

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 1, стр. 18-27
Влияние тромбина на культивируемые астроциты крысы, подвергнутые кислородно-глюкозной депривации
Е. А. Абрамов a, А. Е. Иванова b, Э. Б. Дашинимаев b, c, d, А. Г. Камкин b, Л. Р. Горбачева a, b, *
a Московский государственный университет им. М.В. Ломоносова,
биологический факультет
119234 Москва, Россия
b Российский национальный исследовательский медицинский университет
им. Н.И. Пирогова Минздрава России
117997 Москва, Россия
c Институт биологии развития им Н.К. Кольцова РАН
119334 Москва, Россия
d Московский физико-технический институт (Государственный университет)
141701 Долгопрудный, Россия
* E-mail: gorbi67@mail.ru
Поступила в редакцию 19.04.2021
После доработки 15.05.2021
Принята к публикации 31.05.2021
- EDN: TLEQGX
- DOI: 10.31857/S0233475522010042
Аннотация
Ишемия мозга представляет собой сложный комплекс повреждающих факторов, к которым относятся депривация кислорода и глюкозы, а также возможность повторных поражений в период реперфузии ткани. Тромбин, ключевая сериновая протеаза гемостаза, может быть обнаружена в паренхиме мозга при ишемических и геморрагических инсультах, куда попадает непосредственно из кровеносного русла через поврежденный гематоэнцефалический барьер (ГЭБ). В настоящей работе оценено влияние тромбина на состояние глиальных клеток мозга в условиях кислородно-глюкозной депривации и последующей нормоксии (модель ишемии/реперфузии). Обнаружено, что тромбин в низких концентрациях (10 нМ) обладает защитным действием на астроциты крысы при ишемии/реперфузии. Предварительная инкубация астроцитов с протеазой в низкой концентрации вызывала повышение выживаемости клеток в данных условиях, в то время как повышение концентрации (50 нМ) снижало ее защитное действие. Выявлено, что тромбин в обеих концентрациях не влияет на индуцированную ишемией/реперфузией пролиферацию астроцитов. Было показано, что кислородно-глюкозная депривация приводит к перераспределению актиновых филаментов в астроцитах из-под мембранного пространства в область ядра клетки. Тромбин в высокой концентрации потенцировал влияние кислородно-глюкозной депривации на актиновый цитоскелет астроцитов. Таким образом, тромбин является эндогенной регуляторной молекулой для астроцитов мозга, и его действие на фоне кислородно-глюкозной депривации и последующей реперфузии носит дозозависимый характер.
ВВЕДЕНИЕ
Инсульт, занимая лидирующую позицию среди причин смертности и инвалидизации населения, является одной из главных современных медико-социальных проблем [1]. В связи с этим разработка новых подходов к терапии и профилактике инсульта является одной из главных задач как фундаментальной физиологии, так и современной медицины. Ишемический инсульт наиболее часто встречающийся тип данного заболевания, связанный с сокращением или полным прекращением кровоснабжения ткани вследствие тромбирования, резкого сужения или эмболии сосудов. Эти события непосредственно связаны с системой гемостаза и в первую очередь с ее ключевой протеазой – тромбином. Тромбин регулирует клеточные функции через активацию специфических рецепторов, активируемых протеазами – PAR, посредством расщепления внеклеточного N-конца с образованием укороченной аминокислотной последовательности (“привязанного лиганда”) [2]. Показано, что при ишемии мозга экспрессия данного вида рецепторов увеличивается [3], при этом появление тромбина в очаге повреждения преимущественно происходит за счет проникновения через поврежденный гематоэнцефалический барьер (ГЭБ) [4, 5]. Протромбин, прекурсор тромбина, экспрессируется клетками мозга [6]. Имеющиеся в литературе данные и наши собственные исследования указывают на способность тромбина оказывать противоположное действие на клетки в зависимости от его концентрации и типа воздействия [7]. Тромбин в высоких концентрациях обладает провоспалительными свойствами, стимулирует выброс цитокинов, вызывает гибель нейронов и активирует астро- и микроглию [8–10]. В связи с этим целью данного исследования было изучение влияния тромбина на первичную культуру астроцитов крыс при кислородно-глюкозной депривации (КГД) in vitro.
МАТЕРИАЛЫ И МЕТОДЫ
Реактивы, использованные в работе: тромбин (Sigma, США), HBSS с Ca2+ и Mg2+ (Gibco, США), HBSS без Ca2+ и Mg2+ (Gibco), 100 мМ пируват натрия, 1М HEPES, L-Cystein-HCl, BSA, глюкоза, папаин (Sigma), ДНКаза (Sigma); среды для культивирования клеток DMEM/F12, DMEM и DMEM без глюкозы (ПанЭко, Россия); инактивированная телячья сыворотка HI FBS (Invitrogen, США), GlutaMax (Invitrogen), пенициллин/стрептомицин (Invitrogen), трипсин-EDTA 0.5% (Invitrogen); набор реактивов для измерения содержания LDH (CytoTox 96 Non-Radioactive Cytotoxicity Assay, Promega, США), фаллоидин (Cell Signaling, США), ДНК-тропный краситель DAPI, PSA (персульфат аммония, Sigma), 12% раствор хлоралгидрата (Sigma).
Получение культуры астроцитов. Работа выполнена на первичной культуре астроцитов, полученных из коры головного мозга новорожденных крысят по ранее описанному протоколу [11]. Полученную суспензию астроцитов на 10-ый день культивации in vitro высеивали на чашки с прикрепленными стеклами (glassbottom) и на 48-луночные планшеты в количестве 7 × 104 клеток на ячейку. Далее астроциты инкубировали 4 дня при 37°C, 5% CO2. Однородность культуры астроцитов составляла более 90% при окрашивании антителами к специфическому глиальному фибриллярному кислому белку GFAP. На момент проведения эксперимента первичной культуре астроцитов крыс было 14 дней.
Моделирование ишемии in vitro. Кислородно-глюкозная депривация (КГД) – это модель in vitro, имитирующая эффекты инсульта [12]. КГД моделировали инкубированием клеток в среде DMEM без глюкозы в течение 5 ч при 5% О2, 5% СО2, 90% N2, при 37°С. После 5-часовой КГД производили реперфузию, заменяя в культуре астроцитов DMEM без глюкозы на DMEM с 4.5 г/л глюкозы, культивируя их при 21% О2, 5% СО2, 74% N2 и 37°С последующие 24 ч до регистрации исследуемых параметров. В качестве контрольной группы были выбраны интактные клетки без тромбина в аналогичных условиях.
Оценка уровня гибели астроцитов. При нарушении целостности цитоплазматической мембраны клеток высвобождается лактатдегидрогеназа (LDH) [13]. Под действием LDH соли тетразолия (иодонитротетразолий, INT) превращаются в красный формазан. Интенсивность окрашивания культуральной среды пропорциональна количеству LDH – доле клеток с нарушенной мембраной, что позволяет оценить уровень некроза. Количество LDH оценивали с использованием CytoTox 96 Non-Radioactive Cytotoxicity Assay. Для анализа отбирали 30 мкл клеточной среды и смешивали с 30 мкл CytoTox 96 Reagent. Смесь инкубировали 30 мин при комнатной температуре в темноте. После чего реакцию останавливали добавлением 30 мкл Stop Solution. Оптическую плотность измеряли при 490 нм на фотометре iMark (Bio-Rad, США).
Оценка пролиферации астроцитов (МТТ-тест). МТТ (3-(4,5-диметилтиазол-2-)ил)-2,5-дифенилтетразолий бромид) восстанавливается до формазана дегидрогеназами митохондрий живых клеток [14]. Водный раствор МТТ добавляли в культуральную среду с астроцитами до конечной концентрации 500 мкг/мл и инкубировали 1 час при 37°С, 5% СО2. Далее культуральную среду отбирали и к клеткам добавляли 150–300 мкл диметилсульфоксида (DMSO, Sigma) для растворения формазанов. Оптическую плотность измеряли с помощью фотометра iMark (Bio-Rad) при 530 нм.
При обработке данных оптическую плотность контрольной группы (интактные астроциты) принимали за 100%.
Иммуноцитохимическое окрашивание актина. Для оценки реорганизации актина астроциты, культивируемые на стеклах, промывали фосфатно-солевым буфером и фиксировали в 4% параформальдегиде (15 мин). После отмывки 120 мМ Na-фосфатом, 1 × 10 мин низкосолевым буфером (150 мМ NaCl, 10 мМ Na2HPO4), 2 × 10 мин высокосолевым буфером (0.5 М NaCl, 20 мМ Na2HPO4) блокировали неспецифическое связывание инкубацией клеток в блокирующем буфере, содержащим 0.1% Triton X-100, 5% FBS в фосфатно-солевом буфере. Далее астроциты инкубировали с фаллоидином (фаллоидин Alexa 488, Cell Signaling, 1:40), токсином, связывающим F-актин, в течение 1.5 ч. После чего клетки промывали 1 × 10 мин высокосолевым буфером (0.5 М NaCl, 20 мМ Na2HPO4), 1 × 10 мин 120 мМ Na-фосфатом, 1 × 10 мин 5 мМ Na-фосфатом. Для детекции ядер использовали ДНК-тропный краситель DAPI (1 : 500). Регистрацию флуоресценции фаллоидина осуществляли при помощи конфокального микроскопа (ZEISS LSM 700, Германия), получая не менее 5 снимков с каждого образца.
Статистический анализ. Статистический анализ данных проводили в программе GraphPad Prism 6 (GraphPad Software Inc., США). Для анализа использовали параметрический критерий One-way ANOVA и непараметрические критерии Манна–Уитни (Mann–Whitney test) и Краскела-Уоллиса (Kruskal–Wallis test). Результаты представлены как среднее ± стандартная ошибка среднего (mean ± SEM). Различия считались статистически значимыми при p < 0.05.
РЕЗУЛЬТАТЫ
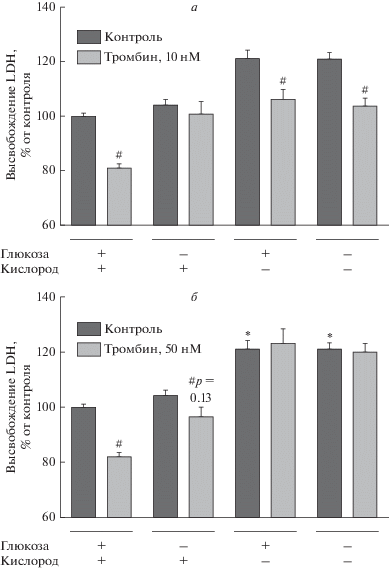
Влияние тромбина на выживаемость культивируемых астроцитов при пятичасовой кислородно-глюкозной депривации (КГД). Известно, что депривация глюкозы и кислорода вызывает гибель клеток при ишемии головного мозга. В первой серии экспериментов мы исследовали влияние тромбина в низкой (10 нМ) и высокой (50 нМ) концентрациях на жизнедеятельность культивируемых астроцитов мозга крыс при КГД. При нарушении целостности мембраны клеток происходит высвобождение в среду культивирования устойчивого фермента, LDH, по количеству которого можно оценить уровень некроза клеток. Изолированная депривация глюкозы в течение 5 ч не вызывала гибель клеток; в то время как 5-часовая гипоксия (5% O2) приводила к повышению данного показателя на 23% по сравнению с нормоксией (4.5 г/л глюкозы, 21% O2) (рис. 1). 30-минутное воздействие тромбина в концентрации 10 нМ (рис. 1a) перед КГД снижало гибель клеток до контрольных значений, тогда как тромбин в концентрации 50 нМ не обладал защитным действием (рис. 1б).
Рис. 1.
Влияние тромбина (10 нМ (a) и 50 нМ (б)) на гибель астроцитов после 5-часовой КГД. Mean ± SEM. *p < 0.05 относительно контрольной группы в условиях нормоксии (4.5 г/л глюкозы и 21% O2 в среде), Kruskal–Wallis test; #p < < 0.05 относительно группы без тромбина в тех же условиях.
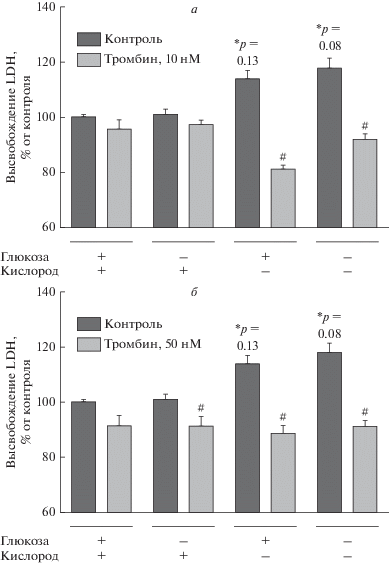
Влияние тромбина на выживаемость культивируемых астроцитов крыс после реперфузии. Основные современные терапевтические подходы при инсульте головного мозга направлены на возобновление кровотока в области, подвергшейся ишемии. В связи с этим в данной серии экспериментов мы исследовали влияние тромбина в концентрациях 10 и 50 нМ на выживаемость астроцитов мозга крыс после 24-часовой реперфузии (после КГД клетки помещали в среду DMEM с 4.5 г/л глюкозой, 21% O2). В ходе реперфузии тромбин добавляли к клеткам повторно в тех же концентрациях, что и при КГД. Уровень некроза клеток оценивали по высвобождению LDH. Показано, что после реперфузии в группах, подвергнутых гипоксии (5% O2), гибель клеток на 15% выше, чем при нормоксии (рис. 2а). Во всех используемых концентрациях тромбин снижал уровень некроза астроцитов до значений близких к контрольным при нормоксии.
Рис. 2.
Влияние тромбина в концентрациях 10 нМ (а) и 50 нМ (б) на гибель астроцитов мозга крыс после 24-часовой реперфузии. После 5-часовой депривации (глюкозы, кислорода или кислородно-глюкозной) или нормоксии клетки помещали в среду DMEM с 4.5 г/л глюкозой, при 21% O2, через 24 ч оценивали гибель астроцитов по высвобождению LDH. Контроль – группа без тромбина при соответствующих условиях (нормоксии, депривации глюкозы, депривации кислорода (5% О2) и КГД). Mean ± SEM. *p < 0.05 относительно контрольной группы в условиях нормоксии (4.5 г/л глюкозы и 21% O2 в среде), Kruskal-Wallis test; #p < 0.05 относительно группы без тромбина в тех же условиях.
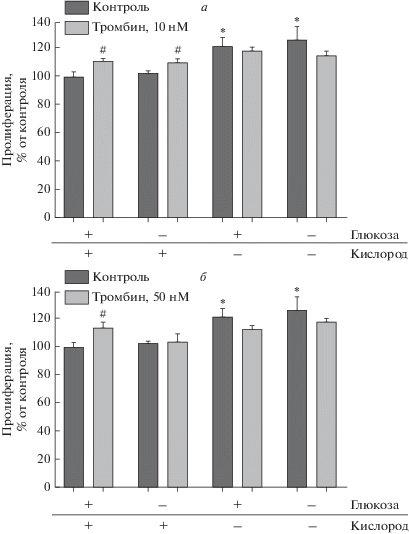
Влияние тромбина на пролиферацию астроцитов после ишемии/реперфузии. Ранее было показано, что ишемия стимулирует развитие реактивного астроглиоза [14, 15]. На следующем этапе нашей работы мы исследовали влияние тромбина в концентрациях 10 и 50 нМ на пролиферацию астроцитов после 24-часовой реперфузии. Мы обнаружили, что гипоксия стимулировала повышение пролиферации астроцитов на 21% по сравнению с нормоксией, в то время как депривация глюкозы подобного эффекта не вызывала (рис. 3). Тромбин в используемых концентрациях не оказывал влияния на пролиферацию клеток в условиях КГД. Однако в условиях нормоксии тромбин 10 и 50 нМ способствовал повышению уровня пролиферации астроцитов по сравнению с контролем.
Рис. 3.
Влияние тромбина в концентрациях 10 нМ (а) и 50 нМ (б) на пролиферацию астроцитов в постишемический реперфузионный период через 24 ч. Контроль – контрольная группа при нормоксии. Mean ± SEM, Mann–Whitney test. *p < 0.05 относительно контрольной группы в условиях нормоксии (4.5 г/л глюкозы и 21% O2 в среде), #p < 0.05 относительно группы без тромбина в в тех же условиях.
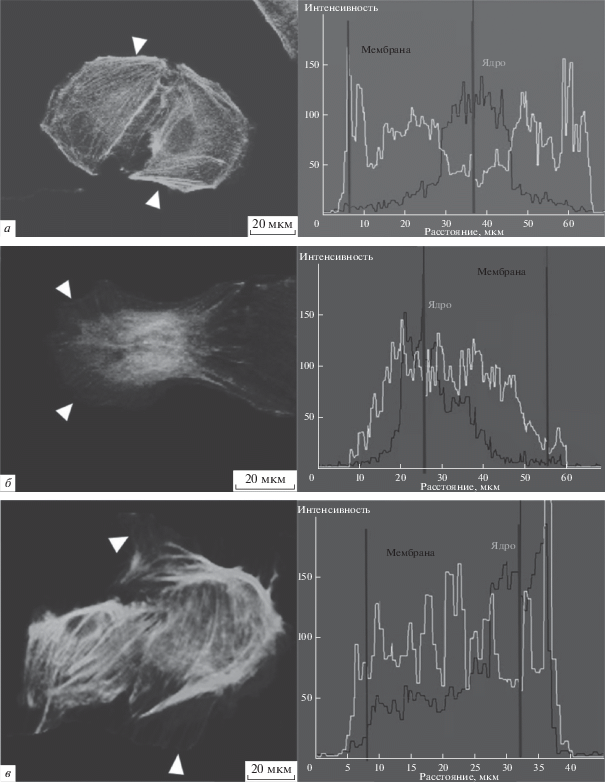
Актиновый цитоскелет культивируемых астроцитов при КГД и на фоне действия тромбина. Известно, что одним из маркеров астроглиоза является изменение морфологии клеток, что определяется состоянием цитоскелета, поэтому нами было оценено состояние F-актина в условиях КГД с использованием фаллоидина. Установлено, что при нормоксии F-актин располагается преимущественно в примембранном пространстве, стабилизируя форму клетки. Пятичасовая КГД вызывала перераспределение актина из примембранной области в область ядра и ретракцию цитоплазмы. Тромбин 50 нМ потенцировал формирование стресс-фибрилл и усиливал ретракцию цитоплазмы при КГД.
ОБСУЖДЕНИЕ
Мозговые инсульты как геморрагического, так и ишемического типов тесно связаны с системой гемостаза [16–20]. Ишемия мозга, спровоцированная образованием тромба, сопровождается изменением локальной концентрации сериновых протеаз гемостаза, в первую очередь тромбина [21, 22]. Известно, что протеазы гемостаза участвуют не только в процессе коагуляции, но и в регуляции пролиферации, миграции, воспаления, апоптоза и других клеточных функциях через специфические рецепторы, активируемые протеазами (PAR) [2, 11, 19, 23, 24].
Основная доля (80%) всех инсультов имеет ишемический характер и только около 15% вызваны геморрагическим повреждением кровотока. Несмотря на запуск реперфузии областей мозга, подвергнутых ишемии, даже краткосрочное (до 60 мин) снижение кровотока в ишемической области до 20 мл/г и ниже запускает процессы, ведущие к гибели клеток мозга [25]. Астроциты, наряду с нейронами, остро реагируют на ишемическое воздействие [14, 15]. Результаты наших исследований подтверждают эти данные. Более того, согласно представленным результатам, ключевым повреждающим фактором при 5-часовой КГД для культивируемых астроцитов является именно гипоксия, а не депривация глюкозы (рис. 1). Согласно литературным данным устойчивость глиальных клеток к снижению уровня глюкозы в среде может определяться тем, что астроциты, являясь депо гликогена в мозге, обеспечивают глюкозой и молочной кислотой при гипоксии не только собственное функционирование, но и жизнедеятельность нейронов [14, 15].
Ранее было показано, что тромбин – ключевая сериновая протеаза гемостаза, через специфический тип рецепторов (PAR) модулирует выживаемость клеток мозга при разных патологических состояниях, в том числе и при ишемии [7, 22, 26, 27]. Повышение содержания тромбина в тканях мозга, по-видимому, возможно не только в результате нарушения ГЭБ [17, 28], но и из протромбина, экспрессия которого показана в мозге [21]. Протеаза может оказывать на клетки мозга как нейродегенеративное, так и нейропротекторное действие, что определяется ее концентрацией [7, 29]. Рецепторы тромбина уже давно обнаружены на астроцитах, что обеспечивает возможность их активации тромбином и запуска внутриклеточных сигнальных каскадов [30, 31]. В настоящем исследовании установлено, что влияние тромбина на гибель клеток в культуре астроцитов при КГД определяется концентрацией протеазы, что согласуется с полученными ранее в нашей лаборатории данными на нейрональной культуре в условиях глутаматной эксайтотоксичности [32]. На основе полученных и уже имеющихся данных можно заключить, что концентрация тромбина 10 нМ обладает защитным действием, как на культивируемых нейронах, так и на астроцитах; в то время как 50 нМ тромбина обладает токсическим действием на нейроны и не изменяет выживаемость астроцитов при КГД (рис. 1) [32].
Известно, что одним из наиболее важных событий после ишемии головного мозга является восстановление притока крови – реперфузия. Показано, что именно в этот период появляется большое количество активных форм кислорода, приводящих к новой волне некроза в ишемической области [25]. В настоящей работе, мы предположили, что тромбин в низкой концентрации должен снижать некроз, вызванный реперфузией, подобно своему действию во время гипоксии. Полученные данные подтвердили наше предположение (рис. 2a). Кроме того, мы обнаружили, что тромбин в высокой концентрации также обладал защитным действием на астроциты мозга крыс после ишемии/реперфузии (рис. 2б). Таким образом, тромбин в диапазоне концентраций 10–50 нМ обладает защитным действием на астроглию во время ишемии/реперфузии, но данный эффект наиболее выражен при низких концентрациях протеазы, что согласуется с литературными данными о защитном действии тромбина на клетки мозга [7, 22, 26, 27].
В последнее время особое внимание исследователей направлено на процесс, именуемый как реактивный астроглиоз – явление патологической активации астроглии, для которого характерно развитие функциональных и морфологических изменений в клетках, усиление пролиферации [14, 15, 33]. Многие патологические состояния, такие как нейровоспаление, нейродегенеративные заболевания и ишемия головного мозга, сопровождаются активацией астроглии и развитием реактивного астроглиоза [34]. Подобный феномен при нарушениях мозговой деятельности, усугубляя течение ишемии, а также способствуя прогрессированию хронических нейродегенеративных заболеваний, остается серьезной проблемой медицинской практики, поскольку на данный момент нет эффективных способов его модуляции. Тромбин – один из факторов, стимулирующих активную пролиферацию астроцитов и способствующий формированию глиального рубца при повреждении мозга [35]. Нами обнаружено, что тромбин в диапазоне концентраций 10–50 нМ не изменяет пролиферацию при гипоксии (рис. 3) в отличие от своего пролиферативного действия в условиях нормоксии.
Считается, что зрелые астроциты в норме не пролиферируют [36]. В связи с этим для экспериментов in vitro астроциты выделяют из новорожденных животных. Кортикальные астроциты грызунов созревают в течение первых недель постнатального развития, пролиферируя и увеличивая размер популяции в 6–8 раз [37]. Общепринятой практикой является использование в экспериментах астроцитов на 7–25 день культивирования [38–40].
При патологических состояниях головного мозга астроглия активируется [5]. Показано, что астроглиоз характеризуется не только повышением пролиферации клеток, но также изменением морфологии астроцитов, что определяется состоянием цитоскелета [14, 15]. Ранее были опубликованы работы о том, что в нормальных условиях F-актин располагается преимущественно в примембранном пространстве, стабилизируя форму клетки [41, 42]. Наши исследования подтверждают эти данные (рис. 4a). Пятичасовая ишемия стимулировала перераспределение актина из примембранной области в область ядра и ретракцию цитоплазмы (рис. 4б). Высокие концентрации тромбина потенцировали формирование стресс-фибрилл и усиливали ретракцию цитоплазмы при КГД (рис. 4в). Такое изменение, вызванное тромбином, может выступать одним из возможных механизмов его влияния на выживаемость/гибель клеток. Так ранее было показано, что стабилизация F-актина в астроцитах приводит к уменьшению размера и количества лизосом, подавляет их созревание и разрыв при аутофагии, предотвращая тем самым гибель клеток [43].
Рис. 4.
Распределение интенсивности окрашивания F-актина в астроцитах в условиях нормоксии (а), КГД (б), КГД с тромбином (50 нМ) (в). Ядерный маркер – DAPI, маркер актина – Phalloidin Alexa 488. Интенсивность флуоресценции в подмембранной области – линия “мембрана”, в области ядра – “ядро”. Белый треугольник – граница клетки (клеточная мембрана).
Таким образом, представленные результаты демонстрируют, что сериновая протеаза тромбин является эндогенной регуляторной молекулой для клеток мозга. Действие протеазы носит дозозависимый характер и определяется типом повреждения. Установлено, что для культивируемых астроцитов крыс защитным действием при ишемии обладает тромбин в концентрации 10 нМ, предотвращающий гибель клеток, вызванную КГД. Протеаза в более высокой концентрации вызывают активацию астроглии, что влечет за собой потенцирование гибели клеток мозга при КГД. Итак, представленные результаты позволяют понять особенности развития повреждений мозговой ткани в условиях ишемии и реперфузии и установить динамику изменения регулирующей роли тромбина при данной патологии. Эти данные представляют интерес не только для фундаментальной физиологии и медицины, но и указывают на необходимость учета действия тромбина вне системы гемостаза при определении тромболитической и антикоагулянтной терапии для пациентов с ишемическим инсультом и другими нарушениями мозгового кровотока.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа выполнена в рамках государственного задания ИБР РАН 2020 года, № 0108-2019-0004 и с использованием оборудования Центра высокоточного геномного редактирования и генетических технологий для биомедицины РНИМУ им. Н.И. Пирогова.
Соответствие принципам этики. Все применимые международные, национальные и/или институциональные принципы ухода и использования животных были соблюдены.
Список литературы
Walter J., Oyere O., Mayowa O., Sonal S. 2016. Stroke: A global response is needed. Bull. World Health Organ. 94, 634–634.
Strukova S.M. 2001. Thrombin as a regulator of inflammation and reparative processes in tissues. Biochemistry (Moscow), 66, 8–18.
Wang J., Jin H., Hua Y., Keep R., Xi G. 2012. Role of protease-activated receptor-1 in brain injury after experimental global cerebral ischemia. Stroke, 43, 2476–2482.
Sokolova E., Reiser G. 2008. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost. 100, 576–581.
Nicole O., Goldshmidt A., Hamill C.E., Sorensen S.D., Sastre A., Lyuboslavsky P., Hepler J.R., McKeon R.J., Traynelis S.F. 2005. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J. Neurosci. 25, 4319–4329.
Rajput S.P., Lamb J., Kothari S., Pereira B., Soetkamp D., Wang Y., Tang J., Van Eyk E.J., Mullins S.E., Lyden D.P. 2019. Neuron-generated thrombin induces a protective astrocyte response via protease activated receptors. Glia. 68 (2), 246–262.
Hu H., Yamashita S., Hua Y., Keep R.F., Liu W., Xi G. 2010. Thrombin-induced neuronal protection: role of the mitogen activated protein kinase/ribosomal protein S6 kinase pathway. Brain Res. 1361, 93–101.
Simmons S., Lee R. V., Moller T., Weinstein J.R. 2013. Thrombin induces release of proinflammatory chemokines interleukin-8 and interferon-g-induced protein-10 from cultured human fetal astrocytes. Neuroreport. 24 (1), 36–40.
Suo Z., Citron B.A., Festoff B.W. 2004. Trombin: A potential proinflammatory mediator in neurotrauma and neurodegenerative disorders. Curr. Drug Targets Inflamm. Allergy. 3 (1), 105–114.
Xi G., Reiser G., Keep F.R. 2002. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective. J. Neurochem. 84(1), 3–9.
Ivanova A.E., Gorbacheva L.R., Strukova S.M., Pinelis V.G., Reiser G. 2014. Activated protein C and thrombin participate in the regulation of astrocyte functions. Biochemistry (Moscow). Suppl. Ser. A. Membr. Cell Biol. 8, 50–59.
Holloway P.M., Gavins F.N.E. 2016. Modeling ischemic stroke in vitro: Status quo and future perspectives. Stroke. 47 (2), 561–569.
Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 65, 55–63.
Sofroniew M.V., Vinters H.V. 2010. Astrocytes: Biology and pathology. Acta Neuropathol. 119, 7–35.
Горбачева Л.Р., Помыткин И.А., Сурин А.М., Абрамов Е.А., Пинелис В.Г. 2018. Астроциты и их роль в патологии центральной нервной системы. Росс. педиатр. журн. 21, 1, 46–53.
Rajput P.S., Lyden P.D., Chen B., Lamb J.A., Pereira B., Lamb A., Zhao L., Lei I.F., Bai J. 2014. Protease activated receptor-1 mediates cytotoxicity during ischemia using in vivo and in vitro models. Neurosci. 281, 229–240.
De Castro Ribeiro M., Badaut J., Price M., Meins M., Bogousslavsky J., Monard D., Hirt L. 2006. Thrombin in ischemic neuronal death. Exp. Neurol. 198, 199–203.
Xi G., Keep R.F., Hua Y., Xiang J., Hoff J.T. 1999. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke. 30, 1247–1255.
Mosnier L.O., Zlokovic B.V., Griffin J.H. 2014. Cytoprotective-selective activated protein C therapy for ischaemic stroke. Thromb. Haemost. 112, 883–892.
Cheng T., Liu D., Griffin J.H., Fernández J.A., Castellino F., Rosen E.D., Fukudome K., Zlokovic B.V. 2003. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat. Med. 9, 338–342.
Krenzlin H., Lorenz V., Danckwardt S., Kempski O., Alessandri B. 2016. The importance of thrombin in cerebral injury and disease. Int. J. Mol. Sci. 17, 77–85.
Striggow F., Riek M., Breder J., Henrich-Noack P., Reymann K.G., Reiser G. 2000. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. Proc. Natl. Acad. Sci. USA. 97, 2264–2269.
Strukova S. 2006. Blood coagulation-dependent inflammation. Coagulation-dependent inflammation and inflammation-dependent thrombosis. Front. Biosci. 11, 59–80.
Gorbacheva L.R., Kiseleva E.V., Savinkova I.G., Strukova S.M. 2017. A new concept of action of hemostatic proteases on inflammation, neurotoxicity, and tissue regeneration. Biochemistry (Moscow). 82, 778–790.
Meschia J.F., Brott T. 2018. Ischaemic stroke. Eur. J. Neurol. 25, 35–40.
Jiang Y., Wu J., Hua Y., Keep R.F., Xiang J., Hoff J.T., Xi G. 2002. Thrombin-receptor activation and thrombin-induced brain tolerance. Cereb. Blood Flow Metab. 22, 404–410.
Masada T., Xi G., Hua Y., Keep R.F. 2000. The effects of thrombin preconditioning on focal cerebral ischemia in rats. Brain Res. 867, 173–179.
Machida T., Takata F., Matsumoto J., Miyamura T., Hirata R., Kimura I., Kataoka Y., Dohgu Sh., Yamauchi A. 2017. Contribution of thrombin-reactive brain pericytes to blood-brain barrier dysfunction in an in vivo mouse model of obesity-associated diabetes and an in vitro rat model. PLoS ONE. 12(5), e0177447.
Suo Z., Citron B.A., Festoff B.W. 2004. Thrombin: A potential proinflammatory mediator in neurotrauma and neurodegenerative disorders. Curr. Drug Targets. Inflamm. Allergy. 3, 105–114.
Weinstein J.R., Gold S.J., Cunningham D.D., Gall C.M. 1995. Cellular localization of thrombin receptor mRNA in rat brain: Expression by mesence phalic dopaminergic neurons and codistribution with prothrombin mRNA. J. Neurosci. 15, 2906–2919.
Junge C.E., Lee C.J., Hubbard K.B., Zhang Z., Olson J.J., Hepler J.R., Brat D.J., Traynelis S.F. 2004. Protease-activated receptor-1 in human brain: Localization and functional expression in astrocytes. Exp. Neurol. 188, 94 –103.
Gorbacheva L., Pinelis V., Ishiwata S., Strukova S., Reiser G. 2010. Activated protein C prevents glutamate- and thrombin-induced activation of nuclear factor-kappaB in cultured hippocampal neurons. Neurosci. 165, 1138–1146.
Sofroniew M.V. 2009. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647.
Pekny M., Wilhelmsson U., Pekna M. 2014. The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 565, 30–38.
Wang H., Ubl J.J., Stricker R., Reiser G. 2002. Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am. J. Physiol. Cell Physiol. 283, 1351–1364.
Guizzetti M., Kavanagh T.J., Costa L.J. 2011. Measurements of astrocyte proliferation. Meth. Mol. Biol. 758, 349–359.
Ge W.P., Miyawaki A., Gage F.H., Jan Y.N., Jan L.Y. 2012. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 484, 376–380.
Moreno C., Sampieri A., Vivas O., Pena-Segura C., Vaca L. 2012. STIM1 and Orai1 mediate thrombin-induced Ca2+ influx in rat cortical astrocytes. Cell Calcium. 52, 457–67.
Wang H., Ubl J.J., Stricker R., Reiser G. 2002. Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am. J. Physiol. Cell Physiol. 283, C1351–C1364.
Simmons M.L., Murphy S. 1992. Induction of nitric oxide synthase in glial cells. J. Neurochem. 59, 897–905.
Delbro D., Westerlund A., Björklund U., Hansson E. 2009. In inflammatory reactive astrocytes co-cultured with brain endothelial cells nicotine-evoked Ca2+ transients are attenuated due to interleukin-1β release and rearrangement of actin filaments. Neurosci. 159, 770–779.
O’Shea R.D., Lau C.L., Farso M.C., Diwakarla S., Zagami C.J., Svendsen B.B., Feeney S.J., Callaway J.K., Jones N.M., Pow D.V., Danbolt N.C., Jarrott B., Beart P.M. 2005. Effects of lipopolysaccharide on glial phenotype and activity of glutamate transporters: Evidence for delayed up-regulation and redistribution of GLT-1. Neurochem. Int. 48, 604–610.
Ryu H.J., Kim J.E., Yeo S.I., Kim D.W., Kwon O.S., Choi S.Y., Kang T.C. 2011. F-actin depolymerization accelerates clasmatodendrosis via activation of lysosome-derived autophagic astroglial death. Brain Res. Bull. 85, 368–373.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии