

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 4, стр. 283-291
Нарушение кальциевого гомеостаза и ответные изменения в кальциевой сигнализации нейронов и глиальных клеток при фотодинамическом воздействии
В. А. Дзреян a, А. М. Хайтин a, *, С. В. Демьяненко a
a Южный федеральный университет, Академия биологии и биотехнологии
им. Д.И. Ивановского
344090 Ростов-на-Дону, Россия
* E-mail: andrej.wojt@gmail.com
Поступила в редакцию 28.02.2022
После доработки 13.03.2022
Принята к публикации 14.03.2022
- EDN: ZITIRZ
- DOI: 10.31857/S0233475522040041
Аннотация
Фотодинамическое воздействие на нейроны и глиальные клетки, вызывающее окислительный стресс и ишемическое повреждение, сопровождается нарушением кальциевого гомеостаза и активизацией либо подавлением различных кальций-зависимых механизмов – кальциевых насосов и каналов, кальций-зависимых сигнальных и исполнительных белков, а также других сигнальных систем, взаимодействующих с кальциевым путем – ионных каналов, насосов и обменников, оксида азота, глутамата и прочих. Каскад инициированных окислительным стрессом и ишемией процессов в нервной ткани включает как защитные реакции, так и апоптотический либо некротический сценарий клеточной смерти. В этом мини-обзоре рассматриваются и сопоставляются с литературой данные об этих процессах, полученные в нашей лаборатории на моделях фототромботического инсульта на головном мозге крыс in vivo, и фотодинамической терапии на модели механорецептора речного рака in vitro. Данные направления исследований обусловлены необходимостью поиска методов экстренной нейропротекции при ишемическом инсульте и повышением точности и эффективности фотодинамической терапии опухолей при минимизации повреждений доброкачественной ткани. В протеомном исследовании области пенумбры на модели фототромботического инсульта были обнаружены изменения экспрессии ряда кальций-зависимых белков, связанные с нарушением кальциевого гомеостаза и имеющее либо защитную, либо повреждающую направленность. При ингибиторном анализе воздействия фотоокислительного стресса на модели рецептора растяжения рака было выявлено участие ряда белков кальций-зависимого пути в смерти либо выживании нейронов и глиальных клеток. В этой статье данные анализируются и обобщаются с тем, чтобы выявить перспективные направления дальнейших исследований.
ВВЕДЕНИЕ
Исследования нашей лаборатории имеют пересечения с работами Б.И. Ходорова по исследованию молекулярно-клеточных основ нейротоксичности, в том числе нарушений кальциевого гомеостаза.
Кальций, будучи универсальным вторичным мессенджером в системе сигнальной трансдукции, интегрирует ряд сигнальных путей, контролирующих различные функции и смерть клетки. Повышение цитозольной концентрации Ca2+ до 10–4–10–3 M запускает некротический или апоптотический сценарий клеточной смерти. В клетках работают одновременно разные системы поддержания кальциевого гомеостаза: натрий-кальциевые и другие обменники, пассивные кальциевые каналы с различными механизмами активации, активные кальциевые насосы, специфические митохондриальные механизмы ввода и вывода Ca2+, депо-завиcимые механизмы, кальций-зависимые белки.
Центральная роль Ca2+ в физиологических и патологических процессах обусловливает необходимость изучения их роли, в том числе и в процессах, вызванных окислительным стрессом. В этом обзоре будут затронуты исследования нарушений кальциевого гомеостаза, происходящих при фототромботическом инсульте головного мозга и фотодинамически индуцированном окислительном стрессе нейронов и прилегающей глии.
ФОТОТРОМБОТИЧЕСКИЙ ИНСУЛЬТ И КАЛЬЦИЕВЫЙ ГОМЕОСТАЗ
Инсульт представляет собой многоступенчатый процесс, при котором повреждение клеток распространяется от очага инфаркта к окружающим тканям, в результате чего формируется переходная зона, так называемая ишемическая пенумбра, где смерть клеток развивается медленней, в течение нескольких часов [1, 2].
Ионы кальция являются универсальными внутриклеточными мессенджерами как во время ишемического повреждения, так и в постишемический период. В частности, они играют ключевую роль в механизме эксайтотоксичности, активации протеолитических ферментов, инициации некроза и апоптоза, распространении повреждающих процессов за пределы ядра инфаркта и формировании пенумбры. Предыдущие исследования показали, что повышенные уровни Ca2+ влияют на размер инфаркта, а также определяют исход и рецидив ишемического инсульта [3]. Такая многогранная роль Ca2+ объясняет необходимость дальнейших исследований роли кальция в инициации гибели клеток при ишемической атаке.
Согласно современной парадигме, в очаге ишемии дефицит кислорода и глюкозы вызывает истощение ATP, образование активных форм кислорода (АФК), окислительное повреждение мембран, потерю ионных градиентов, приток Ca2+, высвобождение K+, окислительный стресс и отек тканей. K+-опосредованная деполяризация способствует открытию каналов NMDA в соседних нейронах, дополнительному притоку Ca2+ и высвобождению K+ и глутамата [4, 5]. Такой саморазвивающийся эксайтотоксический процесс приводит к распространению повреждения за пределы очага инфаркта [6, 7].
Внутриклеточный Ca2+ индуцирует митохондриальную дисфункцию, образование АФК и продукцию NO. Ca2+-активируемая нейрональная NO-синтаза, которая часто пространственно связана с NMDA-рецепторами, продуцирует оксид азота, который реагирует с O2– с образованием мощного окислителя пероксинитрита (ONOO–). Кроме того, цитозольный Ca2+ активирует различные гидролитические ферменты, такие как протеиназы, липазы и нуклеазы, которые разрушают клеточные компоненты, что приводит к смерти клеток [8]. В частности, Ca2+-активируемые кальпаин и катепсины играют существенную роль в нейродегенерации. Ca2+-опосредованные сигнальные пути стимулируют различные факторы транскрипции, такие как NF-kB, AP-1, CREB, STAT и др., которые регулируют экспрессию белков, участвующих в выживании или гибели клеток [9–11]. Избыток внутриклеточного Са2+ может проникать в ядра нейронов и активировать экспрессию специфических генов в зависимости от пути проникновения. Приток Ca2+ через синаптические рецепторы NMDA может индуцировать кальциевую волну, которая распространяется к ядру. Ядерный кальций стимулирует путь CaMKIV/CREB, который индуцирует экспрессию генов, участвующих в регуляции физиологических функций: метаболизма, синаптической передачи и выживания клеток. Напротив, гиперактивация экстрасинаптических NMDA-рецепторов при стрессе или ишемии индуцирует патогенную Ca2+-перегрузку и экспрессию генов, связанных с апоптозом и нейродегенерацией. В этом случае CREB может быть функционально инактивирован быстрым дефосфорилированием и последующей деградацией. Активация этих рецепторов вызывает накопление в ядре гистондеацетилаз HDAC4 и HDAC5, которые репрессируют транскрипцию. Он также стимулирует накопление в ядре проапоптотического фактора транскрипции FOXO3A. Кроме того, перегрузка Ca2+ быстро снижает потенциал митохондриальной мембраны [9, 10, 12].
В нашей лаборатории изучена роль Ca2+ в клеточно-молекулярных механизмах очагового фототромботического инфаркта (ФТИ) в коре головного мозга крыс и мышей, индуцированного локальным фотодинамическим воздействием [9–11]. Фотодинамический эффект основывался на фотовозбуждении красителя бенгальского розового в окрашенных клетках с последующей генерацией синглетного кислорода и других АФК, окислительным стрессом и, наконец, некрозом или апоптозом клеток. Фотооблучение вызывает локальное окислительное повреждение эндотелия сосудов, с последующей агрегацией тромбоцитов и окклюзией микрососудов.
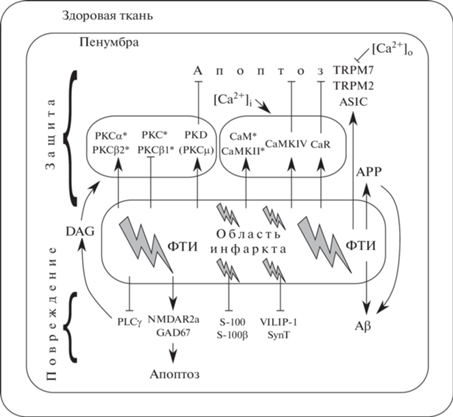
Уже в течение первых 10 мин после ФТИ отмечается резкое повышение уровня Ca2+ в цитозоле [6, 13]. Выделяют следующие механизмы повышения внутриклеточного Ca2+: проникновение через NMDA-рецепторы, потенциалзависимые Ca2+-каналы, [12, 14]; высвобождение из митохондрий и эндоплазматического ретикулума (ЭР); ингибирование Са2+-ATP-аз в ЭР и плазматической мембране и нарушение работы Na+/Ca2+-обменника [7, 12]; долговременная активация белков каналов транзиентного рецепторного потенциала (transient receptor potential, TRP) [12, 15], неселективных катион-зависимых каналов, способных активироваться независимо от деполяризации мембраны. В ядре инсульта гибель клеток наступает немедленно, но в пенумбре многие клетки стремятся защитить себя с помощью различных внутренних механизмов, в том числе путем регулирования обмена Ca2+. Падение внеклеточного Ca2+ и Mg2+ приводит к активации TRPM7. Снижение внеклеточного Ca2+ активирует также кислото-чувствительные ионные каналы (ASICs), которые активируются во время ишемии благодаря продукции молочной кислоты и протонов. Было показано, что TRPM7, TRPM2 и ASIC способствуют отсроченной гибели клеток центральной нервной системы (ЦНС) при инсульте [15, 16].
Показано, что в условиях ишемии при связывании глутамата рецептор NMDAR2a обеспечивает приток Na+ и Ca2+ в нейроны и высвобождение K+, что деполяризует соседние клетки и опосредует распространение эксайтотоксичности. Са2+-зависимый эксайтотоксический ответ приводит к апоптозу. Наблюдаемая в пенумбре после ФТИ гиперэкспрессия NMDAR2a и глутаматдекарбоксилазы (GAD65/67), отвечающей за превращение L-глутамата в гамма-аминомасляную кислоту (ГАМК), была еще одним проапоптотическим эффектом, связанным с метаболизмом глутамата [11]. Этот эффект согласуется с наблюдением, что эмбриональный вариант сплайсинга GAD67 индуцирует апоптоз во взрослом мозге при инсульте [17–19].
Комплекс Ca2+/кальмодулин регулирует многочисленные функции, такие как производство и высвобождение нейротрансмиттеров, ремоделирование цитоскелета, аксональный транспорт, экспрессию генов, выживаемость клеток, обучение и память. Кальмодулин и кальмодулин-зависимые киназы, CaMKII и CaMKIV, широко распространены в мозге млекопитающих. Наши исследования показали, что их уровень в ишемической пенумбре повышался после ФТИ. CaMKII-опосредованная нейропротекция связана с фосфорилированием и ингибированием некоторых про-апоптических белков, таких как NO-синтаза и GSK-3β. С другой стороны, CaMKII фосфорилирует глутаматные рецепторы NMDA и AMPA. Это увеличивает приток Ca2+ и усугубляет эксайтотоксичность. Ингибиторы CaMKII защищают мозг от ишемического повреждения. CaMKIV ингибирует эксайтотоксичность за счет активации антиапоптотического пути PI3K/Akt и фосфорилирования CREB, который опосредует экспрессию некоторых антиапоптотических генов [11].
Другой Ca2+-зависимый белок кальретикулин регулирует гомеостаз кальция и контролирует сворачивание белка в ЭР. Он модулирует экспрессию и активность р53 и, таким образом, регулирует апоптоз. Показано, что на ранних стадиях инсульта кальретикулин ингибирует апоптоз нейронов в пенумбре [7]. Таким образом, наблюдаемая нами в результате протеомного анализа активация кальретикулина в пенумбре через 4 ч после ФТИ может играть защитную роль [9].
Вместе с тем уровень некоторых сигнальных белков в ФТИ-индуцированной пенумбре существенно снижался. Возможно, массовое проникновение Ca2+ в клетки пенумбры при повреждении и отеке мозга вызывает протеолиз и снижение экспрессии Ca2+-зависимых белков. Например, уровень таких Ca2+-зависимых сигнальных белков как фосфолипаза Cγ (PLCγ) и протеинкиназа C (PKC), снижался в ФТИ-индуцированной пенумбре. PLCγ при повреждении плазматической мембраны или активации тирозинкиназных рецепторов расщепляет мембранные фосфолипиды и высвобождает инозитол-1,4,5-трифосфат (IP3), который стимулирует высвобождение Ca2+ из ЭР, и диацилглицерол, который активирует PKC [11]. Активируемая ионами Ca2+ PKC и ее изоформы α, γ, δ, ε, η и μ управляют множеством функций клеток, таких как регуляция гомеостаза, синаптические процессы, пролиферация, апоптоз и реакции мозга на ишемическое повреждение [9, 10]. При изучении экспрессии PKC и ее изоформ с помощью сигнальных и нейрональных микрочипов [9] мы получили противоречивые результаты. Применение нейрональных микрочипов показало более чем 4–6-кратное снижение общего уровня PKC в пенумбре через 1 и 4 ч после ФТИ. Эти результаты согласуются с быстрой потерей общего уровня PKC и ее активности после ишемического повреждения головного мозга вследствие, вероятно, ее деградации. Ингибиторы PKC защищали нейроны от постишемического и эксайтотоксического повреждения, что свидетельствовало об участии PKC в нейродегенерации. Поэтому наблюдавшееся снижение уровня PKC могло соответствовать уменьшению повреждения нервных клеток в пенумбре. Экспрессия ее изоформы PKCβ1 в пенумбре тоже значительно снижалась в 1.65–2 раза в течение всего изученного периода от 1 до 24 ч после ФТИ, но уровень другой изоформы PKCβ2 через 4 ч, напротив, повышался. Однако с помощью сигнальных микрочипов [11] наблюдалось повышение уровня PKCβ и PKCα на 34–36 и 31–54% соответственно, через 4–24 ч после ФТИ. Можно предположить, что в ткани пенумбры больше изоформы PKCβ2, чем PKCβ1. Поэтому именно изоформа PKCβ2 вносила решающий вклад в повышение общего уровня PKCβ. Уровни обеих протеинкиназ, PKCα и PKCβ, в мозге крыс также повышались после неполной глобальной ишемии, что авторы связывают с увеличением проницаемости гематоэнцефалического барьера. Протеинкиназа Cμ (PKCµ, ее также называют PKD) выступает в качестве нейропротектора при ишемии головного мозга. Она фосфорилирует шаперон Hsp27, который подавляет нейрональный апоптоз, опосредованный каскадом ASK1/JNK. Мы наблюдали активацию PKCµ (PKD) в ФТИ-индуцированной пенумбре, что в целом согласуется с литературными данными [9].
Уровни Са2+-связывающего белка S-100 и его β-цепи (S-100β) более чем в 2 раза снижались в пенумбре через 1–24 ч после ФТИ. Белок S-100β специфичен для нервной ткани, в частности для глиальных клеток. В наномолярном диапазоне он демонстрирует нейропротекторные свойства, но при микромолярных концентрациях он может вызвать апоптоз. Появление S-100 и S-100β в сыворотке крови может являться маркером таких нейропатологий, как острый инсульт, нейротравма, опухоль головного мозга и нейродегенеративные заболевания [9–11]. Ca2+ также косвенно контролирует экспрессию генов посредством Ca2+-зависимого взаимодействия между кальмодулином и S-100 с димерными факторами транскрипции, что предотвращает связывание этих факторов с ДНК [9].
Через 1 и 4 ч после ФТИ уровень белка VILIP1 в пенумбре снижался на 66–71%. Этот Са2+-связывающий белок служит одним из датчиков Са2+ в клетке. Он широко распространен в головном мозге. VILIP1 оказывает влияние на различные внутриклеточные сигнальные каскады, включая системы циклических нуклеотидов и МАР-киназный путь. При увеличении цитозольной концентрации Са2+ VILIP1 связывается с клеточной мембраной и регулирует везикулярный трафик и рециркуляцию рецепторов и ионных каналов. После инсульта VILIP1 высвобождается из поврежденных нервных клеток в спинномозговую жидкость и сыворотку крови, являясь маркером инсульта [9]. Ca2+-сенсор cинаптотагмин отвечает за слияние синаптических пузырьков с синаптической мембраной и высвобождение нейромедиатора. ФТИ воздействие приводило к подавлению его экспрессии. Это соответствовало ультраструктурным данным о деструкции и дезорганизации синаптических пузырьков. У крыс мРНК синаптотагмина подавлялась во время ранней рециркуляции после фокальной церебральной ишемии [9, 11].
Протеомный анализ показал повышение экспрессии белка-предшественника β-амилоида (β-amyloid precursor protein, APP) в ФТИ-индуцированной пенумбре в первые 24 ч после воздействия [11]. Наши результаты согласуются с данными об активации АРР после церебральной ишемии, что рассматривается как адаптивный и нейропротекторный ответ [20]. На разных животных моделях ишемического инсульта были показаны защитные эффекты APP или его растворимых фрагментов (APPsa). Основные механизмы такого защитного эффекта включают APP-опосредованную регуляцию гомеостаза кальция через NMDA-рецепторы, а также потенциал-зависимые кальциевые каналы (VGCC) и внутриклеточные кальциевые депо [12, 20]. Исследования на нокаутных мышах показали, что в условиях ишемии мутации пресенилина-1 (PS1), участвующего в протеолизе APP, приводят к нарушению клеточного кальциевого гомеостаза в нейронах, что связано с высвобождением кальция из ЭР, и тем самым повышают их уязвимость к ишемическому повреждению [20]. Наши исследования также продемонстрировали повышение уровня β-амилоида [9], фрагмента амилоидного пептида (Aβ), стимулирующего приток Ca2+ в клетку и обладающего тем самым нейротоксичностью. Механизмы АРР-опосредованной нейропротекции через регуляцию уровней клеточного кальция с помощью блокирования кальциевых каналов L-типа (LTCС) и NMDA-рецепторов определяют новые терапевтические мишени в противоинсультной терапии [20].
Данные о влиянии фототромботической ишемии на экспрессию связанных с кальцием функциональных белков обобщены на рис. 1. Так как Ca2+ является одним из ключевых факторов ишемического повреждения клеток, в ходе многочисленных поисков нейропротекторов были испытаны различные блокаторы Ca2+-каналов, а также ингибиторы Ca2+-связывающих белков. Однако эффективные нейропротекторы пока не найдены. Даже соединения, которые проявляли положительное действие на экспериментальных животных, не были эффективны в организме человека, либо вызвали нежелательные побочные эффекты [1, 2, 8]. Данные о влиянии Ca2+ на процессы выживания и гибели клеток при ишемическом инсульте все еще неполны и местами противоречивы, однако их достаточно для понимания важности этого иона в выживании и смерти клеток. Поэтому для разработки новых подходов к лечению последствий инсульта необходимо глубокое, всестороннее изучение молекулярных механизмов нейродегенерации и нейропротекции в ткани пенумбры с участием Ca2+.
Рис. 1.
Защитные и повреждающие эффекты фототромботического инсульта на экспрессию кальций-зависимых белков в пенумбре. Стрелки с острыми концами указывают на стимулирующее воздействие, стрелки с тупыми концами – на подавляющее воздействие. ФТИ – фототромботический инсульт; PKC – протеинкиназа С; CaM – кальмодулин; CaMK – кальмодулинкиназа; CaR – кальретикулин; APP – белок-прекурсор амилоида; Aβ – β-амилоид; DAG – диацилглицерин; PLC – фосфолипаза С; SynT – синаптотагмин; [Ca2+]o – внеклеточный кальций; [Ca2+]i – внутриклеточный кальций. Звездочками (*) указаны противоречивые данные о защитном/повреждающем характере эффекта. Остальные обозначения см. в тексте.
ФОТОДИНАМИЧЕСКОЕ ВОЗДЕЙСТВИЕ И КАЛЬЦИЕВЫЙ ГОМЕОСТАЗ В НЕЙРОНАХ И ГЛИАЛЬНЫХ КЛЕТКАХ
Фотодинамическая терапия – медицинский метод, при котором световое излучение разрушает патологически измененные ткани (например, опухолевые), избирательно окрашенные специальными фотосенсибилизирующими составами. Фотодинамический (ФД) эффект основан на фотохимическом превращении молекул, находящихся в электронно-возбужденном состоянии, в ходе которого происходит образование АФК, среди которых наиболее важен синглетный кислород, способный повреждать только биоструктуры, находящиеся в непосредственной близости от молекул фотосенсибилизатора. В ЦНС опухоли может образовывать только способная к пролиферации глия, но не нейроны. В нашей лаборатории на фундаментальном уровне были изучены различные сигнальные пути, в том числе кальций-зависимые механизмы, участвующие в клеточном ответе нейронов и глии на ФД воздействие в изолированном абдоминальном механорецепторе речного рака [21, 22], а также совместно с лабораторией А.Ю. Абрамова на первичных культурах нейронов и астроцитов крыс.
Участие кальциевого сигнального пути в фотодинамическом повреждении нейронов и глиальных клеток
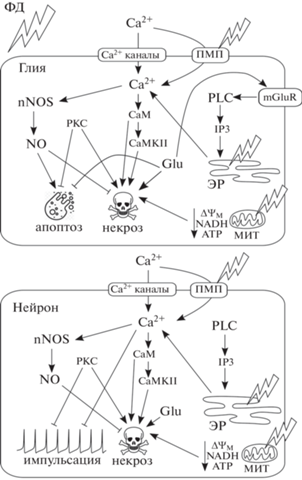
Было изучено участие Ca2+ и PKC в электрофизиологическом ответе нейрона на фотодинамическое повреждение с широко клинически применяемым препаратом Фотосенс (гидрокси-алюминия трисульфофталоцианин), на изолированном механорецепторе речного рака. Активизация PKС 12-O-тетрадеканоилфорбол-13-ацетатом (TPA) и повышение цитозольной концентрации Ca2+ с помощью иономицина и тапсигаргина ускоряли ФД-индуцированное прекращение импульсной активности. Ингибирование PKC стауроспорином, гиперицином и хелеритрином, наоборот, продлевало время функциональной активности. Это показывает участие Ca2+ и PKC в ФД-индуцированной инактивации и последующей смерти нейрона [23]. ФД воздействие не только приводит к прекращению импульсной активности и некрозу нейронов, но и вызывает некроз и апоптоз, а также пролиферацию окружающей их глии. Было изучено участие в этом эффекте белков кальций-зависимого сигнального пути – кальмодулина, CaMKII и PKC, c помощью их специфических ингибиторов. Было выяснено, что кальмодулин и CaMKII участвуют в ФД-индуцированном некрозе нейрона и глии, а PKC защищает глию от апоптоза и некроза при ФД воздействии [24].
Кальциевый ответ на фотодинамическое воздействие радахлорина
В модели рецептора растяжения рака (РРР) перспективный фотосенсибилизатор радахлорин накапливался преимущественно в глиальной оболочке и относительно слабо проникал в тело и аксон механорецепторного нейрона (МРН). Фотоактивированный радахлорин сокращал продолжительность импульсации нейрона и индуцировал некроз и апоптоз сателлитной глии при концентрациях 10−9 M и ниже [25]. ФД эффект радахлорина использовался для выявления воздействия фотоиндуцированного окислительного стресса на кальциевую сигнализацию и перекисное окисление липидов (ПОЛ) в первичных культурах нейронов и астроцитов с помощью прижизненной клеточной визуализации. Облучение в присутствии 200 нМ радахлорина индуцировало кальциевый сигнал в нейронах и астроцитах, который зависит от внутриклеточных кальциевых депо, т.к. может блокироваться истощением ЭР и ингибированием Ca2+-ATP-азы ЭР тапсигаргином. Также кальциевый ответ блокировался ингибированием PLС. При этом радахлорин индуцировал ПОЛ в нейронах и астроцитах. Можно предположить, что ФД-индуцированное ПОЛ ведет к активации PLC, что ведет к продукции IP3, который, в свою очередь, вызывает выход Ca2+ из ЭР в цитозоль через IP3-рецепторы [26]. Также было обнаружено, что ФД воздействие на клетки с радахлорином приводит к митохондриальной деполяризации как в нейронах, так и в астроцитах, и снижает уровень митохондриального NADH [27].
Кальций-зависимая регуляция продукции оксида азота при фотодинамическом воздействии
Оксид азота (NO) регулирует многие физиологические и патофизиологические процессы, включая нейротрансмиссию, вазодилатацию, стрессовый ответ и др. Он обладает защитным действием при апоптозе и ПОЛ как антиоксидант. Фотодинамическое воздействие с Фотосенсом на модели РРР индуцировало генерацию NO в глиальных клетках и дендритах и в меньшей степени в соме и аксоне нейрона. С помощью модуляторов уровня цитозольного кальция было показано, что Ca2+ и NF-kB регулируют генерацию NO в фотосенсибилизированных нейронах и глие. Производство NO стимулировалось четырехкратным повышением концентрации внеклеточного Ca2+, иономицином и ингибированием Ca2+-ATP-азы ЭР. Блокирование кальциевых каналов плазмалеммы, наоборот, снижало продукцию NO. По-видимому, Ca2+-зависимая нейрональная NO-синтаза участвует, наряду с независимой от Ca2+ индуцибельной NO-синтазой, в производстве NO в нейронах и глиальных клетках речного рака [28].
Действие глутамата при фотодинамическом воздействии
Ионотропные глутаматные рецепторы (в частности, NMDA) являются ионными каналами, пропускающими в том числе Ca2+. Метаботропные глутаматные рецепторы участвуют в регуляции потенциал-зависимых кальциевых каналов и IP3-рецепторов ЭР. Добавление глутамата усиливало пронекротический эффект ФД воздействия на нейроны и глию, однако снижало ФД-индуцированный апоптоз глии. Нейроны рака выделяют N-ацетиласпартилглутамат (NAAG), расщепление которого карбоксипептитазой II (GCPII) во внеклеточном пространстве высвобождает глутамат. NAAG также подавлял ФД-индуцированный апоптоз. Ингибирование же GCPII, соответственно, напротив, стимулировало апоптоз глии. Ингибирование метаботропных рецепторов глутамата, но не NMDA, снижало ФД-индуцированный апоптоз глии. Таким образом, высвобождаемый посредством GCPII глутамат защищает глию от апоптоза, по-видимому, через метаботропные, но не ионотропные рецепторы [29]. Полученные результаты проиллюстрированы на рис. 2.
Рис. 2.
Участие Ca2+-зависимых путей, включая NO- и глутаматную сигнализацию, в реакциях нейронов и глиальных клеток на фотодинамическое воздействие. Потенциальные мишени ФД воздействия обозначены молниями. Стрелки с острыми концами указывают на стимулирующее воздействие, стрелки с тупыми концами – на подавляющее воздействие. Сокращения: МИТ – митохондрии; ЭР – эндоплазматический ретикулум; СаМ – кальмодулин; CaMKII – кальмодулин-зависимая киназа II; nNOS – нейрональная NO-синтаза; IP3 – инозитолтрифосфат; PKC – протеинкиназа C; PLC – фосфолипаза С; ПМП – поры в плазматической мембране; Glu – глутамат; mGluR – метаботропные рецепторы глутамата; ΔΨM – митохондриальный потенциал.
ЗАКЛЮЧЕНИЕ
Нарушение кальциевого гомеостаза нейронов и глиальных клеток может быть спровоцировано различными факторами, включая вызванную фототромботическим инсультом ишемию и вызванный фотодинамическим повреждением окислительный стресс. Ионы кальция, в свою очередь, опосредуют различные механизмы клеточного ответа на повреждение, направленные как на защиту, так и на дегенерацию клеток. Центральным звеном этих процессов является концентрация кальция в цитозоле, которая в физиологических условиях поддерживается на низком уровне, а в патологических повышается на несколько порядков и зачастую запускает необратимые сигнальные каскады, ведущие к клеточной смерти. Исследование на модели РРР также подтвердило противоапоптотическую активность глутамата, предположительно связанную с метаботропными рецепторами, и кальций-зависимое повышение продукции оксида азота в условиях окислительного стресса на физиологическую активность нейрона и выживание нейронов и глиальных клеток. В сочетании с данными, полученными на других моделях, эти результаты дают вклад в фундаментальную базу для дальнейшей разработки методов нейропротекции и терапии опухолей.
Конфликт интересов. Авторы заявляют, что у них нет конфликта интересов.
Источники финансирования. Работа выполнена при поддержке Министерства науки и высшего образования Российской Федерации (проект № 0852-2020-0028).
Соответствие принципам этики. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
Campbell B.C.V., De Silva D.A., Macleod M.R., Coutts S.B., Schwamm L.H., Davis S.M., Donnan G.A. 2019. Ischaemic stroke. Nat. Rev. Dis. Prim. 5, 70.
Chavez J.C., Hurko O., Barone F.C., Feuerstein G.Z. 2009. Pharmacologic interventions for stroke: Looking beyond the thrombolysis time window into the penumbra with biomarkers, not a stopwatch. Stroke. 40 (10), e558–e563. https://doi.org/10.1161/STROKEAHA.109.559914
Ludhiadch A., Sharma R., Muriki A., Munshi A. 2022. Role of calcium homeostasis in ischemic stroke: A review. CNS Neurol. Disord. – Drug Targets. 21, 52–61.
Khodorov B.I. 2000. Mechanisms of destabilization of Ca2+-homeostasis of brain neurons caused by toxic glutamate challenge. Membr. Cell Biol. 14, 149–162.
Khodorov B. 2004. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Prog. Biophys. Mol. Biol. 86, 279–351.
Uzdensky A.B. 2018. Photothrombotic stroke as a model of ischemic stroke. Transl. Stroke Res. 9, 437–451.
Узденский А.Б., Демьяненко С.В. 2016. Фототромботический инсульт: биохимия пенумбры. Ростов-на-Дону: Изд-во Южного федерального университета. 127 с.
Radak D., Katsiki N., Resanovic I., Jovanovic A., Sudar-Milovanovic E., Zafirovic S., Mousad S.A., Isenovic E.R. 2017. Apoptosis and acute brain ischemia in ischemic stroke. Curr. Vasc. Pharmacol. 15, 115–122.
Uzdensky A., Demyanenko S., Fedorenko G., Lapteva T., Fedorenko A. 2017. Protein profile and morphological alterations in penumbra after focal photothrombotic infarction in the rat cerebral cortex. Mol. Neurobiol. 54, 4172–4188.
Uzdensky A.B. 2019. Apoptosis regulation in the penumbra after ischemic stroke: Expression of pro- and antiapoptotic proteins. Apoptosis. 24, 687–702.
Demyanenko S., Uzdensky A. 2017. Profiling of signaling proteins in penumbra after focal photothrombotic infarct in the rat brain cortex. Mol. Neurobiol. 54, 6839–6856.
Singh V., Mishra V.N., Chaurasia R.N., Joshi D., Pandey V. 2019. Modes of calcium regulation in ischemic neuron. Indian J. Clin. Biochem. 34, 246–253.
Watson B.D., Dietrich W.D., Busto R., Wachtel M.S., Ginsberg M.D. 1985. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 17, 497–504.
Wang M., Tan J., Miao Y., Li M., Zhang Q. 2018. Role of Ca2+ and ion channels in the regulation of apoptosis under hypoxia. Histol. Histopathol. 33, 237–246.
MacDonald J.F., Xiong Z.-G., Jackson M.F. 2006. Paradox of Ca2+ signaling, cell death and stroke. Trends Neurosci. 29, 75–81.
Aarts M., Iihara K., Wei W.-L., Xiong Z.-G., Arundine M., Cerwinski W., MacDonald J.F., Tymianski M. 2003. A key role for TRPM7 channels in anoxic neuronal death. Cell. 115, 863–877.
Ginsberg M.D. 2008. Neuroprotection for ischemic stroke: Past, present and future. Neuropharmacology. 55, 363–389.
Zhou X., Ding Q., Chen Z., Yun H., Wang H. 2013. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J. Biol. Chem. 288, 24151–24159.
Jaenisch N., Popp A., Guenther M., Schnabel J., Witte O.W., Frahm C. 2014. Pro-apoptotic function of GABA-related transcripts following stroke. Neurobiol. Dis. 70, 237–244.
Hefter D., Draguhn A. 2017. APP as a protective factor in acute neuronal insults. Front. Mol. Neurosci. 10, 22. https://doi.org/10.3389/fnmol.2017.00022
Uzdensky A.B., Rudkovskii M.V., Fedorenko G.M., Berezhnaya E.V., Ischenko I.A., Kovaleva V.D., Komandirov M.A., Neginskaya M.A., Khaitin A.M., Sharifulina S.A. 2014. Responses of crayfish neurons and glial cells to photodynamic impact: Intracellular signaling, ultrastructural changes, and neuroglial interactions. Biochem. Suppl. Ser. A Membr. Cell Biol. 8, 1–15.
Uzdensky A., Berezhnaya E., Khaitin A., Kovaleva V., Komandirov M., Neginskaya M., Rudkovskii M., Sharifulina S. 2015. Protection of the crayfish mechanoreceptor neuron and glial cells from photooxidative injury by modulators of diverse signal transduction pathways. Mol. Neurobiol. 52, 811–825.
Bragin D.E., Kolosov M.S., Uzdensky A.B. 2003. Photodynamic inactivation of isolated crayfish neuron requires protein kinase C, PI 3-kinase and Ca2+. J. Photochem. Photobiol. B. 70, 99–105.
Uzdensky A., Lobanov A., Bibov M., Petin Y. 2007. Involvement of Ca2+- and cyclic adenosine monophosphate-mediated signaling pathways in photodynamic injury of isolated crayfish neuron and satellite glial cells. J. Neurosci. Res. 85, 860–870.
Neginskaya M.A., Berezhnaya E.V., Rudkovskii M.V., Demyanenko S.V., Uzdensky A.B. 2014. Photodynamic effect of Radachlorin on nerve and glial cells. Photodiagnosis Photodyn. Ther. 11, 357–364.
Neginskaya M., Berezhnaya E., Uzdensky A.B., Abramov A.Y. 2018. Reactive oxygen species produced by a photodynamic effect induced calcium signal in neurons and astrocytes. Mol. Neurobiol. 55, 96–102.
Berezhnaya E., Neginskaya M., Uzdensky A.B., Abramov A.Y. 2018. Photo-induced oxidative stress impairs mitochondrial metabolism in neurons and astrocytes. Mol. Neurobiol. 55, 90–95.
Rodkin S.V., Kovaleva V.D., Berezhnaya E.V., Neginskaya M.A., Uzdensky A.B. 2019. Ca2+- and NF‑κB-dependent generation of NO in the photosensitized neurons and satellite glial cells. J. Photochem. Photobiol. B Biol. 199, 111603.
Rudkovskii M.V., Romanenko N.P., Berezhnaya E.V., Kovaleva V.D., Uzdensky A.B. 2010. Glutamate-mediated protection of crayfish glial cells from PDT-induced apoptosis. Saratov Fall Meet. 2010 Opt. Technol. Biophys. Med. XII. 7999, 79990Q.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии