

Биологические мембраны: Журнал мембранной и клеточной биологии, 2021, T. 38, № 2, стр. 139-151
Влияние глюкокортикоида дефлазакорта на дыхание и кальций-зависимую проницаемость митохондрий печени крыс
М. В. Дубинин a, *, А. А. Семенова a, Е. И. Хорошавина a, К. Н. Белослудцев a, b
a Марийский государственный университет
424000 Республика Марий Эл, Йошкар-Ола, Россия
b Институт теоретической и экспериментальной биофизики РАН
142290 Пущино, Россия
* E-mail: dubinin1989@gmail.com
Поступила в редакцию 19.11.2020
После доработки 30.11.2020
Принята к публикации 01.12.2020
Аннотация
Изучено влияние глюкокортикоида дефлазакорта на функционирование митохондрий печени крыс. Показано, что дефлазакорт в концентрациях до 100 мкМ не оказывает влияния на дыхание и окислительное фосфорилирование митохондрий печени крыс, энергизованных как в присутствии глутамат/малата (субстраты комплекса I дыхательной цепи), так и сукцината (субстрат комплекса II дыхательной цепи). Выяснено, что дефлазакорт не влияет на проницаемость внутренней мембраны митохондрий печени, но снижает устойчивость органелл к индукции кальций-зависимой MPT поры. Кроме того, установлено, что этот глюкокортикоид способен индуцировать снижение уровня митохондриального NAD(P)H, а также ингибировать продукцию перекиси водорода органеллами. Обсуждается, как влияние дефлазакорта на функционирование митохондрий может быть связано с терапевтическими эффектами этого агента.
ВВЕДЕНИЕ
Природные и синтетические глюкокортикоиды влияют на большинство физиологических систем и являются наиболее часто используемыми агентами для терапии заболеваний различной этиологии, а также благодаря своим иммуносупрессивным свойствам активно используются при трансплантации органов и тканей. Общепризнано, что глюкокортикоиды участвуют в поддержании гиперметаболического состояния в ответ на стрессовые условия и травмы, что проявляется в повышении интенсивности метаболизма белков в скелетных мышцах, глюконеогенеза в печени, а также в развитии гипергликемии и резистентности к инсулину [1].
Действие глюкокортикоидов в организме в основном проявляется через глюкокортикоидные рецепторы. Такие рецепторы обнаружены в мембранах клеток и их ядер, а также на мембранах митохондрий [2–5]. Известно, что их активация глюкокортикоидами приводит к запуску внутриклеточных сигнальных путей, оказывающих в том числе значительное влияние на биогенез и функциональную активность митохондрий [6–9]. Так, показано, что основным эффектом глюкокортикоидов является активация ядерных генов, приводящая к усилению митохондриального биогенеза и массы органелл [9]. Кроме того, известно, что глюкокортикоиды оказывают значительное влияние на экспрессию ядерных генов, кодирующих белки системы окислительного фосфорилирования митохондрий, и, таким образом, влияют на дыхательную активность этих органелл [6–8, 10]. Наряду с этим выяснено, что глюкокортикоиды способны оказывать непосредственное влияние на митохондриальную биоэнергетику, не связанное с участием специализированных рецепторов. Так, было показано, что природные и синтетические глюкокортикоиды способны ингибировать дыхание изолированных митохондрий почек и мозга, которое, как предполагается, обусловлено снижением активности АТР-синтазы и цитохром-с–оксидазы [11–13]. Сообщается о способности кортикостерона подавлять транспорт электронов от NADH к комплексу I дыхательной цепи в кортикальных митохондриях [14]. Кроме того, известно, что глюкокортикоиды оказывают модулирующее влияние на работу мембранных белков, участвующих в том числе в транспорте ионов кальция в клетке и митохондриях [15].
В настоящей работе нами изучено влияние глюкокортикоида дефлазакорта, активно применяемого для коррекции нервно-мышечных заболеваний и, в частности, мышечной дистрофии Дюшенна, на функционирование митохондрий печени крыс в условиях in vitro. Дефлазакорт (рис. 1) представляет собой оксазолон-производное глюкокортикоида преднизона, обладающее сравнимыми с ним противовоспалительными и имунносупрессивными эффектами, но менее выраженными побочными эффектами по сравнению с предшественниками [16, 17]. Дефлазакорт, метаболизируясь в печени до активного метаболита – 21-дезацетилдефлазакорта, воздействует на глюкокортикоидные рецепторы, что приводит к подавлению высвобождения цитокинов и пролиферации CD4+ лимфоцитов, но в то же время увеличению количества CD8+ клеток, что способствует подавлению иммунного ответа и воспалительных процессов в мышечной ткани [16, 17]. Показано, что его использование продлевает двигательную способность пациентов, страдающих дистрофией Дюшенна, на срок от 2 до 5 лет и улучшает мышечную силу и сердечно-легочную функцию [17]. Однако его использование, тем не менее, сопровождается побочными эффектами, такими как синдром Кушинга, эритема, гирсутизм, увеличение веса, назофарингит, задержка полового созревания и катаракта [18–20].
Ранее нами было показано, что терапия с использованием дефлазакорта может быть использована для коррекции митохондриальной дисфункции, характерной для скелетной мускулатуры модельных мышей, страдающих дистрофией Дюшенна [21, 22]. Было предположено, что такое действие этого агента опосредовано его действием на глюкокортикоидные рецепторы, активирующие внутриклеточные сигнальные пути, затрагивающие, в том числе, и митохондрии. В то же время практически ничего не известно о непосредственном влиянии дефлазакорта на функционирование митохондрий. Поэтому в настоящей работе нами изучено влияние этого глюкокортикоида на параметры дыхания и окислительного фосфорилирования митохондрий печени крыс, проницаемость внутренней мембраны органелл, а также индукцию Са2+-зависимой митохондриальной поры (mitochondrial permeability transition pore, MPT-поры). Показано, что 1) дефлазакорт не оказывает влияния на дыхание и окислительное фосфорилирование митохондрий печени крыс, а также на активность комплексов дыхательной цепи органелл; 2) дефлазакорт индуцирует снижение уровня митохондриального пула NAD(P)H и оказывает ингибирующее влияние на генерацию перекиси водорода органеллами; 3) дефлазакорт снижает кальциевую емкость митохондрий печени и устойчивость органелл к индукции Са2+-зависимой MPT-поры.
МАТЕРИАЛЫ И МЕТОДЫ
Выделение митохондрий печени крыс. Митохондрии из печени половозрелых самцов крыс линии Вистар (весом 210–250 г) выделяли методом дифференциального центрифугирования с последующим освобождением от эндогенных жирных кислот с помощью бычьего сывороточного альбумина (БСА) фракции V в соответствии с описанной ранее методикой [23]. Среда выделения содержала 250 мМ сахарозы, 1 мМ EGTA, 5 мМ HEPES–KOH, pH 7.4. Концентрацию белка митохондрий определяли биуретовым методом, в качестве стандарта использовали раствор БСА. Во время проведения эксперимента суспензию митохондрий (70–80 мг митохондриального белка в 1 мл) хранили на льду.
Оценка дыхания и окислительного фосфорилирования митохондрий печени крыс. Дыхание митохондрий регистрировали полярографическим методом при 25°C в ячейке объемом 0.5 мл с помощью кислородного электрода типа Кларк и установки Oxygraph Plus (Hansatech Instruments, Великобритания) [24]. Среда инкубации содержала 200 мМ сахарозу, 20 мМ KCl, 5 мМ КН2РО4, 0.5 мМ EGTA, 2 мМ MgCl2, 10 мМ HEPES–KOH, pH 7.4. Применяли следующие концентрации субстратов и других реагентов: 2.5 мМ малат калия, 2.5 мМ глутамат калия, 5 мМ янтарная кислота, 0.2 мМ ADP, 50 мкМ 2,4-динитрофенол и 1 мкМ ротенон. Оценивали дыхание митохондрий в основном метаболическом состоянии (т.е. в присутствии экзогенных субстратов или состояние 2), в состоянии 3 (в присутствии экзогенных субстратов и ADP), в состояние 4 (после того, как весь добавленный ADP был израсходован в процессе синтеза АТР), скорость дыхания митохондрий в состоянии 3UДНФ (в присутствии протонофорного разобщителя 2,4-динитрофенола (ДНФ) в концентрации 50 мкМ, вызывающей максимальную стимуляцию дыхания). Скорость окисления субстратов выражена в нмоль O2 × мин−1 мг−1 белка митохондрий. Коэффициент дыхательного контроля (ДК = состояние 3/состояние 4). ADP/O – стехиометрический коэффициент, показывающий эффективность окислительного фосфорилирования. Значение коэффициента ADP/O определяли пульсовым методом [25]. Концентрация митохондриального белка в ячейке составляла ~1.0 мг/мл.
Оценка активности комплексов дыхательной цепи митохондрий печени крыс. Влияние дефлазакорта на активность комплексов дыхательной цепи митохондрий печени крыс оценивали спектрофотометрически согласно общепринятым протоколам [26, 27] с использованием планшетного спектрофотометра Multiskan GO (Thermo, США). Для разрушения митохондрий, освобождения индивидуальных комплексов дыхательной цепи и достижения их максимальной ферментативной активности изолированные митохондрии (10 мг/мл митохондриального белка) предварительно подвергали трехкратной процедуре замораживания/оттаивания при –20/+30°C в гипотоническом буфере, содержавшем 10 мМ Трис–HCl, pH 7.6. Состав буферов, использованных для анализа активности индивидуальных комплексов дыхательной цепи, приведен в работах [27]. Активность комплекса I оценивалась при 25°C по эффективности восстановления 2,6-дихлорфенолиндофенола разрушенными митохондриями (50 мкг митохондриального белка/мл), которое оценивалось по скорости уменьшения оптической плотности при 600 нм после внесения 100 мкМ NADH [26]. Активность комплекса II дыхательной цепи оценивали при 37°C по эффективности восстановления 2,6-дихлорфенолиндофенолята натрия суспензией разрушенных митохондрий (50 мкг митохондриального белка/мл) в присутствии субстрата окисления сукцината, которое определялось по снижению светопоглощения суспензии при длине волны 600 нм после внесения 50 мкМ децилубихинона. Активность комплекса III дыхательной цепи оценивали при 25°C по эффективности восстановления добавленного цитохрома с суспензией разрушенных митохондрий (25 мкг митохондриального белка/мл), которое определялось по снижению оптической плотности суспензии при длине волны 550 нм после внесения 100 мкМ децилубихинола. Активность комплекса IV дыхательной цепи оценивали при 25°C по эффективности окисления добавленного цитохрома с (восстановленного в соответствии с [27]) суспензией разрушенных митохондрий (50 мкг митохондриального белка/мл), которое определялось по снижению оптической плотности суспензии при длине волны 550 нм после внесения суспензии разрушенных органелл. Суммарная активность комплексов I и III оценивалась при 25°C по эффективности восстановления добавленного цитохрома с суспензией разрушенных митохондрий (50 мкг митохондриального белка/мл), что выражалось в снижении поглощения при длине волны 550 нм после внесения 200 мкМ NADH. Суммарная активность комплексов II и III была оценена при 37°C по эффективности восстановления добавленного цитохрома c суспензией разрушенных митохондрий (50 мкг митохондриального белка/мл), что выражалось в снижении поглощения при длине волны 550 нм после внесения 50 мкМ цитохрома с (окисленной формы). Активность комплексов дыхательной цепи оценивали в течение первых 1–2 мин с начала редокс-реакции (нмоль/мин на 1 мг белка). Влияние дефлазакорта на активность комплексов дыхательной цепи выражали в процентах от средней активности, регистрируемой в серии контрольных экспериментов.
Оценка разности электрических потенциалов (Δψ) на внутренней мембране митохондрий. Разность электрических потенциалов (Δψ) на внутренней мембране митохондрий оценивали по распределению катиона тетрафенилфосфония (ТФФ+) через внутреннюю мембрану, концентрацию которого регистрировали с помощью ТФФ+-чувствительного электрода и многоканальной электрометрической системы Record 4usb (разработка ИБК РАН, Россия) в кювете объемом 1.2 мл при 25°C и постоянной аэрации, и перемешивании [28]. Среда инкубации содержала 210 мМ маннитол, 70 мМ сахарозу, 1 мМ KH2PO4, 10 мкМ EGTA, 10 мМ HEPES–KOH, pH 7.4. Применяли следующие концентрации субстратов и других реагентов: 2.5 мМ малат калия, 2.5 мМ глутамат калия, 5 мМ янтарная кислота, 50 мкМ 2,4-динитрофенол и 1 мкМ ротенон. Концентрация митохондриального белка в ячейке составляла ~1.0 мг/мл.
Оценка кальциевой емкости митохондрий печени крыс. Транспорт Ca2+ через внутреннюю митохондриальную мембрану оценивали спектрофотометрически с помощью индикатора арсеназо III (3,6-бис-[(2-арсонофенил)азо]-4,5-дигидрокси-2,7-нафталиндисульфокислота) индикатора при 675–685 нм с помощью планшетного спектрофотометра Multiskan GO (Thermo, США) при 25°C и постоянном перемешивании [29]. Среда инкубации содержала 210 мМ маннитол, 70 мМ сахарозу, 1 мМ KH2PO4, 50 мкМ арсеназо III, 10 мкМ EGTA, 10 мМ HEPES–KOH, pH 7.4. Применяли следующие концентрации субстратов и других реагентов: 2.5 мМ малат калия, 2.5 мМ глутамат калия, 5 мМ янтарная кислота, 1 мкМ ротенон. Концентрация митохондриального белка в ячейке составляла ~ 0.5 мг/мл. Для определения способности митохондрий удерживать Ca2+, 5 мкМ CaCl2 последовательно добавлялись в реакционную среду. После нескольких добавлений происходило увеличение внешней [Ca2+], что указывало на массивное высвобождение иона из органелл вследствие открытия кальций-зависимой MPT-поры. Способность Ca2+ индуцировать открытие поры в митохондриях выражалась количественно как кальциевая емкость (КЕ) митохондрий, т.е. то максимальное количество Ca2+, которое может быть аккумулировано в матриксе без последующего открытия поры.
Оценка интенсивности набухания митохондрий. Открытие митохондриальной поры оценивали по набуханию митохондрий, которое регистрировали по изменению оптической плотности суспензии митохондрий при длине волны 540 нм (A540) при постоянном перемешивании и термостатировании при 25°C на планшетном спектрофотометре Multiskan GO (Thermo). Среда инкубации содержала 210 мМ маннитол, 70 мМ сахарозу, 1 мМ KH2PO4, 5 мМ сукцинат, 1 мкМ ротенон, 10 мкМ EGTA, 10 мМ HEPES-KOH, pH 7.4. Концентрация митохондриального белка в ячейке составляла ~ 0.5 мг/мл. Скорость набухания митохондрий (Vmax = ΔА540/мин на 1 мг белка) рассчитывали как изменение оптической плотности суспензии в течение первых 30 с от начала высокоамплитудного набухания.
Оценка состояния пула NAD(P)H. Автофлуоресценцию NAD(P)H измеряли при длине волны возбуждения 340 нм и длине волны излучения 450 нм с использованием спектрофлуориметра Fluorat-02-Panorama (Lumex Instruments, Россия). Среда инкубации содержала 210 мМ маннитол, 70 мМ сахарозу, 1 мМ KH2PO4, 10 мкМ EGTA, 10 мМ HEPES–KOH, pH 7.4. Применяли следующие концентрации субстратов и других реагентов: 2.5 мМ малат калия, 2.5 мМ глутамат калия, 5 мМ янтарная кислота, 1 мкМ ротенон. Концентрация митохондриального белка в ячейке составляла ~ 0.5 мг/мл. 5 мкМ n-трифторометоксикарбонилцианидфенилгидразона (FCCP) добавляли в конце измерений, чтобы вызвать максимальное окисление NAD(P)H.
Оценка скорости образования H2O2 митохондриями печени. Скорость образования Н2О2 измеряли с помощью тест-системы, включающей в себя флуоресцентный индикатор Amplex Red и пероксидазу хрена на планшетном флуориметре Tecan Spark 10M (Tecan, Швейцария) при длине волны возбуждения 560 нм и длине волны излучения 590 нм. Среда инкубации содержала 210 мM маннитол, 70 мM сахарозу, 5 мM сукцинат, 1 мM KH2PO4, 10 мкM ЭГTA, 1 мкM ротенон и 10 мM HEPES–KOH (pH 7.4), 10 мкМ Amplex Red и пероксидазу хрена (акт. 1 ед/мл). Концентрация митохондриального белка составляла 0.15 мг/мл. Измерения проводились при 37°C и постоянном перемешивании. Количество образованной перекиси водорода рассчитывалось по калибровочной кривой. Стандартный раствор H2O2 был приготовлен непосредственно в день проведения эксперимента; его концентрацию определяли с использованием коэффициента молярной экстинкции Е240 = 43.6 M−1 ⋅ см–1.
Статистическая обработка результатов. Данные были проанализированы с использованием программ Graph Pad Prizm 5 и Microsoft Excel и представлены как среднее ± стандартная ошибка средней. Полученные данные были обработаны статистически с использованием t-критерия. Для оценки значимости различий использовался уровень вероятности p < 0.05.
Материалы. Дефлазакорт и другие реактивы, используемые в работе, были приобретены в Sigma-Aldrich (США). Дефлазакорт растворяли в ДМСО (10 мМ сток-раствор). В контрольных пробах к митохондриям добавляли ДМСО в том же объеме, как в добавках дефлазакорта. Во всех случаях ДМСО не влиял на исследуемые параметры митохондрий.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Влияние различных стероидов in vitro на функциональную активность митохондрий ранее было подробно исследовано. В частности, было показано, что предшественник дефлазакорта, глюкокортикоид преднизон, дозозависимо в концентрациях 10–100 мкМ и более существенно снижал скорость дыхания митохондрий печени и почек в состоянии 3 (ADP-стимулированном) [11, 12, 30, 31]. Подобным эффектом обладали и другие природные и синтетические глюкокортикоиды (кортизон, кортизол, гидрокортизон, преднизолон, дексаметазон и др.) [12]. Такое действие указанных стероидов сопровождалось существенным снижением дыхательного контроля (ДК) и коэффициента ADP/O, что свидетельствовало об угнетении функциональной активности органелл.
В настоящей работе мы оценили влияние глюкокортикоида дефлазакорта, применяемого для коррекции мышечной дистрофии Дюшенна, на параметры дыхания и окислительного фосфорилирования митохондрий печени крыс. В табл. 1 представлены результаты, описывающие эффект дефлазакорта на дыхание митохондрий печени крыс в разных функциональных состояниях в присутствии глутамата и малата, субстратов комплекса I дыхательной цепи. Можно видеть, что дефлазакорт в концентрациях 20–100 мкМ не влияет на скорости дыхания митохондрий печени крыс во всех метаболических состояниях (состояние 2, состояние 3, состояние 4 и состояние 3UДНФ), а также не влияет на параметр ДК и эффективность синтеза ATP, что оценивалось по коэффициенту ADP/O. Аналогичные результаты получены в присутствии сукцината, субстрата комплекса II дыхательной цепи митохондрий (табл. 2), можно видеть, что дефлазакорт и в этом случае не влияет на параметры дыхания и окислительного фосфорилирования органелл во всем диапазоне изучаемых концентраций. В то же время другой глюкокортикоид – преднизолон в концентрации 100 мкМ достоверно снижал скорость ADP-стимулированного дыхания митохондрий печени и, напротив, увеличивал скорость дыхания органелл в состоянии 4 вне зависимости от используемого субстрата дыхания (табл. 1 и 2). Такое действие преднизолона сопровождалось снижением параметра дыхательного контроля ДК и коэффициента ADP/O. Это соответствует известным данным, свидетельствующим об угнетающем влиянии преднизолона на функциональную активность митохондрий печени [30].
Таблица 1.
Влияние дефлазакорта и преднизолона на дыхание митохондрий в разных функциональных состояниях в присутствии глутамата и малата
| Дефлазакорт, мкМ | Состояние 2 | Состояние 3 | Состояние 4 | Состояние 3UДНФ | ДК | ADP/O |
|---|---|---|---|---|---|---|
| V дыхания, нмоль O2/мин/мг белка митохондрий | отн. ед. | |||||
| 0 | 4.68 ± 0.44 | 24.01 ± 0.23 | 5.94 ± 0.30 | 27.53 ± 1.40 | 4.08 ± 0.26 | 2.61 ± 0.11 |
| 20 | 3.89 ± 0.28 | 21.87 ± 1.33 | 5.19 ± 0.25 | 25.48 ± 1.61 | 4.21 ± 0.05 | 2.63 ± 0.09 |
| 50 | 4.50 ± 0.49 | 25.45 ± 0.56 | 6.17 ± 0.27 | 30.35 ± 0.71 | 4.13 ± 0.22 | 2.64 ± 0.07 |
| 100 | 4.01 ± 0.13 | 23.94 ± 1.56 | 6.12 ± 0.64 | 27.71 ± 0.57 | 3.94 ± 0.14 | 2.52 ± 0.13 |
| 100 мкМ преднизолон | 5.21 ± 0.32 | 21.75 ± 0.53* | 8.26 ± 0.44* | 25.41 ± 1.09 | 2.71 ± 0.16* | 2.15 ± 0.08* |
Примечание. Дыхание митохондрий в состоянии 3 индуцировали добавлением 200 мкМ ADP. Скорость разобщенного дыхания измеряли в присутствии 50 мкМ ДНФ (состояние 3UДНФ). Дыхательный контроль (ДК) рассчитан как отношение скорости дыхания в состоянии 3 к скорости дыхания в состоянии 4. Приведены средние значения ± стандартная ошибка среднего (n = 3). * – различия между данными опытов (присутствие указанной концентрации преднизолона) и контролем (отсутствие преднизолона) статистически значимы, р < 0.05.
Таблица 2.
Влияние дефлазакорта и преднизолона на дыхание митохондрий в разных функциональных состояниях в присутствии сукцината
| Дефлазакорт, мкМ | Состояние 2 | Состояние 3 | Состояние 4 | Состояние 3UДНФ | ДК | ADP/O |
|---|---|---|---|---|---|---|
| V дыхания, нмоль O2/мин/мг белка митохондрий | отн. ед. | |||||
| 0 | 9.44 ± 1.05 | 45.61 ± 1.98 | 11.97 ± 0.62 | 63.41 ± 2.38 | 3.82 ± 0.17 | 1.63 ± 0.08 |
| 20 | 7.92 ± 0.65 | 48.89 ± 3.30 | 12.84 ± 0.35 | 65.65 ± 2.92 | 3.81 ± 0.16 | 1.61 ± 0.11 |
| 50 | 7.90 ± 0.34 | 45.47 ± 0.68 | 11.78 ± 0.22 | 61.01 ± 0.28 | 3.86 ± 0.09 | 1.61 ± 0.05 |
| 100 | 8.96 ± 0.61 | 47.12 ± 1.12 | 12.95 ± 0.21 | 65.04 ± 0.95 | 3.65 ± 0.09 | 1.60 ± 0.09 |
| 100 мкМ преднизолон | 10.17 ± 0.88 | 37.86 ± 0.92* | 15.81 ± 0.52* | 57.24 ± 0.78 | 2.43 ± 0.11* | 1.21 ± 0.09* |
Примечание. В качестве субстрата дыхания применяли 5 мМ янтарную кислоту. Дыхание митохондрий в состоянии 3 индуцировали добавлением 200 мкМ ADP. Скорость разобщенного дыхания измеряли в присутствии 50 мкМ ДНФ (состояние 3UДНФ). Дыхательный контроль (ДК) рассчитан как отношение скорости дыхания в состоянии 3 к скорости дыхания в состоянии 4. Приведены средние значения ± стандартная ошибка среднего (n = 3). * – различия между данными опытов (присутствие указанной концентрации преднизолона) и контролем (отсутствие преднизолона) статистически значимы, р < 0.05.
Из данных табл. 3 видно, что дефлазакорт в концентрации 50 мкМ не оказывает действия на активность как отдельных комплексов дыхательной цепи органелл (комплексы I, II, III и IV), так и суммарную активность комплексов I + III и II + III, что свидетельствует об отсутствии влияния этого агента на мобильность коэнзима Q и его доступность для комплексов дыхательной цепи.
Таблица 3.
Активность комплексов дыхательной цепи митохондрий печени крыс в присутствии 50 мкМ дефлазакорта (в % от контроля)
| Комплекс I | Комплекс II | Комплекс III | Комплекс IV | Комплекс I + III | Комплекс II + III |
|---|---|---|---|---|---|
| 100.5 ± 2.5 | 101.5 ± 3.0 | 96.5 ± 6.7 | 97.7 ± 3.3 | 103.1 ± 1.9 | 103.3 ± 1.1 |
Ранее было показано, что кортикостероиды и, в частности, предшественник дефлзакорта преднизон, оказывают ингибирующее влияние на активность комплексов дыхательной цепи митохондрий и, прежде всего, цитохром с оксидазы (комплекс IV), которое сопровождается подавлением дыхания органелл [12]. При этом предполагается, что такое действие глюкокортикоидов может лежать в основе многочисленных побочных эффектов этих терапевтических соединений и, прежде всего, психических расстройств [12]. В нашем случае можно отметить, что дефлазакорт, в отличие от других природных и синтетических глюкокортикоидов не оказывает влияния на активность цитохром с оксидазы и других комплексов дыхательной цепи и ее компонентов. В то же время следует отметить наши недавние данные, свидетельствующие о том, что in vivo введение дефлазакорта модельным мышам, страдающим мышечной дистрофией Дюшенна, приводит к увеличению уровня комплексов III и IV в митохондриях скелетных мышц, а также АТР-синтазы [22]. Однако в этом случае можно полагать, что действие дефлазакорта обусловлено известной способностью этих агентов активировать глюкокортикоидные рецепторы и внутриклеточные сигнальные пути, затрагивающие, в том числе, ядерные гены и гены митохондриальной ДНК, кодирующие белки комплексов дыхательной цепи митохондрий [5]. С другой стороны, введение этого агента здоровым мышам не приводило к достоверному изменению уровня комплексов дыхательной цепи органелл [22]. Полученные результаты можно рассматривать как свидетельство того, что дефлазакорт, в отличие от своего предшественника преднизона, не способен оказывать прямое влияние на функционирование системы окислительного фосфорилирования митохондрий. Возможно, что это также обуславливает менее выраженные психиатрические побочные эффекты, характерные для терапии с использованием дефлазакорта по сравнению с преднизоном и другими глюкокортикоидами [16, 17].
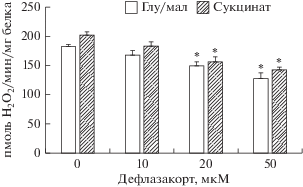
Известно, что глюкокортикоиды в концентрациях порядка 1 мМ индуцируют набухание митохондрий, что может свидетельствовать о способности этих соединений влиять на проницаемость внутренней мембраны органелл [30]. Однако меньшие концентрации этих агентов не эффективны [12]. В настоящей работе мы также оценили влияние дефлазакорта на проницаемость внутренней мембраны митохондрий печени крыс, которое может быть зарегистрировано по снижению оптической плотности суспензии органелл, свидетельствующему об их набухании. Из рис. 2 можно видеть, что дефлазакорт в концентрациях до 100 мкМ не оказывает влияния на оптическую плотность суспензии митохондрий печени вне зависимости от используемого субстрата дыхания (глутамат/малат или сукцинат). В этом случае лишь дальнейшее добавление известного каналообразующего агента аламетицина индуцировало высокоамплитудное набухание органелл. Кроме того, мы изучили влияние дефлазакорта на изменение Δψ на внутренней мембране митохондрий, которое оценивали по распределению ТФФ+. Как видно на рис. 3, пульсовое внесение дефлазакорта к суспензии органелл практически не оказывает влияния на изменение Δψ на внутренней мембране митохондрий. Лишь в конечной концентрации 100 мкМ дефлазакорт вызывает незначительное снижение мембранного потенциала органелл, энергизованных NAD-зависимыми субстратами. Наряду с этим можно отметить, что дефлазакорт в концентрациях 50 и 100 мкМ, добавленный к митохондриям печени крыс, энергизованным глутамат/малатом, вызывает дозозависимое уменьшение интенсивности автофлуоресценции NAD(P)H, что свидетельствует о снижение уровня восстановленной формы нуклеотидов (рис. 4а). Подобного не наблюдается при внесении к митохондриям эквивалентного количества растворителя – ДМСО (рис. 4а). Аналогичные результаты получены в случае энергизации митохондрий сукцинатом – дефлазакорт также индуцировал дозозависимое уменьшение интенсивности автофлуоресценции NAD(P)H, свидетельствующее о снижении пула восстановленной формы последних (рис. 4б). Стоит отметить, что такое действие дефлазакорта проявлялось только в присутствии субстрата окисления и не наблюдалось в случае неэнергизованных митохондрий (рис. 4в). Кроме того, в контрольных экспериментах (при внесении восстановленной формы пиридиновых нуклеотидов в среду инкубации в отсутствие митохондрий) выяснено, что дефлазакорт не оказывает непосредственного влияния на редокс-состояние пиридиновых нуклеотидов (данные не приведены). Исходя из этого, можно предполагать, что дефлазакорт способен активировать митохондриальные процессы, требующие участия NAD(P)H. К таким процессам могут относиться NAD(P)H-зависимые антиоксидантные системы митохондрий и, в частности, тиол-дисульфидная глутатионовая редокс-система и ее ферменты: глутатионпероксидаза и глутатионредуктаза, участвующие в утилизации перекиси водорода, генерируемой, прежде всего, дыхательной цепью митохондрий [32]. В этом случае стоит ожидать снижения уровня H2O2 в митохондриях. Действительно, можно видеть, что дефлазакорт дозозависимо снижает скорость генерации H2O2 митохондриями как в присутствии субстратов комплекса I дыхательной цепи (глутамата/малат), так и субстрата комплекса II дыхательной цепи (сукцинат в присутствии ротенона) (рис. 5).
Рис. 2.
Влияние 50 мкМ дефлазакорта на изменения оптической плотности суспензии митохондрий печени, энергизованных глутамат/малатом (а) и сукцинатом (б). Субстраты и реагенты: 2.5 мМ малат калия, 2.5 мМ глутамат калия (а), 5 мМ янтарная кислота, 1 мкМ ротенон (б). Концентрация митохондриального белка в кювете – 0.5 мг/мл. 5 мкг/мл аламетицина (Ала) добавлено в конце каждого эксперимента для оценки максимального набухания органелл. На каждом из рисунков представлены данные типичного эксперимента, полученные на одном препарате митохондрий. В каждом случае аналогичные результаты были получены еще в двух независимых экспериментах.
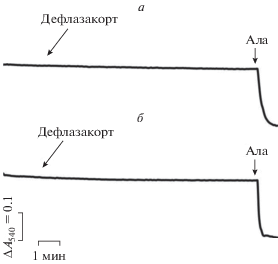
Рис. 3.
Влияние дефлазакорта (ДФК) на мембранный потенциал митохондрий печени крыс, энергизованных глутамат/малатом (а) и сукцинатом (б). Субстраты и реагенты: 2.5 мМ малат калия, 2.5 мМ глутамат калия (а), 5 мМ янтарная кислота, 1 мкМ ротенон (б), 50 мкМ ДНФ (а, б). На каждом из рисунков представлены данные типичного эксперимента, полученные на одном препарате митохондрий. В каждом случае аналогичные результаты были получены еще в двух независимых экспериментах.
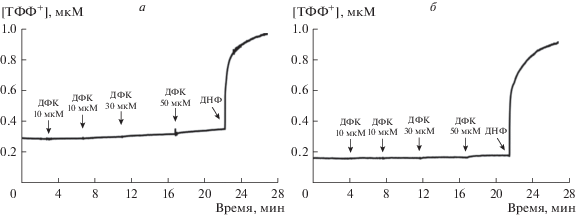
Рис. 4.
Изменение редокс-состояние пула NAD(P)H в митохондриях печени крыс, энергизованных глутамат/малатом (а), сукцинатом (б) и в отсутствии субстратов окисления (в) при добавлении 50 мкМ (точечная линия) и 100 мкМ дефлазакорта (пунктирная линия) или эквивалентного количества растворителя – ДМСО (сплошная линия). Субстраты и реагенты: 2.5 мМ малат калия, 2.5 мМ глутамат калия (а), 5 мМ янтарная кислота, 1 мкМ ротенон (б), 1 мкМ ротенон (в). Концентрация митохондриального белка в кювете – 0.5 мг/мл. 5 мкМ FCCP добавлено в конце каждого эксперимента для оценки максимального окисления пиридиновых нуклеотидов. На каждом из рисунков представлены данные типичного эксперимента, полученные на одном препарате митохондрий. В каждом случае аналогичные результаты были получены еще в двух независимых экспериментах.
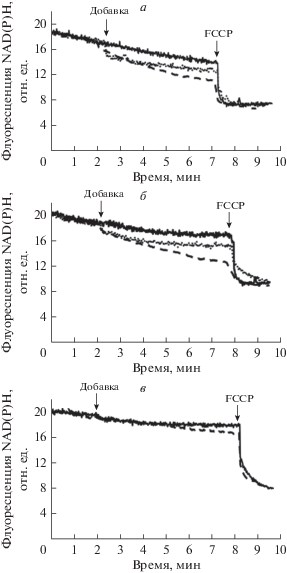
Рис. 5.
Влияние различных концентраций дефлазакорта на продукцию H2O2 митохондриями печени крысы, энергизованными глутамат/малатом или сукцинатом. Приведены средние ± ошибка среднего (n = 4). * – различия между данными опытов (присутствие указанных концентраций дефлазакорта) и контролем (отсутствие дефлазакорта) статистически значимы, р < 0.05.
Общепризнано, что митохондрии не только обеспечивают клетку АТР и теплом, но играют ведущую роль в процессах регуляции ее функций при участии свободного Ca2+ [33–35]. Известно, что изолированные митохондрии из различных органов и тканей животных в энергизованном состоянии обладают способностью аккумулировать и удерживать Ca2+ в матриксе [34, 35]. Однако при критической перегрузке матрикса органелл ионами кальция наблюдается индукция неспецифической проницаемости внутренней мембраны для ионов и растворимых в воде веществ с молекулярной массой до 1500 Да по их градиенту концентрации (или открытие митохондриальной поры) [33–35]. Образование такой поры приводит к нарушению энергетических функций митохондрий, прежде всего синтеза АТР, а также может вызвать набухание матрикса митохондрий, разрыв внешней мембраны и, как следствие, выход находящихся в межмембранном пространстве цитохрома c и других, так называемых апоптогенных белков [33–35]. В связи с этим индукция поры во внутренней мембране митохондрий рассматривается как один из факторов гибели клеток при различных патологических состояниях [33–36].
Известно, что природные и синтетические глюкокортикоиды способны модулировать внутриклеточные потоки ионов кальция [15]. В тоже время в случае митохондрий действие глюкокортикоидов достаточно специфично. Так было показано, что дексаметазон в высоких концентрациях (до 200 мкМ) ингибирует открытие кальций-зависимой MPT-поры в митохондриях печени и почек в экспериментах in vitro [37]. С другой стороны, в подобных экспериментальных условиях другие глюкокортикоиды (преднизолон, гидрокортизон и др.) в концентрациях менее 100 мкМ не оказывали значительного влияния на кальций-зависимое набухание изолированных митохондрий почек [12].
В настоящей работе мы также оценили влияние дефлазакорта на эффективность индукции кальций-зависимой поры в митохондриях печени крыс. На рис. 6 представлены результаты сравнительного изучения кинетики поглощения Ca2+ митохондриями печени крыс, энергизованными глутамат/малатом или сукцинатом в инкубируемых в отсутствие и в присутствии дефлазакорта. Можно видеть, что митохондрии печени крыс, энергизованные глутамат/малатом, полностью поглощают Ca2+ при условии добавления CaCl2 8 раз по 10 нмоль на 1 мг белка (рис. 6а, кривая 1) и удерживают его в матриксе в течение как минимум 10 мин (данные не приведены). В этом случае только после девятой добавки CaCl2 наблюдается выход Ca2+ из митохондрий (рис. 6а, кривая 1). Предварительная инкубация митохондрий в присутствии 20 мкМ дефлазакорта не оказывает существенного влияния на способность митохондрий поглощать ионы кальция (рис. 6а, кривая 2). Однако внесение дефлазакорта в концентрации 50 и 100 мкМ приводит к тому, что выход кальция из митохондрий происходит, соответственно, уже после 8 и 7 добавки CaCl2 в том же количестве (рис. 6а, кривые 3 и 4). Подобная картина наблюдается и в случае митохондрий, энергизованных сукцинатом (рис. 4б), – предварительная инкубация органелл с 50 и 100 мкМ дефлазакорта приводит к снижению способности митохондрий поглощать ионы кальция.
Рис. 6.
Влияние дефлазакорта (ДФК) на транспорт кальция в митохондриях печени крыс. а – Поглощение добавок Са2+ (пульсы по 5 мкМ) митохондриями печени, энергизованными глутамат/малатом, в отсутствие добавок (1) и в присутствии 20 мкМ (2), 50 мкМ (3) и 100 мкМ (4) дефлазакорта. б – Поглощение добавок Са2+ (пульсы по 5 мкМ) митохондриями печени, энергизованными сукцинатом, в отсутствие добавок (1) и в присутствии 20 мкМ (2), 50 мкМ (3) и 100 мкМ (4) дефлазакорта. в – Кальциевая емкость митохондрий печени крыс в отсутствие (контроль) и в присутствии различных концентраций дефлазакорта. Субстраты и реагенты: 2.5 мМ малат калия, 2.5 мМ глутамат калия (а), 5 мМ янтарная кислота, 1 мкМ ротенон (б). Концентрация митохондриального белка в кювете – 0.5 мг/мл. На панелях а и б представлены данные типичного эксперимента, полученные на одном препарате митохондрий. В каждом случае аналогичные результаты были получены еще в трех независимых экспериментах. Панель в – приведены средние значения ± стандартная ошибка среднего (n = 4). * – различия между данными опытов (присутствие указанных концентраций дефлазакорта) и контролем (отсутствие дефлазакорта) статистически значимы, р < 0.05.
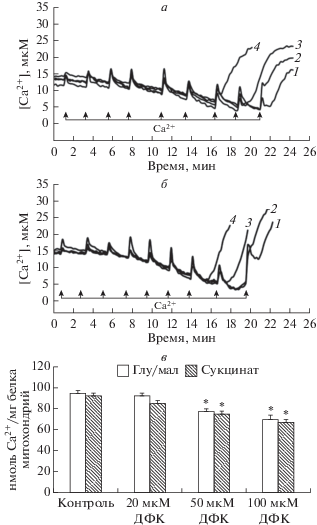
Способность Ca2+ индуцировать открытие поры в митохондриях можно выразить количественно как кальциевая емкость (КЕ) митохондрий, т.е. то максимальное количество Ca2+, которое может быть аккумулировано в матриксе без последующего открытия поры [34]. Очевидно, что, исходя из кинетики поглощения Ca2+ (рис. 6а и 6б), можно определить КЕ митохондрий печени в отсутствие и в присутствии различных концентраций дефлазакорта. Как показано на рис. 6в, дефлазакорт в концентрации 20 мкМ не оказывает существенного влияния на этот параметр митохондрий вне зависимости от субстрата окисления. В то же время 50 и 100 мкМ дефлазакорта вызывают достоверное снижение КЕ митохондрий печени крыс по сравнению с контролем.
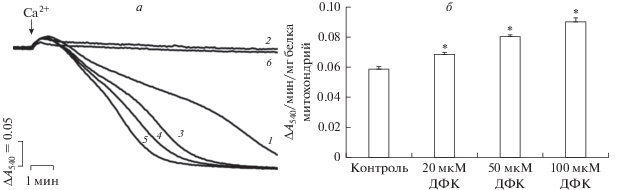
Другим параметром, позволяющим оценить влияние дефлазакорта на эффективность индукции Ca2+-зависимой митохондриальной поры, является регистрация набухания органелл в ответ на добавку ионов кальция. Известно, что набухание митохондрий приводит к уменьшению рассеивания проходящего через суспензию света, и это может быть зарегистрировано как снижение оптической плотности суспензии этих органелл [21]. Как видно на рис. 7а (кривая 1), добавление 60 нмоль на 1 мг белка CaCl2 к митохондриям печени крыс, инкубируемым в среде с Pi и энергизованным путем окисления сукцината, приводит к существенному снижению оптической плотности суспензии. В то время как в присутствии циклоспорина А (ЦсА), ингибитора митохондриальной MPT-поры, такого эффекта не наблюдается (рис. 7а, кривая 2). Предварительная инкубация органелл с дефлазакортом в концентрациях 20, 50 и 100 мкМ приводит к дозозависимому увеличению эффективности Ca2+-индуцированного набухания органелл (рис. 7а, кривые 3–5), которое также блокируется ЦсА (рис. 7а, кривая 6). Полученные данные суммированы на рис. 7б, где представлены скорости Ca2+-индуцированного набухания митохондрий печени крыс в отсутствие и в присутствии дефлазакорта. Можно видеть, что дефлазакорт дозозависимо увеличивает эффективность Ca2+ как индуктора MPT-поры. Таким образом, этот глюкокортикоид способен снижать резистентность органелл к индукции ЦсА-чувствительной митохондриальной поры. Стоит отметить, что подобные результаты были получены нами ранее и в экспериментах in vivo – терапия с использованием дефлазакорта достоверно снижала КЕ митохондрий скелетных мышц и, соответственно, устойчивость органелл к индукции кальций-зависимой MPT-поры [22]. Однако в этом случае действие этого агента было также обусловлено запуском внутриклеточных сигнальных путей и увеличением экспрессии генов, кодирующих белки – предполагаемые компоненты MPT-поры и, прежде всего, изоформы ADP/ATP-антипортера ANT2 [22]. Можно полагать, что снижение устойчивости митохондрий к индукции кальций-зависимой поры в присутствии дефлазакорта также может быть связано с редокс-состоянием пула NAD(P)H. Действительно, известно, что снижение уровня митохондриального NAD(P)H может приводить к уменьшению резистентности органелл к индукции кальций-зависимой MPT-поры [38–40].
Рис. 7.
Влияние дефлазакорта (ДФК) на Са2+-зависимое набухание митохондрий печени. а – кинетика изменения оптической плотности суспензии митохондрий печени в присутствии 30 мкМ Са2+ (1); 1 мкМ ЦсА и 30 мкМ Са2+ (2); 20 мкМ ДФК и 30 мкМ Са2+ (3); 50 мкМ ДФК и 30 мкМ Са2+ (4); 100 мкМ ДФК и 30 мкМ Са2+ (5); 1 мкМ ЦсА, 100 мкМ ДФК и 30 мкМ Са2+ (6). Концентрация митохондриального белка в кювете – 0.5 мг/мл. б – зависимость скорости Са2+-зависимого набухания митохондрий печени от концентрации ДФК. На панели а представлены данные типичного эксперимента, полученные на одном препарате митохондрий. В каждом случае аналогичные результаты были получены еще в трех независимых экспериментах. Панель б –приведены средние значения ± стандартная ошибка среднего (n = 4). * – различия между данными опытов (присутствие указанных концентраций дефлазакорта) и контролем (отсутствие дефлазакорта) статистически значимы, р < 0.05.
Суммируя полученные данные, можно полагать, что действие дефлазакорта в организме, с одной стороны, может реализовываться через рецепторы и активацию сигнальных клеточных путей, затрагивающих митохондрии. С другой стороны, этот глюкокортикоид, по всей видимости, способен оказывать непосредственное влияние на функционирование митохондриальных ферментов и индукцию кальций-зависимой MPT-поры.
Работа выполнена при поддержке гранта Российского научного фонда № 20-75-10 006.
Список литературы
Brillon D.J., Zheng B., Campbell R.G., Matthews D.E. 1995. Effect of cortisol on energy expenditure and amino acid metabolism in humans. Am. J. Physiol. 268 (3), 501–513.
Gregory M.C., Duval D., Meyer P. 1976. Changes in cardiac and hepatic glucocorticoid receptors after adrenalectomy. Clin. Sci. Mol. Med. 51, 487–493.
Demonacos C., Tsawdaroglou N.C., Djordjevic-Markovic R., Papalopoulou M., Galanopoulos V., Papadogeorgaki S., Sekeris C.E. 1993. Import of the glucocorticoid receptor into rat liver mitochondria in vivo and in vitro. J. Steroid Biochem. Mol. Biol. 46, 401–413.
Tsiriyotis C., Spandidos D.A., Sekeris C.E. 1997. The mitochondrion as a primary site of action of glucocorticoids: mitochondrial nucleotide sequences, showing similarity to hormone response elements, confer dexamethasone inducibility to chimaeric genes transfected in LATK-cells. Biochem. Biophys. Res. Commun. 235, 349–354.
Lee S.R., Kim H.K., Song I.S., Youm J., Dizon L.A., Jeong S.H., Ko T.H., Heo H.J., Ko K.S., Rhee B.D., Kim N., Han J. 2013. Glucocorticoids and their receptors: Insights into specific roles in mitochondria. Prog. Biophys. Mol. Biol. 112 (1–2), 44–54.
Kittas C., Chaniotis F.J., Antonakopoulos G., Papacharalampous N.X. 1984. The effects of bilateral adrenalectomy on the fine structure of the rat myocardium. Zentralbl. Allg. Pathol. 129, 225–229.
Djouadi F., Bastin J., Gilbert T., Rotig A., Rustin P., Merlet-Benichou C. 1994. Mitochondrial biogenesis and development of respiratory chain enzymes in kidney cells: Role of glucocorticoids. Am. J. Physiol. 267, 245–254.
Prieur B., Bismuth J., Delaval E. 1998. Effects of adrenal steroid hormones on mitochondrial maturation during the late fetal period. Eur. J. Biochem. 252, 194–199.
Goffart S., Wiesner R.J. 2003. Regulation and co-ordination of nuclear gene expression during mitochondrial biogenesis. Exp. Physiol. 88, 33–40.
Desquiret V., Gueguen N., Malthiery Y., Ritz P., Simard G. 2008. Mitochondrial effects of dexamethasone imply both membrane and cytosolic-initiated pathways in HepG2 cells. Int. J. Biochem. Cell Biol. 40, 1629–1641.
Morin C., Zini R., Simon N., Charbonnier P., Tillement J. P., Le Louet H. 2000. Low glucocorticoid concentrations decrease oxidative phosphorylation of isolated rat brain mitochondria: An additional effect of dexamethasone. Fundam. Clin. Pharmacol. 14 (5), 493–500.
Simon N., Jolliet P., Morin C., Zini R., Urien S., Tillement J.P. 1998. Glucocorticoids decrease cytochrome c oxidase activity of isolated rat kidney mitochondria. FEBS Lett. 435, 25–28.
Katyare S.S., Balasubramanian S., Parmar D.V. 2003. Effect of corticosterone treatment on mitochondrial oxidative energy metabolism in developing rat brain. Exp. Neurol. 183, 241–248.
Fujita C., Ichikawa F., Teratani T., Murakami G., Okada T., Shinohara M., Kawato S., Ohta Y. 2009. Direct effects of corticosterone on ATP production by mitochondria from immortalized hypothalamic GT1–7 neurons. J. Steroid Biochem. Mol. Biol. 117, 50–55.
Du J., Wang Y., Hunter R., Wei Y., Blumenthal R., Falke C., Khairova R., Zhou R., Yuan P., Machado-Vieira R., McEwen B.S., Manji H.K. 2009. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. USA. 106, 3543–3548.
Markham A., Bryson H.M. 1995. Deflazacort. A review of its pharmacological properties and therapeutic efficacy. Drugs. 50 (2), 317–333.
Bylo M., Farewell R., Coppenrath V.A., Yogaratnam D. 2020. A review of deflazacort for patients with Duchenne muscular dystrophy. Ann. Pharmacother. 54 (8), 788–794.
Bello L., Gordish-Dressman H., Morgenroth L.P., Henricson E.K., Duong T., Hoffman E.P., Cnaan A., McDonald C.M. 2015. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne natural history study. Neurology. 85 (12), 1048–1055.
Griggs R.C., Miller J.P., Greenberg C.R., Fehlings D.L., Pestronk A., Mendell J.R., Moxley R.T., King W., Kissel J.T., Cwik V., Vanasse M., Florence J.M., Pandya S., Dubow J.S., Meyer J.M. 2016. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology. 87 (20), 2123–2131.
McDonald C.M., Henricson E.K., Abresch R.T., Duong T., Joyce N.C., Hu F., Clemens P.R., Hoffman E.P., Cnaan A., Gordish-Dressman H. 2018. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet. 391 (10 119), 451–461.
Dubinin M.V., Talanov E.Y., Tenkov K.S., Starinets V.S., Mikheeva I.B., Sharapov M.G., Belosludtsev K.N. 2020. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta. Mol. Basis Dis. 1866 (5), 165 674.
Dubinin M.V., Talanov E.Y., Tenkov K.S., Starinets V.S., Belosludtseva N.V., Belosludtsev K. N. The effect of deflazacort treatment on the functioning of skeletal muscle mitochondria in Duchenne muscular dystrophy. Int. J. Mol. Sci. 2020, 21 (22), 8763.
Dubinin M.V., Samartsev V.N., Stepanova A.E., Khoroshavina E.I., Penkov N.V., Yashin V.A., Starinets V.S., Mikheeva I.B., Gudkov S.V., Belosludtsev K.N. 2018. Membranotropic effects of ω-hydroxypalmitic acid and Ca2+ on rat liver mitochondria and lecithin liposomes. Aggregation and membrane permeabilization. J. Bioenerg. Biomembr. 50 (5), 391–401.
Dubinin M.V., Svinin A.O., Vedernikov A.A., Starinets V.S., Tenkov K.S., Belosludtsev K.N., Samartsev V.N. 2019. Effect of hypothermia on the functional activity of liver mitochondria of grass snake (Natrix natrix): inhibition of succinate-fueled respiration and K+ transport, ROS-induced activation of mitochondrial permeability transition. J. Bioenerg. Biomembr. 51 (3), 219–229.
Chance B., Williams G.R. 1955. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 217 (1), 383–393.
Pollard A.K., Craig E.L., Chakrabarti L. 2016. Mitochondrial complex I activity measured by spectrophotometry is reduced across all brain regions in ageing and more specifically in neurodegeneration. PLoS One. 11 (6), e0157405.
Spinazzi M., Casarin A., Pertegato V., Salviati L., Angelini C. 2012. Assessment of itochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 7 (6), 1235–1246.
Kamo N., Muratsugu M., Hongoh R., Kobatake Y. 1979. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenylphosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membr. Biol. 49, 105–121.
Dubinin M.V., Talanov E.Y., Tenkov K.S., Starinets V.S., Mikheeva I.B., Belosludtsev K.N. 2020. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta. Bioenergetics. 1861 (10), 148250.
Symons A.M., Lewis D.A., Ancill R.J. 1974. The actions of anti-inflammatory steroids on isolated rat liver mitochondrial function. J. Ster. Biochem. 5 (7), 639–644.
Pandya J.D., Agarwal N.A., Katyare S.S. 2004. Effect of dexamethasone treatment on oxidative energy metabolism in rat liver mitochondria during postnatal developmental periods. Drug Chem. Toxicol. 27 (4), 389–403.
Ribas V., García-Ruiz C., Fernández-Checa J. C. 2014. Glutathione and mitochondria. Front. Pharmacol. 5, 151.
Chalmers S., Nicholls D.G. 2003. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 278, 19 062–19 070.
Rasola A., Bernardi P. 2011. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium. 50, 222–233.
Белослудцев К.Н., Дубинин М.В., Белослудцева Н.В., Миронова Г.Д. 2019. Транспорт ионов Ca2+ митохондриями: механизмы, молекулярные структуры и значение для клетки. Биохимия. 6 (84), 759–775.
Malhi H., Guicciardi M.E., Gores G.J. 2010. Hepatocyte death: A clear and present danger. Physiol. Rev. 90, 1165–1194.
Campbell P.I., Al-Nasser I.A. 1995. Dexamethasone inhibits inorganic phosphate stimulated Ca2+-dependent damage of isolated rat liver and renal cortex mitochondria. Comp. Biochem. Physiol. 111, 221–225.
Nieminen A.L., Byrne A.M., Herman B., Lemasters J.J. 1997. Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am. J. Physiol. 272(4), 1286–1294.
Kowaltowski A.J., Castilho R.F., Vercesi A.E. 2001. Mitochondrial permeability transition and oxidative stress. FEBS letters. 495 (1–2), 12–15.
Bernardes C.F., Meyer-Fernandes J.R., Basseres D.S., Castilho R.F., Vercesi A.E. 1994. Ca2+-dependent permeabilization of the inner mitochondrial membrane by 4,4'-diisothiocyanatostilbene-2,2'-disulfonic acid (DIDS). Biochim. Biophys. Acta. 1188 (1–2), 93–100.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии