

Биологические мембраны: Журнал мембранной и клеточной биологии, 2021, T. 38, № 3, стр. 209-216
Малоновый диальдегид, но не метилглиоксаль, нарушает инсулиновый каскад, синтез NO и эндотелиальный барьер
М. В. Самсонов a, Н. В. Подкуйченко a, В. З. Ланкин b, А. В. Воротников a, *, В. П. Ширинский a, **
a Национальный медицинский исследовательский центр кардиологии,
Институт экспериментальной кардиологии
121552 Москва, Россия
b Национальный медицинский исследовательский центр кардиологии,
Институт клинической кардиологии
121552 Москва, Россия
* E-mail: a.vorotnikov@icloud.com
** E-mail: shirinsky@gmail.com
Поступила в редакцию 12.12.2020
После доработки 27.01.2021
Принята к публикации 28.01.2021
Аннотация
Дислипидемию и гипергликемию можно рассматривать как причину и следствие развития сахарного диабета 2 типа (СД2Т). Эти состояния связаны с образованием малонового диальдегида (МДА) и метилглиоксаля (МГО), которые представляют собой результат перекисного окисления липидов биологических мембран и окислительных превращений глюкозы. Сравнение эффектов МДА и МГО на эндотелиальные клетки пупочной вены человека, выполненное в данной работе, показало, что МДА, в отличие от МГО, нарушает барьерную функцию эндотелия, активацию инсулином PI3-киназного каскада и синтеза оксида азота (NO). МДА подавлял активацию протеинкиназы В (Akt) и эндотелиальной NO-синтазы (eNOS), но не затрагивал активацию субстрата инсулинового рецептора (IRS). Полученные результаты свидетельствуют о том, что МДА может вносить вклад в дисфункцию эндотелия на ранних этапах развития СД2Т.
ВВЕДЕНИЕ
Ожирение и дислипидемия выступают ведущими факторами риска развития инсулиновой резистентности и сахарного диабета 2 типа (СД2Т) [1, 2]. Предполагается, что дисфункция сосудистого эндотелия может связывать ожирение и ранние патологические процессы СД2Т с развитием сердечно-сосудистых осложнений [3, 4]. При ожирении и СД2Т уровень циркулирующих свободных жирных кислот (СЖК) значительно повышен и сосудистый эндотелий первым испытывает контакт с СЖК. В результате развивается липотоксичность, окислительный стресс и возникают воспалительные реакции в эндотелии сосудов [5–7]. При этом нарушается активация инсулином рецепторного каскада, включающего субстрат инсулинового рецептора (IRS), PI3-киназу, протеинкиназу В (Akt) и эндотелиальную NO-синтазу (eNOS); падает синтез NO и усиливается вазоконстрикция [8–11].
Окислительный стресс и воспалительные реакции связаны с появлением в стенке сосуда активных форм кислорода (АФК), малонового диальдегида (МДА) и продуктов окислительного превращения глюкозы, таких как метилглиоксаль (МГО) [7, 12–14]. Повышенный уровень МДА характерен для пациентов с СД2Т [15–17] и сердечно-сосудистыми заболеваниями [18, 19]. МДА является продуктом перекисного окисления липидов, которое усиливается при окислительном стрессе и продукции избыточных АФК [20]. В отличие от МДА причиной накопления МГО, глиоксаля и 3‑дезоксиглюкозона служит высокий уровень глюкозы и других углеводов в крови [21]. Несмотря на то, что для СД2Т характерны как гиперлипидемия, так и гипергликемия, повышение уровня СЖК характерно для первичного ожирения, как основного фактора риска развития СД2Т [22].
Ранее мы обнаружили, что МДА влияет на цитоскелет и целостность эндотелиального барьера, вызывая дисфункцию модельного эндотелия человека EA.hy926 [23]. Однако остался невыясненным вопрос, влияет ли МДА на активность инсулинового каскада, синтез NO и эндотелиальный барьер первичного сосудистого эндотелия человека. В данной работе мы провели сравнение прямых эффектов МДА и МГО на активность инсулинового каскада и синтез NO в эндотелиальных клетках пупочной вены человека (HUVEC). Мы обнаружили, что, в отличие от МГО, МДА нарушает барьерную функцию покоящегося эндотелиального монослоя, активацию инсулином Akt, eNOS и синтеза NO. По-видимому, МДА не нарушает активацию инсулинового рецептора и его непосредственной мишени, белка IRS. Полученные результаты согласуются с гипотезой о том, что МДА может вносить вклад на ранних этапах развития дисфункции эндотелия при СД2Т и ожирении, в то время как действие МГО реализуется на его более поздних стадиях, связанных с хронической гипергликемией.
МАТЕРИАЛЫ И МЕТОДЫ
Материалы. Общелабораторные реактивы, желатин, 1,1,4,4-тетраэтоксипропан (для получения МДА путем кислотного гидролиза) и МГО приобретали у фирмы Sigma-Aldrich (США) и использовали, как описано ранее [23]. Стандартизированная базовая питательная среда для культивирования эндотелиальных клеток (EBM), стандартизированная питательная среда с факторами роста эндотелиальных клеток, содержащая 3.5 мМ глюкозы (EGM-2MV) и карнитин приобретали у фирмы Lonza (Швейцария). Пенициллин и стрептомицин – у фирмы Gibco (США). Для нормировки общего белка в образцах для иммуноблотинга использовали набор реактивов BCA Protein Assay Kit (ThermoFisher Scientific, США). В работе использовали следующие первичные антитела фирмы Cell Signaling (США): фосфо-IRS1 (Y612, #3203), фосфо-Akt (Thr308, #9275), фосфо-Akt (Ser473, #4060), Akt (#4691). Антитела против фосфо-eNOS (Ser1177, #612392) приобретали у фирмы BD Biosciences (США), антитела против винкулина (#ab18058) у фирмы Abcam (США), антитела против белков, модифицированных МДА (клон 12E7) или МГО (клон 6D8) были описаны ранее [23]. Использовали вторичные, конъюгированные с пероксидазой хрена, антитела козы против антител кролика (Cell Signaling, #7074) и антитела кролика против антител мыши (Sigma, #A9044).
Культивирование HUVEC. Эндотелий из вены пупочного канатика человека выделяли путем обработки ткани коллагеназой. Выделенные клетки высаживали на покрытые желатином чашки петри и культивировали в среде Игла, модифицированной по способу Дульбекко (DMEM), содержащей 10% фетальной бычьей сыворотки, 200 мкг/мл фактора роста эндотелия, 50 ед/мл пенициллина, 50 мкг/мл стрептомицина и 5 ед/мл гепарина. При достижении конфлюэнтности клетки пересевали с использованием 0.05% раствора трипсина. В экспериментах использовали клетки 2–3 пассажа, особенности условий культивирования указаны в соответствующих разделах.
Проницаемость монослоя HUVEC. Для оценки проницаемости измеряли трансэндотелиальное электрическое сопротивление монослоя HUVEC (ТЭС). Клетки культивировали в атмосфере 5% CO2 при 37°С в питательной среде EGM-2MV, дополнительно содержавшей 2 мМ глутамина, 50 мкМ карнитина, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина. Клетки высаживали на покрытые 0.2–1.0% раствором желатина золотые электроды в 8-луночные планшеты 8W1E и измеряли ТЭС в режиме реального времени с помощью прибора ECIS-z (Applied Biophysics, США). Питательную среду ежедневно заменяли на свежую до формирования монослоя и последующей стабилизации в течение 24 ч. Перед внесением МДА или МГО питательную среду удаляли и инкубировали клетки в течение 2 ч при 37°C в растворе Хенкса.
Измерение NO. Клетки HUVEC высаживали в 96-луночные планшеты с прозрачным дном и затемненными стенками (μclear black, F-bottom, Greiner Bio-One, Австрия), покрытые 0.2% желатином, и культивировали в среде EGM-2MV, как указано выше. Для измерения NO к клеткам вносили 2 мкМ DAF-FM диацетата в присутствии 1 мМ пробенецида (оба – Invitrogen, США) и 15 мМ HEPES в базовой питательной среде EBM и стимулировали клетки 100 нМ инсулином (Novo Nordisk, Дания). Флуоресценцию DAF регистрировали с помощью микроскопа AxioVert 200M (Zeiss, Германия), оборудованного высокоскоростной камерой AxioCam HSm с CCD-матрицей с охлаждением. Перед внесением DAF-FM определяли фоновые значения флуоресценции (Fb), после внесения – начальное значение (F0), затем добавляли инсулин или растворитель и измеряли флуоресценцию (F) в тех же полях зрения, где и Fb и F0, с интервалом в 10 мин и минимальными временами экспозиции во избежание эффектов выгорания. Для каждой точки регистрировали 4 различных поля зрения. Накопление NO рассчитывали по изменению флуоресценции DAF, используя формулу (F – ‒ Fb)/(F0 – Fb), в линейном участке зависимости повышения флуоресценции от времени инкубации клеток с инсулином (как правило, 30 или 40 мин). Полученные значения нормализовали к средним значениям флуоресценции в контроле без добавок.
Иммуноблотинг. Монослой HUVEC выращивали в покрытых 0.2% желатином 6-луночных планшетах как описано выше. Клетки стимулировали 100 нМ инсулина без предварительной депривации от сыворотки. Клетки промывали охлажденным на льду раствором Хенкса и лизировали в 2-кратном буфере для образцов по Лэммли, дополнительно содержавшем коктейль ингибиторов протеаз и фосфатаз (ThermoFisher Scientific, США). Клеточную ДНК разрушали механически, пропуская образец 5 раз через иглу 30G, образцы прогревали при 60°C в течение 25 мин, центрифугировали при 16 000 g при 4°C в течение 10 мин. Иммуноблотинг и регистрацию хемилюминесценции проводили, как описано ранее [23], с использованием набора реагентов Clarity ECL reagents (Bio-Rad, США) и системы для визуализации Fusion-SL 3500WL (Vilber Lourmat, Франция). Мембраны блокировали 5% раствором обезжиренного молока (для анти-МДА антител) или 0.2% раствором казеина (I-Block™, Fisher Scientific, США) (для анти-МГО антител) в трис-солевом буфере с добавлением 0.1% Tween-20 (TBST) и окрашивали антителами, растворенными в 1% растворе обезжиренного молока в TBST.
Статистическая обработка данных. Данные анализировали с помощью критерия Стьюдента и представляли как средние ± стандартное отклонение (SD). Различия считали статистически достоверными при значениях p < 0.05. Все эксперименты проводили не менее трех раз с 3–4 параллельными измерениями.
РЕЗУЛЬТАТЫ
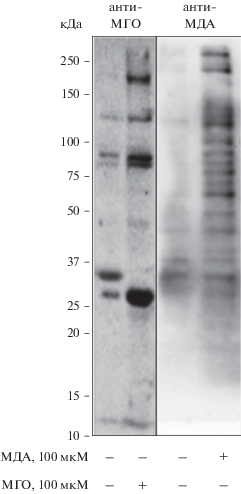
МДА и МГО модифицируют разные белки в клетках HUVEC. Чтобы подтвердить функциональность МДА и МГО и различие их белков-мишеней в клетках HUVEC, мы использовали антитела 12Е7 и 6D8, избирательно реагирующие с белками, модифицированными МДА или МГО, соответственно. Иммуноблотинг лизатов HUVEC, обработанных 100 мкМ МДА или 100 мкМ МГО, выявил различные наборы модифицируемых белков (рис. 1). В контрольных клетках был обнаружен некий фоновый сигнал, который значительно усиливался после обработки клеток МДА или МГО. Перекрестного взаимодействия антител не наблюдалось. Эти результаты означают, что в клетках HUVEC имеются разные мишени прямого действия МДА и МГО.
Рис. 1.
МДА и МГО модифицируют различный набор белков в клетках HUVEC. Клетки были обработаны 100 мкМ МДА или 100 мкМ МГО в течение 4 ч, как показано внизу, лизированы и проанализированы с помощью иммуноблотинга, как обозначено сверху. Показаны репрезентативные мембраны из трех независимых экспериментов.
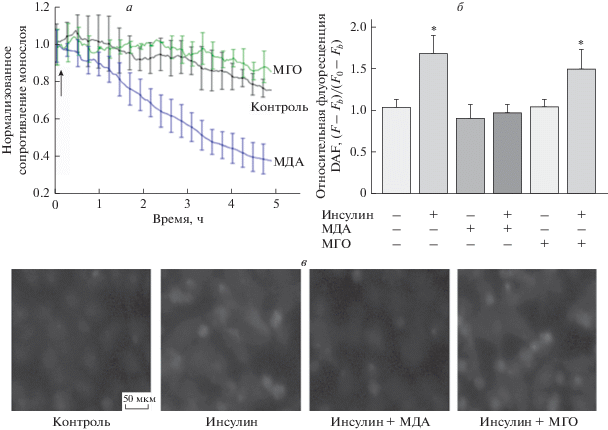
МДА, но не МГО, нарушает эндотелиальный барьер и стимулированный инсулином синтез NO в клетках HUVEC. Измерение ТЭС показало, что прямое воздействие МДА (100 мкМ) ведет к быстрому повышению проницаемости монослоя HUVEC, которое усиливалось с течением времени; в то же время МГО (100 мкМ) такого действия не оказывал (рис. 2а). При этом использованная концентрация МДА соответствует максимальным значениям, а концентрация МГО в 40 раз превышает максимальные значения, достигаемые в сосудистой системе [23]. При измерении через 4 ч инкубации в присутствии 100 мкМ МДА или 100 мкМ МГО стимулированный инсулином синтез NO нарушался только под действием МДА (рис. 2б и 2в). Эти наблюдения согласуются с нашими ранее полученными данными о нарушении барьерной функции культивируемых эндотелиальных клеток EA.hy926 под действием МДА [23].
Рис. 2.
Влияние МДА и МГО на проницаемость монослоя и синтез NO в клетках HUVEC. а – МДА (100 мкМ) снижает сопротивление эндотелиального барьера в отличие от МГО (100 мкМ). Значения ТЭС нормированы по базовым значениям ТЭС, которые составляли 12 кОм; стрелкой обозначено время добавления МДА / МГО. Показаны репрезентативные результаты из 5 независимых экспериментов, p < 0,01 для МДА против контроля. б, в – МДА (100 мкМ) в отличие от МГО (100 мкМ) блокирует синтез NO в ответ на стимуляцию клеток инсулином. б – Изменения флуоресценции DAF в клетках (средние значения ± SD, * p < 0.05 в сравнении с контролем без обработки клеток, n = 3); условия обработки клеток указаны внизу. в – Репрезентативные изображения, иллюстрирующие изменения флуоресценции DAF в клетках после стимуляции и использованные для количественного анализа, приведенного на панели б.
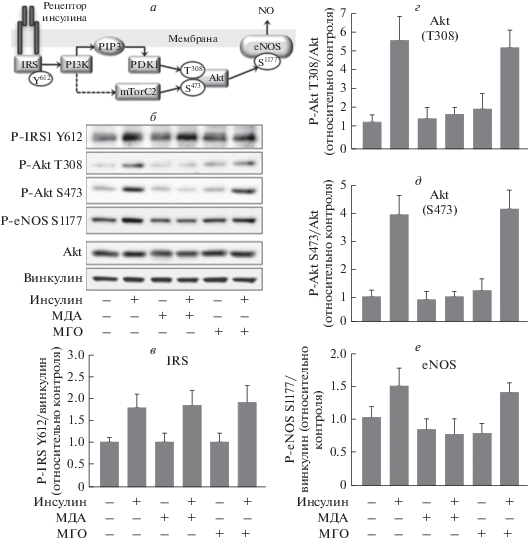
МДА, но не МГО, подавляет активацию eNOS инсулином. В клетках эндотелия ключевую роль в инсулиновом сигнальном каскаде в направлении синтеза оксида азота играют IRS, Akt и eNOS (рис. 3а). Чтобы выяснить влияние МДА и МГО на активность инсулинового каскада мы оценили уровень активирующего фосфорилирования этих белков в клетках HUVEC. Как видно из рис. 3б и 3в инсулин вызывал фосфорилирование IRS по Tyr-612. Этот остаток распознается регуляторной субъединицей PI3-киназы, связывание которой активирует этот фермент. Аналогично инсулин повышал уровень фосфорилирования остатка Thr-308 в активационной петле Akt (рис. 3б и 3г), а также остатка Ser-473, расположенного в гидрофобной последовательности фермента и необходимого для максимальной активации Akt (рис. 3б и 3д). Инсулин также стимулировал фосфорилирование eNOS по остатку Ser-1177, участвующему в активации NO-синтазы (рис. 3б и 3в), и усиливал синтез NO (рис. 2б и 2в). МГО (100 мкМ) существенно не влиял на активацию инсулинового каскада. Следует отметить, что при инкубации клеток с МДА (100 мкМ) активация IRS сохранялась (рис. 3в), но степень фосфорилирования Akt и eNOS не увеличивалась (рис. 3б). Это значит, что МДА, скорее всего, не влияет на активацию инсулиновых рецепторов, но прерывает передачу сигнала на последующих за IRS этапах, по-видимому, на участке PI3-киназа > PDK1 (см. рис. 3а).
Рис. 3.
Влияние МДА и МГО на фосфорилирование белков инсулинового сигнального каскада в клетках HUVEC. а – Схема активации синтеза оксида азота (NO) за счет передачи сигнала от рецептора инсулина (mTorC2 – второй комплекс mTOR, PIP3 – фосфатидилинозитол-3,4,5-трисфосфат, PDK1 – фосфоинозитид-зависимая киназа-1). б – Репрезентативные мембраны лизатов клеток HUVEC, стимулированных 100 нМ инсулином после инкубации в присутствии 100 мкМ МДА или 100 мкМ МГО, после окраски антителами, специфичность которых указана слева. в–е – Статистический анализ результатов, представленных в б. Показаны средние значения ± SD из трех независимых экспериментов. Условия инкубации обозначены в нижней части панелей.
ОБСУЖДЕНИЕ
В данной работе мы показали, что МДА нарушает эндотелиальный барьер, инсулиновый сигнальный каскад и синтез NO в клетках сосудистого эндотелия. В то же время МГО, накопление которого связывают с нарушениями углеводного обмена (гипергликемией) при СД2Т, не вызывает существенных изменений ни барьерной функции HUVEC, ни активности инсулинового каскада, ни синтеза NO. Учитывая, что максимальные концентрации МГО, обнаруженные in vivo, примерно в 40 раз ниже использованных [23], представляется маловероятным, что МГО может прямо влиять на эндотелиальный барьер и инсулиновый каскад при СД2Т. Обнаруженные нами различия по влиянию МДА и МГО на сосудистый эндотелий согласуются с точкой зрения о том, что дислипидемия и/или ожирение могут быть более значимы для дисфункции эндотелия и нарушения инсулиновой сигнализации при развитии сердечно-сосудистых осложнений на начальных этапах развития СД2Т.
Считается, что МДА возникает в результате действия АФК на полиненасыщенные жирные кислоты в составе биологических мембран [20]. Однако есть данные о том, что и насыщенные СЖК также могут приводить к образованию МДА. В частности, мы обнаружили, что длительная инкубация первичного эндотелия HUVEC с пальмитатом, типичным представителем СЖК, приводит к индукции митохондриальных АФК и накоплению МДА (данные не представлены). Несмотря на отсутствие ясного понимания механизма, который связывает высокий уровень внешнего пальмитата с образованием МДА внутри клеток, сходные наблюдения были сделаны в работах с макрофагами [24], сосудистыми [25] и гладкомышечными клетками человека [26]. Кроме того, обнаружена выраженная корреляция между уровнем МДА в крови и весом тела у пациентов с СД2Т [27]. Совокупность этих данных свидетельствует о наличии связи между повышенным уровнем СЖК и уровнем МДА.
Конститутивная активность eNOS и генерация NO играют важную роль в поддержании барьерной функции микрососудов [27–32], а также базовой барьерной функции культивируемого эндотелия [33, 34]. Активность eNOS регулируется взаимодействием с различными белками, например, кавеолином-1 и Ca2+-кальмодулином, а также фосфорилированием и внутриклеточной локализацией eNOS [32]. Инсулин усиливал барьерные свойства эндотелия микрососудов, активируя PI3-киназу и синтез NO [35], и препятствовал их нарушению под действием окислительного стресса [36]. В связи с этим мы исследовали влияние МДА и МГО на активность инсулинового каскада и синтез NO в клетках HUVEC. МДА, но не МГО, подавлял активирующее фосфорилирование ключевых компонентов инсулинового каскада, в том числе eNOS по остатку Ser-1177 (рис. 3), и подавлял стимулируемый инсулином синтез NO (рис. 2). В то же время сохранялась стимуляция инсулином фосфорилирования IRS, что указывает на сохранение активности инсулинового рецептора. Кроме того, МДА подавлял только реакции на стимуляцию инсулином, но существенно не влиял на базовую активность инсулинового каскада и базовый синтез NO. Учитывая неспецифическое воздействие МДА на клеточные белки, можно предполагать, что его эффект не ограничивается подавлением активации eNOS, но может затрагивать активность и других белков-мишеней, в том числе белков межклеточных контактов. Для их идентификации нужны дальнейшие исследования.
Работа выполнена при финансовой поддержке РНФ (проект № 19-15-00361) в части, касающейся экспериментов с измерениями проницаемости эндотелия, и РФФИ (проект № 18-315-00377) в остальной работе. Авторы выражают благодарность Т.Н. Власик и Е.Е. Ефремову за предоставление антител против белков, модифицированных МДА или МГО.
Список литературы
Boden G., Chen X., Ruiz J., White J.V., Rossetti L. 1994. Mechanisms of fatty acid-induced inhibition of glucose uptake. J. Clin. Invest. 93, 2438–2446.
Petersen M.C., Shulman G.I. 2018. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 98, 2133–2223.
Sena C.M., Pereira A.M., Seica R. 2013. Endothelial dysfunction – a major mediator of diabetic vascular disease. Biochim. Biophys. Acta. 1832, 2216–2231.
Shi Y., Vanhoutte P.M. 2017. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes. 9, 434–449.
Betteridge D.J. 2000. What is oxidative stress? Metabolism. 49, 3–8.
Evans J.L., Goldfine I.D., Maddux B.A., Grodsky G.M. 2002. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocrine Rev. 23, 599–622.
Paneni F., Costantino S., Cosentino F. 2015. Role of oxidative stress in endothelial insulin resistance. World J. Diab. 6, 326–332.
Bakker W., Eringa E.C., Sipkema P., van Hinsbergh V.W.M. 2009. Endothelial dysfunction and diabetes: Roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res. 335, 165–189.
Del Turco S., Gaggini M., Daniele G., Basta G., Folli F., Sicari R., Gastaldelli A. 2013. Insulin resistance and endothelial dysfunction: a mutual relationship in cardiometabolic risk. Curr. Pharm. Des. 19, 2420–2431.
Jansson P.A. 2007. Endothelial dysfunction in insulin resistance and type 2 diabetes. J. Intern. Med. 262, 173–183.
Potenza M.A., Addabbo F., Montagnani M. 2009. Vascular actions of insulin with implications for endothelial dysfunction. Am. J. Physiol. 297, E568–E577.
Betteridge D.J. 2000. What is oxidative stress? Metabolism. 49, 3–8.
Del Rio D., Stewart A.J., Pellegrini N. 2005. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 15, 316–328.
Wang L., Chen Y., Li X., Zhang Y., Gulbins E., Zhang Y. 2016. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget. 7, 73229–73241.
Dierckx N., Horvath G., Van Gils C., Vertommen J., Van de Vliet J., De Leeuw I., Manuel-y-Keenoy B. 2003. Oxidative stress status in patients with diabetes mellitus: Relationship to diet. Eur. J. Clin. Nutr. 57, 999–1008.
Gunawardena H.P., Silva K.D.R.R., Sivakanesan R., Katulanda P. 2019. Increased lipid peroxidation and erythrocyte glutathione peroxidase activity of patients with type 2 diabetes mellitus: Implications for obesity and central obesity. Obes. Med. 15, 100118.
Martin-Gallan P., Carrascosa A., Gussinye M., Dominguez C. 2003. Biomarkers of diabetes-associated oxidative stress and antioxidant status in young diabetic patients with or without subclinical complications. Free Rad. Biol. Med. 34, 1563–1574.
Polidori M.C., Savino K., Alunni G., Freddio M., Senin U., Sies H., Stahl W., Mecocci P. 2002. Plasma lipophilic antioxidants and malondialdehyde in congestive heart failure patients: Relationship to disease severity. Free Rad. Biol. Med. 32, 148–152.
Tamer L., Sucu N., Polat G., Ercan B., Aytacoglu B., Yücebilgiç G., Ünlu A., Dikmengil M., Atik U. 2002. Decreased serum total antioxidant status and erythrocyte-reduced glutathione levels are associated with increased serum malondialdehyde in atherosclerotic patients. Arch. Med. Res. 33, 257–260.
Ayala A., Munoz M.F., Arguelles S. 2014. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 360438.
Singh R., Barden A., Mori T., Beilin L. 2001. Advanced glycation end-products: A review. Diabetologia. 44, 129–146.
Arner P., Ryden M. 2015. Fatty acids, obesity and insulin resistance. Obes. Facts 8, 147–155.
Samsonov M.V., Khapchaev A.Y., Vorotnikov A.V., Vlasik T.N., Yanushevskaya E.V., Sidorova M.V., Efremov E.E., Lankin V.Z., Shirinsky V.P. 2017. Impact of atherosclerosis- and diabetes-related dicarbonyls on vascular endothelial permeability: A comparative assessment. Oxid. Med. Cell Longev. 2017, 1625130.
Li H., Xiao Y., Tang L., Zhong F., Huang G., Xu J.-M., Xu A.-M., Dai R.-P., Zhou Z.-G. 2018. Adipocyte fatty acid-binding protein promotes palmitate-induced mitochondrial dysfunction and apoptosis in macrophages. Front. Immunol. 9, 81.
Girona J., Rosales R., Saavedra P., Masana L., Vallve J.-C. 2019. Palmitate decreases migration and proliferation and increases oxidative stress and inflammation in smooth muscle cells: Role of the Nrf2 signaling pathway. Am. J. Physiol. 316, C888–C897.
Oberbach A., Schlichting N., Heinrich M., Till H., Stolzenburg J.-U., Neuhaus J. 2012. Free fatty acid palmitate impairs the vitality and function of cultured human bladder smooth muscle cells. PLoS One. 7, e41026.
Volpe C.M.O., Abreu L.F.M., Gomes P.S., Gonzaga R.M., Veloso C.A., Nogueira-Machado J.A. 2014. The Production of nitric oxide, IL-6, and TNF-alpha in palmitate-stimulated PBMNCs is enhanced through hyperglycemia in diabetes. Oxid. Med. Cell Longev. 2014, 479587.
Baldwin A.L., Thurston G., Al Naemi H. 1998. Inhibition of nitric oxide synthesis increases venular permeability and alters endothelial actin cytoskeleton. Am. J. Physiol. 274, H1776–1784.
Kurose I., Kubes P., Wolf R., Anderson D.C., Paulson J., Miyasaka M., Granger D.N. 1993. Inhibition of nitric oxide production. Mechanisms of vascular albumin leakage. Circ. Res. 73, 164–171.
Predescu D., Predescu S., Shimizu J., Miyawaki-Shimizu K., Malik A.B. 2005. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am. J. Physiol. 289, L371–381.
Mehta D., Malik A.B. 2006. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367.
Duran W.N., Breslin J.W., Sanchez F.A. 2010. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc. Res. 87, 254–261.
Cho M.M., Ziats N.P., Pal D., Utian W.H., Gorodeski G.I. 1999. Estrogen modulates paracellular permeability of human endothelial cells by eNOS- and iNOS-related mechanisms. Am. J. Physiol. 276, C337–349.
May J.M., Qu Z.-C. 2011. Nitric oxide mediates tightening of the endothelial barrier by ascorbic acid. Biochem. Biophys. Res. Commun. 404, 701–705.
Gunduz D., Thom J., Hussain I., Lopez D., Hartel F.V., Erdogan A., Grebe M., Sedding D., Piper H.M., Tillmanns H., Noll T., Aslam M. 2010. Insulin stabilizes microvascular endothelial barrier function via phosphatidylinositol 3-kinase/Akt-mediated Rac1 activation. Arterioscler. Thromb. Vasc. Biol. 30, 1237–1245.
Rath S., Kalogeris T., Mai N., Zibari G., Alexander J.S., Lefer D., Turnage R.H. 2006. Insulin prevents oxidant-induced endothelial cell barrier dysfunction via nitric oxide-dependent pathway. Surgery. 139, 82–91.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии