndrome. Physiol. Res. 66 (5), 715–727.
Lu W., Roche K.W. 2012. Posttranslational regulation of AMPA receptor trafficking and function. Curr. Opin. Neurobiol. 22, 470–479.
Wierenga C.J., Ibata K., Turrigiano G.G. 2005. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J. Neurosci. 25, 2895–2905.
Choquet D., Triller A. 2003. The role of receptor diffusion in the organization of the postsynaptic membrane. Nat. Rev. Neurosci. 4, 251–265.
van der Sluijs P., Hoogenraad C.C. 2011. New insights in endosomal dynamics and AMPA receptor trafficking. Semin. Cell. Dev. Biol. 22, 499–505.
Huganir R.L., Nicoll R.A. 2013. AMPARs and synaptic plasticity: The last 25 years. Neuron. 80, 704–717.
Mansour M., Nagarajan N., Nehring R.B., Clements J.D., Rosenmund C. 2001. Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron. 32, 841–853.
Kim K.S., Yan D., Tomita S. 2010. Assembly and stoichiometry of the AMPA receptor and transmembrane AMPA receptor regulatory protein complex. J. Neurosci. 30 (3), 1064–1072.
Soderling T.R., Derkach V.A. 2000. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 23 (2), 75–80.
Barria A., Muller D., Derkach V., Griffith L.C., Soderling T.R. 1997. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 276, 2042–2045.
Benke T., Traynelis S.F. 2019. AMPA-type glutamate receptor conductance changes and plasticity: Still a lot of noise. Neurochem. Res. 44 (3), 539–548.
Rakhade, S.N., Zhou C., Aujla P.K., Fishman R., Sucher N.J., Jensen F.E. 2008. Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J. Neurosci. 28 (32), 7979–7990.
Hollmann M., Hartley M., Heinemann S. 1991. Ca2+ permeability of KA-AMPA – gated glutamate receptor channels depends on subunit composition. Science. 252, 851–853.
Hume R.I., Dingledine R., Heinemann S.F. 1991. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 253, 1028–1031.
Sommer B., Kohler M., Sprengel R., Seeburg P.H. 1991. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 67, 11–19.
Verdoorn T.A., Burnashev N., Monyer H., Seeburg P.H., Sakmann B. 1991. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 252, 1715–1718.
Hollmann M., Boulter J., Maron C., Heinemann S. 1994. Molecular biology of glutamate receptors. Potentiation of N-methyl-D-aspartate receptor splice variants by zinc. Ren. Physiol. Biochem. 17 (3–4), 182–183.
Schwenk J., Harmel N., Brechet A., Zolles G., Berkefeld H., Müller C.S., Bildl W., Baehrens D., Hüber B., Kulik A., Klöcker N., Schulte U., Fakler B. 2012. High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron. 74 (4), 621–633.
Joshi P., Moradipour M., Nerkar A., Sawant S.D. 2012. AMPA receptor: A review. Int. J. Pharm. Pharm. Sci. 4, 39–44.
Bissen D., Foss F., Acker-Palmer A. 2019. AMPA receptors and their minions: Auxiliary proteins in AMPA receptor trafficking. Cell. Mol. Life Sci. 76 (11), 2133–2169.
Blair L.J., Criado-Marrero M., Zheng D., Wang X., Kamath S., Nordhues B.A., Weeber E.J., Dickey C.A. 2019. The disease-associated chaperone FKBP51 impairs cognitive function by accelerating AMPA receptor recycling. eNeuro. 6 (1), ENEURO.0242–18.2019.
Schwenk J., Harmel N., Zolles G., Bildl W., Kulik A., Heimrich B., Chisaka O., Jonas P., Schulte U., Fakler B., Klocker N. 2009. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 323, 1313–1319.
Shanks N.F., Savas J.N., Maruo T., Cais O., Hirao A., Oe S., Ghosh A., Noda Y., Greger I.H., Yates J.R. 3rd., Nakagawa T. 2012. Differences in AMPA and kainate receptor interactomes facilitate identification of AMPA receptor auxiliary subunit GSG1L. Cell Rep. 1, 590–598.
Farrow P., Khodosevich K., Sapir Y., Schulmann A., Aslam M., Stern-Bach Y., Monyer H., von Engelhardt J. 2015. Auxiliary subunits of the CKAMP family differentially modulate AMPA receptor properties. Elife. 4, e09693.
Tomita S., Stein V., Stocker T.J., Nicoll R.A., Bredt D.S. 2005. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 45, 269–277.
Bats C., Groc L., Choquet D. 2007. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 53, 719–734.
Coombs I.D., Cull-Candy S.G. 2009. Transmembrane AMPA receptor regulatory proteins and AMPA receptor function in the cerebellum. Neuroscience. 162, 656–665.
Soto D., Coombs I.D., Kelly L., Farrant M., Cull-Candy S.G. 2007. Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nat. Neurosci. 10, 1260–1267.
Soto D., Coombs I.D., Renzi M., Zonouzi M., Farrant M., Cull-Candy S.G. 2009. Selective regulation of long-form calcium-permeable AMPA receptors by an atypical TARP, γ-5. Nat. Neurosci. 12, 277–285.
Studniarczyk D., Coombs I., Cull-Candy S.G., Farrant M. 2013. TARP γ-7 selectively enhances synaptic expression of calcium-permeable AMPARs. Nat. Neurosci. 16 (9), 1266–1274.
Sumioka A., Yan D., Tomita S. 2010. TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron. 66, 755–767.
Benke T., Traynelis S.F. 2019. AMPA-type glutamate receptor conductance changes and plasticity: Still a lot of noise. Neurochem. Res. 44 (3), 539–548.
Chen L., Chetkovich D.M., Petralia R.S., Sweeney N.T., Kawasaki Y., Wenthold R.J., Bredt D.S., Nicoll R.A. 2000. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 408, 936–943.
Morimoto-Tomita M., Zhang W., Straub C., Cho C.H., Kim K.S., Howe J.R., Tomita S. 2009. Autoinactivation of neuronal AMPA receptors via glutamateregulated TARP interaction. Neuron. 61, 101–112.
Constals A., Penn A.C., Compans B., Toulmé E., Phillipat A., Marais S., Retailleau N., Hafner A.S., Coussen F., Hosy E., Choquet D. 2015. Glutamate-induced AMPA receptor desensitization increases their mobility and modulates short-term plasticity through unbinding from Stargazin. Neuron. 85, 787–803.
Priel A., Kolleker A., Ayalon G., Gillor M., Osten P., Stern-Bach Y. 2005. Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J. Neurosci. 25, 2682–2686.
Opazo P., Labrecque S., Tigaret C.M., Frouin A., Wiseman P.W., De Koninck P., Choquet D. 2010. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 67 (2), 239–252.
Khodosevich K., Jacobi E., Farrow P., Schulmann A., Rusu A., Zhang L., Sprengel R., Monyer H., von Engelhardt J. 2014. Coexpressed auxiliary subunits exhibit distinct modulatory profiles on AMPA receptor function. Neuron. 83, 601–615.
von Engelhardt J., Mack V., Sprengel R., Kavenstock N., Li K.W., Stern-Bach Y., Smit A.B., Seeburg P.H., Monyer H. 2010. CKAMP44: A brain-specific protein attenuating short-term synaptic plasticity in the dentate gyrus. Science. 327, 1518–1522.
von Engelhardt J. 2019. AMPA Receptor Auxiliary Proteins of the CKAMP Family. Int. J. Mol. Sci. 20 (6), E1460.
McGee T.P., Bats C., Farrant M., Cull-Candy S.G. 2015. Auxiliary subunit GSG1L acts to suppress calcium-permeable AMPA receptor function. J. Neurosci. 35 (49), 16171–16179.
Boudkkazi S., Brechet A., Schwenk J., Fakler B. 2014. Cornichon2 dictates the time course of excitatory transmission at individual hippocampal synapses. Neuron. 82, 848–858.
Kato A.S., Gill M.B., Ho M.T., Yu H., Tu Y., Siuda E.R., Wang H., Qian Y.W., Nisenbaum E.S., Tomita S., Bredt D.S. 2010. Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron. 68, 1082–1096.
Shi Y., Suh Y.H., Milstein A.D., Isozaki K., Schmid S.M., Roche K.W., Nicoll R.A. 2010. Functional comparison of the effects of TARPs and cornichons on AMPA receptor trafficking and gating. Proc. Natl. Acad. Sci. USA. 107, 16315–16319.
Gill M.B., Kato A.S., Roberts M.F., Yu H., Wang H., Tomita S., Bredt D.S. 2011. Cornichon-2 modulates AMPA receptor-transmembrane AMPA receptor regulatory protein assembly to dictate gating and pharmacology. J. Neurosci. 31, 6928–6938.
Herring B.E., Shi Y., Suh Y.H., Zheng C.Y., Blankenship S.M., Roche K.W., Nicoll R.A. 2013. Cornichon proteins determine the subunit composition of synaptic AMPA receptors. Neuron. 77, 1083–1096.
Konorski J. 1948. Conditioned reflexes and neuron organization. Cambridge, UK: Hefner Press. 267 p.
Malenka R.C., Nicoll R.A. 1999. Long-term potentiation – a decade of progress? Science. 285, 1870–1874.
Bliss T.V., Lomo T. 1973. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 232, 331–356.
Abbott L.F., Regehr W.G. 2004. Synaptic computation. Nature. 431, 796–803.
Abraham W.C. 2003. How long will long-term potentiation last? Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 735–744.
Martin S.J., Grimwood P.D., Morris R.G.M. 2000. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 23, 649–711.
Turrigiano G. 2011. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 34, 89–103.
Malinow R., Schulman H., Tsien R.W. 1989. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 245, 862–866.
Silva A.J., Stevens C.F., Tonegawa S., Wang Y. 1992. Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science. 257, 201–206.
Reymann K.G., Frey J.U. 2007. The late maintenance of hippocampal LTP: Requirements, phases, “synaptic tagging”, “late-associativity” and implications. Neuropharmacology. 52, 24–40.
Johnstone V.P., Raymond C.R. 2011. A protein synthesis and nitric oxide-dependent presynaptic enhancement in persistent forms of long-term potentiation. Learn. Mem. 18, 625–633.
Malenka R.C., Bear M.F. 2004. LTP and LTD: An embarrassment of riches. Neuron. 44 (1), 5–21.
Malinow R., Malenka R.C. 2002. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126.
Jia Z., Agopyan N., Miu P., Xiong Z., Henderson J., Gerlai R., Taverna F.A., Velumian A., Macdonald J., Carlen P., Abramow-Newerly W., Roder J. 1996. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 17, 945–956.
Harvery S.C., Koster A., Yu H., Skolnick P., Baumbarger P., Nisenbaum E.S. 2001. AMPA receptor function is altered in GluR2-deficient Mice. J. Mol. Neurosci. 17, 35–43.
Meng Y., Zhang Y., Jia Z. 2003. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 39, 163–176.
Swanson G.T., Kamboj S.K., Cull-Candy S.G. 1997. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J. Neurosci. 17, 58 –69.
Geiger J.R., Melcher T., Koh D.S., Sakmann B., Seeburg P.H., Jonas P., Monyer H. 1995. Relative abundance of subunit mRNAs determines gating and Ca 2+-permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 15, 193–204.
Зайцев А.В., Ким К.Х., Магазаник Л.Г. 2012. Роль кальций-проницаемых АМРА рецепторов в механизме дисинаптического торможения в префронтальной коре крысы. Биол. мембраны. 29 (1–2), 114–122.
Cull-Candy S., Kelly L., Farrant M. 2006. Regulation of Ca2+-permeable AMPA receptors: Synaptic plasticity and beyond. Curr. Opin. Neurobiol. 16, 288–297.
Riedel G., Micheau J., Lam A.G., Roloff E.L., Martin S.J., Bridge H., De Hoz L., Poeschel B., Mcculloch J., Morris R.G. 1999. Reversible neural inactivation reveals hippocampal participation in several memory processes. Nature. 2, 898–905.
Wright A., Vissel B. 2012. The essential role of AMPA receptor GluA2 subunit RNA editing in the normal and diseased brain. Front. Mol. Neurosci. 5, 34.
Liu B., Liau M., Mielke J.G., Ning K., Chen Y., Li L., El-Hayek Y.H., Gomez E., Zukin R.S., Fehlings M.G., Wan Q. 2006. Ischemic insults directs glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J. Neurosci. 26, 5309–5319.
Noh K.M., Yokota H., Mashiko T., Castillo P.E., Zukin R.S., Bennet M.V. 2005. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc. Natl. Acad. Sci. USA. 102, 12230–12235.
Brusa R., Zimmermann F., Koh, D.S., Feldmeyer D., Gass P., Seeburg P.H., Sprengel R. 1995. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 270, 1677–1680.
Bellone C., Luscher C. 2006. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat. Neurosci. 9, 636–641.
Park P., Kang H., Sanderson T.M., Bortolotto Z.A., Georgiou J., Zhuo M., Kaang B.K., Collingridge G.L. 2018. The role of calcium-permeable AMPARs in long-term potentiationat principal neurons in the rodent hippocampus. Front. Synaptic Neurosci. 10, 42.
Plant K., Pelkey K.A., Bortolotto Z.A., Morita D., Terashima A., McBain C.J., Collingridge G.L., Isaac J.T.R. 2006. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604.
Yang Y., Wang X.-B., Frerking M., Zhou Q. 2008. Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long term potentiation. Proc. Natl. Acad. Sci. USA. 105, 11388–11393.
Purkey A.M., Woolfrey K.M., Crosby K.C., Stich D.G., Chick W.S., Aoto J., Dell’Acqua M.L. 2018. AKAP150 Palmitoylation regulates synaptic incorporation of Ca2+-permeable AMPA receptors to control LTP. Cell Rep. 25, 974–987.
Jaafari N., Henley J.M. Hanley J.G. 2012. PICK1 mediates transient synaptic expression of GluA2-lacking AMPA receptors during glycine-induced AMPA receptor trafficking J. Neurosci. 32 (34), 11618–11630.
Park P., Sanderson T.M., Amici M., Choi S.L., Bortolotto Z.A., Zhuo M., Kaang B.K., Collingridge G.L. 2016. Calcium-permeable AMPA receptors mediate the induction of the protein kinase A-dependent component of long-term potentiation in the hippocampus. J. Neurosci. 36 (2), 622–631.
Washburn M.S., Dingledine R. 1996. Block of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by polyamines and polyamine toxins. J. Pharmacol. Exp. Ther. 278, 669–678.
Bowie D., Mayer M.L. 1995. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 15, 453–462.
Koh D.S., Burnashev N., Jonas P. 1995. Block of native Ca2+-permeable AMPA receptors in rat brain by intracellular polyamines generates double rectification. J. Physiol. 486, 305–312.
Rozov A., Zakharova Y., Vazetdinova A., Valiullina-Rakhmatullina F. 2018. The role of polyamine-dependent facilitation of calcium permeable AMPARs in short-term synaptic enhancement. Front.Cell Neurosci. 12, 345.
Baronas V.A., Kurata H.T. 2014. Inward rectifiers and their regulation by endogenous polyamines. Front. Physiol. 5, 325.
Lujan B., Dagostin A., von Gersdorff H. 2019. Presynaptic diversity revealed by Ca2+-permeable AMPA receptors at the calyx of held synapse. J. Neurosci. 39 (16), 2981–2994.
Spaethling J.M., Klein D.M., Singh P., Meaney D.F. 2008. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma. 25, 1207–1216.
Szczurowska E., Ergang P., Kubova H., Druga R., Salaj M., Mares P. 2016. Influence of early life status epilepticus on the developmental expression profile of the GluA2 subunit of AMPA receptors. Exp. Neurol. 283, 97–109.
Pellegrini-Giampietro D.E., Zukin R.S., Bennett M.V., Cho S., Pulsinelli W.A. 1992. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc. Natl. Acad. Sci. USA. 89, 10499–10503.
Whitehead G., Regan P., Whitcomb D.J., Cho K. 2017. Ca2+-permeable AMPA receptor: A new perspective on amyloid-β mediated pathophysiology of Alzheimer’s disease. Neuropharmacology. 112, 221–227.
Li S., Hong S., Shepardson N.E., Walsh D.M., Shankar G.M., Selkoe D. 2009. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 62, 788–801.
Miñano-Molina A.J., España J., Martín E., Barneda-Zahonero B., Fadó R., Solé M., Trullás R., Saura C.A., Rodríguez-Alvarez J. 2011. Soluble oligomers of amyloid-β peptide disrupt membrane trafficking of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. J. Biol. Chem. 286, 27311–27321.
Guntupalli S., Jang S.E., Zhu T., Huganir R.L., Widagdo J., Anggono V. 2017. GluA1 subunit ubiquitination mediates amyloid-β-induced loss of surface α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J. Biol. Chem. 292, 8186–8194.
Lehmann K., Steinecke A., Bolz J. 2012. GABA through the ages: regulation of cortical function and plasticity by inhibitory interneurons. Neural Plast. 2012, 892784.
Allen K., Monyer H. 2014. Interneuron control of hippocampal oscillations. Curr. Opin. Neurobiol. 31, 81–87.
Cobb S.R., Buhl E.H., Halasy K., Paulsen O., Somogyi P. 1995. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 378, 75–78.
Aponte Y., Bischofberger J., Jonas P. 2008. Efficient Ca2+ buffering in fast-spiking basket cells of rat hippocampus. J. Physiol. 586, 2061–2075.
Maccaferri G., Roberts J.D., Szucs P., Cottingham C.A., Somogyi P. 2000. Cell surface domain specific postsynaptic currents evoked by identified GABAergic neurones in rat hippocampus in vitro. J. Physiol. 1 (524), 91–116.
Orduz D., Bischop D.P., Schwaller B, Schiffmann S.N., Gall D. 2013. Parvalbumin tunes spike-timing and efferent short-term plasticity in striatal fast spiking interneurons. J. Physiol. 591 (13), 3215–3232.
Franconville R., Revet G., Astorga G., Schwaller B., Llano I. 2011. Somatic calcium level reports integrated spiking activity of cerebellar interneurons in vitro and in vivo. J. Neurophysiol. 106 (4), 793–805.
Cauli B., Audinat E., Lambolez B., Angulo M.C., Ropert N., Tsuzuki K., Hestrin S., Rossier J. 1997. Molecular and physiological diversity of cortical nonpyramidal cells. J. Neurosci. 17 (10), 3894–3906.
Collin T., Chat M., Lucas M.G., Moreno H., Racay P., Schwaller B., Marty A., Llano I. 2005. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J. Neurosci. 25 (1), 96–107.
Goldberg J.H., Tamas G., Aronov D., Yuste R. 2003. Calcium microdomains in aspiny dendrites. Neuron. 40, 807–821.
Goldberg J.H., Yuste R., Tamas G. 2003. Ca2+ imaging of mouse neocortical interneurone dendrites: contribution of Ca2+-permeable AMPA and NMDA receptors to subthreshold Ca2+ dynamics. J. Physiol. 551, 67–78.
Camiré O., Topolnik L. 2014. Dendritic calcium nonlinearities switch the direction of synaptic plasticity in fast-spiking interneurons. J. Neurosci. 34 (11), 3864–3877.
Hainmuller T., Krieglstein K., Kulik A., Bartos M. 2014. Joint CP-AMPA and group I mGlu receptor activation is required for synaptic plasticity in dentate gyrus fast-spiking interneurons. Proc. Natl. Acad. Sci. USA. 111, 13211–13216.
Soler-Llavina G.J., Sabatini B.L. 2006. Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nat. Neurosci. 9, 798–806.
Lamsa K.P., Heeroma J.H., Somogyi P., Rusakov D.A., Kullmann D.M. 2007. Anti-hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science. 315, 1262–1266.
Laezza F., Doherty J.J., Dingledine R. 1999. Long-term depression in hippocampal interneurons: Joint requirement for pre- and postsynaptic events. Science. 285, 1411–1414.
Lalanne T., Oyrer J., Mancino A., Gregor E., Chung A., Huynh L., Burwell S., Maheux J., Farrant• M., Sjöström P.J. 2016. Synapse-specific expression of calcium-permeable AMPA receptors in neocortical layer 5. J. Physiol. 15, 837–861.
Зинченко В.П., Гайдин С.Г., Теплов И.Ю., Косенков А.М., Сергеев А.И., Долгачева Л.П., Тулеуханов С.Т. 2020. Визуализация, свойства и функции ГАМК-ергических нейронов гиппокампа, содержащих кальций-проницаемые каинатные и AMPA-рецепторы. Биол. мембраны. 37 (1), 22–33.
Chávez A.E., Singer J.H., Diamond J.S. 2006. Fast neurotransmitter release triggered by Ca influx through AMPA-type glutamate receptors. Nature. 443 (7112), 705–708.
Xu J., Liu Y., Zhang G.Y. 2008. Neuroprotection of GluR5-containing kainate receptor activation against ischemic brain injury through decreasing tyrosine phosphorylation of N-methyl-D-aspartate receptors mediated by Src kinase. J. Biol. Chem. 283 (43), 29355–29366.
Valiullina F., Zakharova Y., Mukhtarov M., Draguhn A., Burnashev N., Rozov A. 2016. The relative contribution of NMDARs to excitatory postsynaptic currents is controlled by Ca2+-induced inactivation. Front. Cell Neurosci. 10, 12.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии