

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 4, стр. 271-282
Na+,K+-ATP-аза как полифункциональный белок
О. Д. Лопина a, *, О. В. Букач a, С. В. Сидоренко a, Е. А. Климанова a, **
a Московский государственный университет им. М.В. Ломоносова,
биологический факультет, кафедра биохимии
119234 Москва, Россия
* E-mail: od_lopina@mail.ru
** E-mail: klimanova.ea@yandex.ru
Поступила в редакцию 31.01.2022
После доработки 01.03.2022
Принята к публикации 06.03.2022
- EDN: WWCFFG
- DOI: 10.31857/S0233475522040065
Аннотация
С момента открытия Na+,K+-ATP-азы Й. Скоу в 1957 году этот фермент рассматривался исключительно как транспортер, обеспечивающий активный перенос ионов Na+ и K+ через плазматическую мембрану клеток, в связи с чем подробно изучалась его структура и механизм функционирования, а также вовлеченность в работу систем вторичного транспорта ионов. В настоящем обзоре кратко рассмотрены данные о структуре и функционировании фермента. Более подробно описана установленная позднее роль Na+,K+-АТР-азы как рецептора кардиотонических стероидов (КТС), связывание которых с ферментом инициирует разнообразные сигнальные пути за счет белок-белковых взаимодействий, модифицируемых также изменением внутриклеточной концентрации ионов Na+ и K+ за счет ингибирования транспортной функции Na+,K+-АТР-азы, и Са2+ путем опосредованного изменения активности Na+/Ca2+-обменника. Все это обеспечивает разнообразные эффекты КТС, включая их влияние на экспрессию генов, состояние плотных контактов, адгезию клеток, индукцию гипертрофии миокарда, стимуляцию генерации свободно-радикальных форм кислорода, а также инициирует смерть или выживание клеток в зависимости от типа ткани. Приведены данные об открытии эндогенных КТС, а также анализ данных литературы, свидетельствующих о том, что концентрации эндогенных КТС столь малы, что вряд ли они способны вызвать ингибирование Na+,K+-АТР-азы. В связи с этим приведены данные об активации фермента низкими дозами КТС и высказана идея о возможной суммации концентраций различных стероидов. В заключении рассмотрены возможные направления исследования множественных функций Na+,K+-АТР-азы.
ВВЕДЕНИЕ
В 1997 году Нобелевская премия по химии была вручена трем ученым: датчанину Й. Скоу из Орхусского университета, американцу П. Бойеру из Калифорнийского университета в Лос-Анджелесе и англичанину Дж. Уокеру из лаборатории молекулярной биологии Медицинского исследовательского совета Великобритании (Кембридж) за изучение ферментов, обратимо преобразующих энергию АТР в трансмембранный градиент ионов. П. Бойер и Дж. Уокер длительное время занимались изучением АТР-синтетазы митохондрий: П. Бойер предложил трехтактный механизм работы этого фермента и предсказал существование вращения части этой молекулярной машины в процессе работы, а Дж. Уокер с использованием методов электронной микроскопии и рентгеноструктурного анализа выяснил строение этого фермента и подтвердил трехтактную кинетическую схему его работы. Вращение части фермента при его функционировании было позднее подтверждено японскими исследователями.
Третий нобелевский лауреат Й. Скоу был известен научному миру как первооткрыватель и исследователь Na+,K+-АТР-азы, фермента, обеспечивающего перенос ионов Na+ и К+ через плазматическую мембрану клеток животных против электрохимического градиента. В 1957 году, исследуя гомогенат нервов краба, Й. Скоу обнаружил в нем АТР-азную активность, зависящую от соотношения ионов Na+ и К+ в среде, т.е. обнаружил фермент, который специфически распознает два этих одновалентных иона [1].
Еще в конце 30-х годов прошлого века было установлено, что кардиотонические стероиды (класс родственных соединений, в структуру которых входит циклопентанпергидрофенантреновое (стероидное) ядро) вызывают накопление ионов Nа+ и потерю ионов К+ в сердечной мышце [2], а в 1953 году Х. Шатцман установил, что эти соединения подавляют АТР-зависимую аккумуляцию К+ и выведение Na+ из эритроцитов человека [3]. Эти эксперименты дали основание считать, что в клетках имеется Na+,K+-насос, который переносит ионы Na+ и K+ через клеточную мембрану против электрохимического градиента. Уже через три года после обнаружения Na+,K+-зависимой АТР-азы Й. Скоу опубликовал следующую работу, в которой показал, что обнаруженная им Na+,K+-АТР-аза ингибируется уабаином (соединением из класса кардиостероидов) и заключил, что именно она является тем самым Na+-насосом, который выкачивает Na+ из клетки, используя в качестве источника энергии АТР [4].
Na+,K+-ATP-аза: СТРОЕНИЕ И ТРАНСПОРТНЫЕ ФУНКЦИИ
Во второй половине XX века в лаборатории Й. Скоу и еще в нескольких лабораториях США и Европы начали интенсивно исследовать механизм работы Na+-насоса. Этот фермент был получен из почек [5], где он принимает активное участие в реабсорбции ионов Na+ из первичной мочи, а также (за счет создания градиента Na+) обеспечивает энергией системы вторичного транспорта, такие как Na+,K+,Сl–-котранспорт, Na+/Cа2+-обменник, Na+/H+-обменник, а также системы реабсорбции аминокислот, нуклеотидов и др. [6]. Изучение его структуры показало, что Na+,K+-АТР-аза состоит минимум из двух типов субъединиц, одна из которых является каталитической ($\alpha $-субъединица) с молекулярной массой около 100 кДа, а другая ($\beta $-субъединица), белковая часть которой имеет массу около 35 кДа, существенно гликозилируется (до достижения молекулярной массы в 55–60 кДа) и непосредственного участия в катализе не принимает, однако обеспечивает доставку α-субъединицы от места синтеза к плазматической мембране и является регуляторной, влияя на сродство фермента к одновалентным катионам [7, 8].
В настоящее время идентифицировано 4 изоформы α- (α1–α4) и 3 изоформы β-субъединиц (β1–β3), они характеризуются различным распределением в тканях и могут образовывать комплексы в любом сочетании, например, α1β1, α1β2 и т.д. Образованные путем таких комбинаций тканеспецифичные изоферменты Na+,K+-ATP-азы различаются по кинетическим свойствам, в частности имеют различное сродство к уабаину, АТР и одновалентным катионам [7, 9].
Почти сразу после получения очищенной Na+,K+-АТР-азы из почек была обнаружена и ее третья, γ-субъединица [10]. Однако ее функциональное значение долгое время оставалось неясным, пока не было обнаружено, что она формирует в мембране единый комплекс с αβ-димером, влияя на некоторые кинетические характеристики Na+,K+-АТР-азы, в частности на ее чувствительность к одновалентным катионам [11]. γ-Cубъединица Na+,K+-АТР-азы относится к семейству малых мембранных белков FXYD с единственным трансмембранным доменом, по крайней мере 5 из 7 белков этого семейства, имеющие различные названия, а именно FXYD1 (фосфолемман), FXYD2 (γ-субъединица Na+,K+-ATP-азы), FXYD3 (Mat-8), FXYD4 (CHIF) и FXYD7, входят в состав Na+,K+-ATP-азы и принимают участие в регуляции ее активности. Следует, однако, отметить, что белки семейства FXYD взаимодействуют не только с Na+,K+-ATP-азой, они способны образовывать комплексы и с другими мембранными белками, например, в плазматической мембране сердца они находятся рядом и при иммунопреципитации осаждаются совместно с Na+/Ca2+-обменником и Са2+-каналом L-типа [12]. Взаимодействуя с этим транспортером, фосфолемман (FXYD1) подавляет его активность [13].
При изучении ферментативных свойств Na+,K+-АТР-азы было установлено, что в процессе каталитического цикла происходит образование так называемого фосфорилированного интермедиата, возникающего за счет переноса терминального фосфорильного остатка АТР на остаток аспартата, расположенного в активном центре Na+,K+-АТР-азы [14]. Этот процесс происходит одновременно с переносом Na+ из клетки наружу, в то время как гидролиз фосфоинтермедиата – с переносом ионов K+ в противоположном направлении. Почти сразу после обнаружения фосфоинтермедиата Р. Постом была предложена схема каталитического цикла фермента, получившая название схемы Алберса–Поста, по фамилиям авторов, опубликовавших статью с этой схемой [15, 16]. В процессе исследований она уточнялась, несколько модифицировалась, и в результате оказалась пригодной для описания каталитического цикла других ферментов, относящихся к АТР-азам Р-типа (например, Са2+-АТР-азы, Н+,К+-АТР-азы слизистой оболочки желудка и др.) [17].
Итак, уже к началу XXI века были выявлены изоформы всех субъединиц Na+,K+-АТР-азы, исследовано их распределение в различных тканях, выяснено, как именно полипептидные цепи этих субъединиц располагаются внутри мембраны. Благодаря усилиям сотрудников нескольких лабораторий была установлена аминокислотная последовательность отдельных субъединиц. И наконец, фермент был закристаллизован в различных конформациях и были получены 3D-структуры Na+,K+-АТР-азы с высоким разрешением [18]. Следует отметить, что все работы по кристаллизации и определению третичной структуры фермента были выполнены с использованием α1β1γ-изофермента, поскольку только этот изофермент был получен в очищенном виде.
После получения Й. Скоу Нобелевской премии и установления механизма функционирования и трехмерной структуры Nа+,К+-АТР-азы стало казаться, что этот объект уже не представляет интереса для исследователей. Однако ситуация оказалась не столь проста, как выглядела на первый взгляд. Последние данные позволяют считать Na+,K+-АТР-азу белком с множественными функциями.
КАРДИОТОНИЧЕСКИЕ СТЕРОИДЫ – РЕГУЛЯТОРЫ КЛЕТОЧНОГО МЕТАБОЛИЗМА
Работы, вызвавшие вторую волну интереса к Na+,K+-АТР-азе, были начаты еще в 80-е годы прошлого века. Они привели к открытию так называемых эндогенных кардиостероидов. Кардиотонические стероиды, являющиеся ингибиторами Na+,K+-АТР-азы, были известны как вещества растительного происхождения. С давних времен они использовались в качестве ядов и лекарств. Например, название “уабаин” происходит от сомалийского waabaayo, что означает “яд стрелы”. Он в больших количествах содержится в африканских растениях строфант приятный (Strophanthus gratus) и акокантера абиссинская (Acokanthera schimperi) и применялся охотниками для отравления наконечников стрел. Отвары из растений рода наперстянка (Digitalis purpurea и Digitalis lanata), содержащих родственные уабаину кардиостероиды дигоксин и дигитоксин, использовались для лечения водянки. Еще в 1650 г. наперстянка пурпурная была включена в английскую фармакопею, но ее перестали использовать в 1746 г. из-за часто происходящих отравлений. [19]. Однако в 1775 г. английский врач сэр У. Визеринг опубликовал сообщение о том, что экстракт из листьев наперстянки является средством для лечения сердечной недостаточности, и потратил почти 10 лет на его реабилитацию [20]. Дигоксин и в настоящее время используют для лечения хронической сердечной недостаточности, связанной с декомпенсированными пороками клапанов сердца, а также при атеросклеротическом кардиосклерозе и перегрузке миокарда вследствие артериальной гипертензии [21].
Все кардиотонические стероиды по структуре делят на две группы: карденелиды и буфадиенолиды. Соединения обеих групп содержат стероидное ядро, которое в 17 положении имеет в качестве заместителя пятичленное (карденелиды) или шестичленное (буфадиенолиды) лактоновое кольцо. Кроме того, у карденелидов в 3-м положении могут находиться от одного до трех остатков сахаров; буфадиенолиды являются, главным образом, агликонами. Буфадиенолиды обнаружены в слизи жаб рода Bufо (в частности, Bufo rubescens и Bufo marinus), они защищают этих амфибий от поедания хищниками.
В 80-е годы прошлого столетия было обнаружено, что у некоторых экспериментальных животных, например, крыс с гипертензией, сопровождающейся увеличением объема циркулирующей жидкости (модель “one clip, one kidney”), а также у больных гипертонической болезнью и некоторыми другими заболеваниями, в крови обнаруживаются органические соединения с молекулярной массой менее 1000 Да [22]. Использование антител против дигоксина у таких животных снижало у них артериальное давление. Это привело исследователей к выводу, что у млекопитающих в крови присутствуют так называемые эндогенные кардиостероиды, повышение концентрации которых происходит при развитии сердечно-сосудистых, почечных, нейрональных и некоторых других заболеваний [22, 23].
Длительные поиски этих соединений привели к выделению из биологических жидкостей млекопитающих и идентификации сначала уабаина [24], а затем и таких кардиостероидов, как дигоксин, буфалин, маринобуфагенин, телоцинобуфагин и маринобуфатоксин [25]. Есть данные, свидетельствующие в пользу того, что эти эндогенные кардиостероиды синтезируются, предположительно, в коре надпочечников и гипоталамусе [6].
Какова же функция эндогенных стероидов? Что они регулируют в организме млекопитающих? В течение последних двух десятилетий было показано, что длительная инкубация различных клеток с кардиостероидами (в первую очередь с уабаином, который благодаря хорошей растворимости в воде обычно используется в экспериментах in vitro) приводит в разных клетках к различным физиологическим эффектам. Так, было показано, что уабаин влияет на экспрессию генов, синтез белка, состояние плотных контактов, адгезию клеток, вызывает гипертрофию миокарда, активирует генерацию свободно-радикальных форм кислорода, а также инициирует смерть или выживание клеток в зависимости от типа ткани и видовой принадлежности животного [25].
Обнаружено, что кардиотонические стероиды способны активировать различные сигнальные каскады благодаря конформационным переходам α1-субъединицы Na+,K+-АТР-азы. Их связывание с α1-субъединицей и вызванный этим конформационный переход приводит к активации нерецепторной тирозинкиназы Src, фосфатидилинозитол-3-киназы и рецептора инозитол-1,4,5-трифосфата (Ca2+-канал эндоплазматического ретикулума) [26]. Эти данные позволили предположить, что эндогенные кардиостероиды представляют собой новый класс стероидных гормонов.
Достаточно давно было выявлено, что инкубация клеток с уабаином приводит к увеличению экспрессии генов, в частности тех, что кодируют собственные субъединицы Na+,K+-ATP-азы (по крайней мере, в нервной ткани [27–29]). Но одно из обширных пионерских исследований, проведенных в лаборатории А. Аскари с использованием кардиомиоцитов, показало, что после связывания уабаина Na+,K+-ATP-аза взаимодействует с заякоренной в мембране нерецепторной Src-киназой, вызывая ее активацию [30].
Прямое взаимодействие Src-киназы с Na+,K+-АТР-азой впоследствии было подтверждено в экспериментах других исследователей [31], и механизм, лежащий в основе этого явления, был подробно изучен [32]. Установлено, что активированная Src-киназа фосфорилирует остаток тирозина в рецепторе фактора роста эпидермиса (EGFR), находящемся в плазматической мембране, и в нескольких других белках, причем это фосфорилирование устраняется ингибиторами Src-киназы, например, гербимицином A [33]. После фосфорилирования EGFR с остатком фосфотирозина этого белка взаимодействует адапторный белок Shc, содержащий необходимый для такого взаимодействия SH2-домен. В свою очередь с белком Shc взаимодействует белок Grb2, после чего последний связывает белок Sos, являющийся фактором обмена нуклеотидов. После образования комплекса с Grb2 он активируется и обеспечивает обмен GDP на GTP в мономерном G-белке Ras. Эти события происходят независимо от изменения внутриклеточного соотношения ионов Na+ и К+ [30], т.е. для активации Ras достаточно связывания уабаина с Na+,K+-АТР-азой. Однако связывание Ras необходимо, но недостаточно для дальнейшей активации классического каскада митоген-активируемых киназ, включающего Raf (MAP3K), MEK (MAP2K) и p42/p44 MAPK. Последняя в этом ряду протеинкиназа называется также ERK1/2. Она проникает в ядро и путем фосфорилирования регуляторных элементов влияет на экспрессию генов.
Активация Ras оказывается также необходима для увеличения генерации свободно-радикальных форм кислорода митохондриями. Параллельно уабаин, ингибируя Na+,K+-АТР-азу, увеличивает внутриклеточную концентрацию Na+, что приводит к активации Na+/Ca2+-обменника и к локальному повышению внутриклеточной концентрации Ca2+ [34]. Это, с одной стороны, вызывает активацию сокращения сердечной мышцы, с чем и связано, как считается, терапевтическое действие дигоксина при хронической сердечной недостаточности, называемое положительным инотропным эффектом. Предполагается, что этот эффект реализуется в специальных микродоменах, называемых плазмеросомами, основными компонентами которых являются α2- и α3-изоформы Na+,K+-ATP-азы и Na+/Ca2+-обменник [35]. В то же время точный механизм этого явления неясен, и уже давно вызывает дискуссии [36]. Есть сомнения в том, что в основе положительного инотропного эффекта лежит ингибирование Na+,K+-ATP-азы, поскольку концентрация дигоксина в плазме крови пациентов, принимающих этот кардиостероид в дозах, снижающих смертность, не превышает 1–2 нМ [37]. Тогда как концентрация дигоксина, при которой наблюдается полумаксимальное ингибирование Na+,K+-ATP-азы, содержащей α1-субъединицу, составляет 1.2 мкМ [38], а Na+,K+-АТР-азы, содержащей α2- и α3-субъединицы, равна 219 нМ [39]. Даже если учесть, что все изоферменты Na+,K+-АТР-азы человека наиболее чувствительны к кардиостероидам, 1–2 нМ дигоксина недостаточно, чтобы вызвать ингибирование фермента. По мнению З. Cе и А. Аскари, именно повышение концентрации внутриклеточного Са2+, с одной стороны, активирует факторы регуляции транскрипции NFκB и AP-1, что приводит к активации генов раннего ответа (Fos, Jun), а с другой – это второй фактор, необходимый для активации каскада протенкиназ, заканчивающегося активацией ERK1/2 [30]. Все эти события вызывают стимуляцию синтеза белка и, как следствие, гипертрофию миокарда, а также вовлечены в увеличение продукции свободно-радикальных форм кислорода в различных типах клеток [30]. Важно отметить, что невзирая на существующую возможность уабаина инициировать сигнальный каскад МАРК исключительно за счет белок-белковых взаимодействий, на некоторых этапах реализации его действия необходимо также изменение концентрации внутриклеточных ионов Na+ и/или Cа2+ [30].
Однако в дальнейшем не все попытки повторить эти эксперименты оказались успешными. А. Аскари пишет о том, что у него в процессе исследования были разногласия по поводу трактовки результатов экспериментов, проведенных в его лаборатории совместно с З. Се, и подробно обсуждает эту проблему [40]. По мнению А. Аскари, невоспроизводимость результатов обусловлена тем, что результаты экспериментов по изучению взаимодействия Na+,K+-АТР-азы и Src in vitro искажены присутствием детергентов. В дальнейшем было установлено, что активацию Src-киназы вызывает не только уабаин, но и другие ингибиторы Na+,K+-АТР-азы, например, ванадат. Это позволило предположить, что Src-киназа активируется за счет того, что при ингибировании Na+,K+-АТР-азы увеличивается локальная концентрация общего для обоих ферментов субстрата, АТР, и экспериментально было показано, что фосфорилирование EGFR меняется в зависимости от активности Na+,K+-АТР-азы [41]. И наконец, были получены свидетельства, что гипертрофия кардиомиоцитов происходит, скорее всего, с участием одной из изоформ фосфатидилинозитол-3-киназы (PI3K) [40]. Стоит отметить, что исследование сигнальных каскадов, между которыми существует множественные взаимодействия, является довольно сложной технической проблемой. По-видимому, вопрос о механизмах, лежащих в основе действия уабаина, опосредующего гипертрофию сердечной мышцы, до сих пор нельзя считать полностью решенным. Возможно, существуют оба механизма взаимодействия Na+,K+-АТР-азы и Src, которые существуют параллельно, и в определенных условиях преобладает один из них. Для окончательного решения этого вопроса необходимы дальнейшие эксперименты.
Установлено, что кардиотонические стероиды могут через активацию Src инициировать также сигнальный каскад с участием фосфолипазы С (PLC-γ), которая обычно активируется через рецепторы, сопряженные с G-белками. Фосфорилирование Src-киназой PLC-γ крысы по остатку тирозина активирует последнюю, что вызывает гидролиз фосфатидилинозитол-4,5-дифосфата и увеличение концентрации 1,2-диацилглицерина и инозитол-1,4,5-трифосфата [42]. Инозитол-1,4,5-трифосфат в свою очередь связывается с Са2+-каналом эндоплазматического ретикулума, являющегося рецептором этого вторичного мессенджера, вызывая освобождение ионов Ca2+ [42]. Другим сигнальным путем с участием маринобуфагенина, Na+,K+-ATP-азы и фосфолипазы С является каскад, приводящий к ингибированию транскрипционного фактора Fli1, являющегося негативным регулятором синтеза коллагена [43]. Этот механизм может вносить существенный вклад и в патогенез соль-чувствительной гипертонической болезни [44].
Л. Лю и соавторы показали также, что в присутствии низких концентраций уабаина в культуре крысиных кардиомиоцитов наблюдается активация серин-треониновой протеинкиназы В, известной также как Akt. Эта активация устраняется ингибиторами фосфатидилинозитол-3-киназы [45]. Они обнаружили, что уабаин вызывает увеличение концентрации инозитол-1,4,5-трифосфата и приводит к взаимодействию α-субъединицы Na+,K+-ATP-азы и фосфорилированной 85 кДа субъединицы PI3K (р85). Исследуя роль Src-киназы в индукции PI3K/Akt сигнального каскада, Й. У и соавторы провели эксперименты с мышиными фибробластами с нокаутом Src. Они показали, что 10–100 мкМ уабаин индуцирует накопление фосфатидилинозитол-3,4,5-трифосфата, активацию PI3K1A и Akt, а также взаимодействие р85 субъединицы PI3K и Nа+,K+-AТР-азы в обоих типах клеток (нокаутной и нормальной), и все эти процессы нечувствительны к ингибитору Src-киназы PP2 [46]. Эти данные позволяют считать, что активация Akt обусловлена прямым взаимодействием обогащенного пролином участка α-субъединицы Nа+,K+-АТР-азы с SH3-доменом p85 субъединицы PI3K, которое индуцируется связыванием кардиотонических стероидов с Na+,K+-АТР-азой и не связано (или мало) с ингибированием фермента.
Обнаружено, что в клетках проксимального канальца нефрона частичное ингибирование Na+,K+-АТР-азы низкими концентрациями уабаина приводит к возникновению или усилению осцилляций внутриклеточной концентрации Са2+, которые устраняются нифедипином, блокатором Са2+-каналов L-типа [47]. Показано, что это обусловлено прямым взаимодействием N-концевой части полипептидной цепи α-субъединицы Na+,K+-АТРазы с рецептором инозитол-1,4,5-трифосфата, расположенного в прилегающей к плазматической части эндоплазматического ретикулума [47]. Осцилляции внутриклеточной концентрации Ca2+ обеспечивают фосфорилирование большого количества Са2+-зависимых белков. Блокада индуцируемой уабаином активации транскрипционных факторов NF-kB и CREB, устраняющая их активацию, вызвана их фосфорилированием и входом в ядро [48].
Длительная обработка уабаином различных типов клеток собаки, свиньи и человека вызывает их смерть, сочетающую в себе признаки апоптоза и некроза, причем эта смерть не связана с ингибированием Na+,K+-АТР-азы, поскольку такое же по времени выдерживание клеток в среде без K+, обеспечивающее полное ингибирование Na+,K+-AТP-азы, не оказывает аналогичного эффекта. Однако столь же длительная обработка уабаином клеток грызунов, также вызывающая полное ингибирование Na+,K+-AТP-азы, не индуцирует клеточную смерть (см. обзор [49]). Мы предприняли попытку исследовать механизмы, лежащие в основе смерти одних клеток и выживания других. Клетки грызунов отличаются от клеток других млекопитающих “не-грызунов” тем, что они менее чувствительны к уабаину (значения констант диссоциации комплексов уабаин–α1-Na+,K+-ATP-аза для грызунов примерно на три порядка выше, в то же время существенных различий в сродстве других α2–α4-изоформ между грызунами и “не-грызунами” не выявлено. В этой связи далее по тексту термины “уабаин-чувствительная” и “уабаин-резистентная” применяются по отношению к α1-изоформе Na+,K+-ATP-азы). В нашей лаборатории, руководимой профессором Орловым C., было обнаружено, что связывание уабаина с уабаин-чувствительной (“не-грызуны”) и уабаин-резистентной (грызуны) Na+,K+-АТР-азой приводит к различным изменениям конформации фермента, что вызывает активацию в случае “не-грызунов” p38 митоген-активируемой протеинкиназы MAPK [50]. Выживание клеток грызунов обусловлено тем, что уабаин-резистентная Na+,K+-АТР-аза этих животных при связывании уабаина претерпевает иные конформационные изменения, что приводит к активации сигнального каскада, связанного с ERK1/2 [50]. Введение в клетки мыши гена, кодирующего уабаин-чувствительную форму α1-субъединицы, приводит к тому, что клетки мышей начинают умирать при инкубации с уабаином. Таким образом, смерть клеток под действием уабаина обусловлена, по всей видимости, конформационными переходами Na+,K+-АТР-азы, которые приводят к активации тех или иных сигнальных каскадов [49].
Способность кардиотонических стероидов индуцировать гибель клеток позволила рассматривать их в качестве сенолитических компонентов – соединений, вызывающих апоптоз стареющих клеток, у которых нарушены процессы программируемой клеточной смерти [51]. Изучение механизмов этого явления, проведенное А. Шатровой и соавторами на мезенхимальных стволовых клетках эндометрия человека, показало, что старение этих клеток сопровождается одновременным уменьшением и увеличением внутриклеточного содержания K+ ([K+]i) и Na+ ([Na+]i) соответственно. Инкубация этих клеток в присутствии 1 мкМ уабаина в течение 48 ч, приводящая к ингибированию Na+,K+-ATP-азы и сопровождающаяся дальнейшим увеличением соотношения [Na+]i/[K+]i в этих клетках, не влияла на их жизнеспособность [52]. Стоит отметить, что взаимосвязь между внутриклеточным содержанием одновалентных катионов и нарушением механизмов клеточной смерти обсуждается довольно давно. Так, в монографии “Vitamins and Minerals in the Prevention and Treatment of Cancer” под редакцией М. Джейкобс одна из глав посвящена этой проблеме [53]. Авторы подчеркивают, что высокое содержание K+i снижает рост опухоли и риск онкотрансформации, тогда как высокое соотношение [Na+]i/[K+]i (за счет увеличения [Na+]i) оказывает противоположный эффект. Кроме того, приводятся результаты анализа внутриядерного содержания этих ионов в разных типах раковых клеток, который показал, что соотношение [Na+]i/[K+i] в них в несколько раз больше по сравнению с нормальными клетками того же типа.
КАРДИОТОНИЧЕСКИЕ СТЕРОИДЫ КАК АКТИВАТОРЫ Na+,K+-ATP-азы
Еще один важный вопрос связан с тем, какова концентрация эндогенного уабаина (или средняя концентрации других кардиостероидов) в организме млекопитающих по сравнению с теми концентрациями, которые вызывают описанные выше эффекты. Сравнение средних концентраций кардиостероидов, оказывающих действие на различные процессы в клетках, показывает, что большинство эффектов инициируется теми концентрациями, которые в крови млекопитающих не достигаются, т.е. при концентрациях кардиостероидов, существующих в организме млекопитающих, ингибирование Na+,K+-АТР-азы невозможно [25]. Те концентрации уабаина (или других кардиостероидов), которые обнаружены в организмах млекопитающих (менее 1 нМ), могут инициировать лишь часть из описанных выше процессов, а именно: пролиферацию клеток, активацию Src-киназы, оссцилляцию внутриклеточных концентраций Са2+ и активацию Na+,K+-АТР-азы. Таким образом, процессы, для которых необходимо увеличение внутриклеточных концентраций Na+, вызываемое ингибированием Na+,K+-АТР-азы (например, гипертрофия сердечной мышцы), in vivo, по-видимому, не произойдут без воздействия экзогенно добавленных кардиостероидов. Однако нельзя также исключить, что если концентрации разных присутствующих в крови млекопитающих кардиостероидов, например, дигоксина или маринобуфагенина, могут суммироваться и насыщать уабаин-связывающие центры фермента, тогда их суммарной концентрации будет достаточно для инициации процессов, которые обусловлены более высокими концентрациями.
Удивительно, но данные, полученные многими исследовательскими группами, свидетельствуют о том, что уабаин не только ингибирует, но в низких дозах (т.е. в физиологическом диапазоне) активирует Na+,K+-ATP-aзу. Это явление было описано в литературе еще в 60-х годах XX века [54, 55]. Спустя 10 лет после этого сообщения Т. Годфрейнд и Дж. Гизель-Бертон опубликовали данные о том, что инкубация изолированного левого предсердия морской свинки в присутствии 1 и 3 нМ уабаина в течение 3 ч приводила к уменьшению внутриклеточного содержания Na+ и увеличению внутриклеточного содержания K+ по сравнению с контрольными образцами [56]. Авторы предположили существование двух участков связывания уабаина с разным сродством внутри молекулы Na+,K+-ATP-азы. Позднее Д. Лихштейн и соавторы показали, что уабаин в диапазоне концентраций 1–100 нМ оказывает ярко выраженный стимулирующий эффект на Na+,K+-ATP-азу в гомогенате мозга крысы. Однако подобного эффекта не наблюдалось на препарате микросом, полученных из мозга крысы [36]. Авторы также отметили, что активирующий эффект (в отличие от ингибирующего) уабаина зависит от времени хранения препарата при –20°С, что также ранее было показано другими исследователями [54, 55]. Они предложили два возможных объяснения стимулирующего эффекта низких концентраций уабаина. Первое связано с наличием эндогенного уабаино-подобного соединения, которое, связываясь с высокоаффинным участком Na+,K+-ATP-азы, приводит к частичной потере активности фермента. Добавление низких концентраций уабаина в такой ситуации “растормаживает” Na+,K+-ATP-азу. Второе объяснение предполагает изменение мембранного окружения Na+-насоса в результате связывания этого кардиостероида, которое само по себе может оказывать ингибирующее действие на фермент. Т. Хоуген и соавторы в экспериментах с использованием изолированного левого предсердия морской свинки также показали увеличение активности Na+,K+-ATP-азы в присутствии 3 нМ уабаина, и этот эффект устранялся в присутствии пропранолола (ингибитора β-адренергических рецепторов). Это наблюдение (а также эксперименты с использованием резерпина и 6-гидроксидофамина) позволило авторам предположить, что уабаин-опосредованная активация Na+-насоса обеспечивается действием эндогенных катехоламинов [57]. Мы также регистрировали увеличение активности Na+,K+-ATP-азы в микросомах из почек свиньи в присутствии 1–10 нМ уабаина и в клетках эндотелия пупочной вены человека в присутствии 0.3, 1 и 3 нМ уабаина [58, 59].
Возникает вопрос, каков физиологический смысл активации Na+,K+-ATP-азы уабаином, в частности в отношении невозбудимых тканей? Анализ данных литературы относительно физиологических эффектов суб- и наномолярных концентраций КТС в различных типах клеток позволяет сделать следующие заключения. Такие концентрации КТС часто усиливают клеточную пролиферацию [25]. Так, уабаин в концентрации 1–10 нМ, не ингибирующей Na+-насос, увеличивает пролиферацию культивируемых эндотелиальных клеток человека [60], гладкомышечных клеток собаки [61] и эпителиальных клеток почки опоссума [62], содержащих чувствительную к кардиостероидам Nа+,K+-AТР-азу, до 40%. Мы также регистрировали увеличение жизнеспособности HUVEC (клетки эндотелия пупочной вены человека) и синтез ДНК в HREC (клетки почечного эпителия человека) и HUVEC в присутствии 1 и 3 нМ уабаина [58, 59].
Механизм бимодального действия уабаина на активность Na+,K+-АТР-азы (активация и ингибирование при низких и высоких концентрациях соответственно) остается пока не выясненным. Среди возможных, описанных в литературе, можно выделить основные: 1) наличие двух участков связывания уабаина внутри молекулы фермента [56]; 2) изменение мембранного окружения фермента, оказывающего ингибирующее воздействие в результате связывания уабаина [36]; 3) белок-белковые взаимодействия (например, с Na+/H+-обменником [63, 64]), опосредованные связыванием уабаина с Na+,K+-ATP-азой. Экспериментальные данные, полученные в нашей лаборатории с использованием различных культур клеток, а также частично очищенных (фракция микросом) и высокоочищенных препаратов Na+,K+-ATP-азы (солюбилизированный фермент), позволяют предположить следующий механизм активации фермента низкими концентрациями кардиотонических стероидов (в частности, уабаина). Функциональной единицей Na+,K+-ATP-азы является αβ-протомер, который может обратимо олигомеризоваться с образованием более крупных молекулярных комплексов вида (αβ)n [65]. Этот процесс может регулироваться, например, посредством ATP. Так, его связывание с олигомером Na+,K+-ATP-азы приводит к формированию ее закрытой конформации [66], последующей диссоциации комплекса на мономеры и усилением транспортной функции фермента [17]. Образованию закрытой конформации Na+,K+-ATP-азы также способствует связывание уабаина [67]. Таким образом, можно предположить, что уабаин, связываясь с высокоаффинным участком Na+,K+-ATP-азы в составе ее олигомерного комплекса, провоцирует переход фермента в закрытую конформацию, что влечет за собой диссоциацию олигомера на мономеры, которые проявляют усиленную транспортную активность. Увеличение концентрации уабаина приводит к насыщению фермента ингибитором и сопровождается уменьшением его активности.
Резюмируя вышеизложенные факты, можно заключить, что спектр действия кардиотонических стероидов на различные типы клеток весьма обширен и зависит от того, каким образом они влияют на Na+,K+-ATP-азу (рис. 1). Эти влияния могут затрагивать как ионный гомеостаз (причем не только увеличивать соотношение [Na+]i/[K+]i, но и снижать), так и внутриклеточный сигналинг посредством изменения конформации фермента. Таким образом, исследуя эффекты кардиотонических стероидов, следует по возможности оценивать их влияние на транспортную функцию Na+,K+-ATP-азы и внутриклеточный ионный состав.
Рис. 1.
Эффекты связывания кардиотонических стероидов с Na+,K+-АТP-азой: роль изменений соотношения [Na+]i/[K+]i, которые возникают при ингибировании и активации Na+,K+-АТP-азы соответственно, и запуска клеточной сигнализации за счет конформационных изменений фермента.
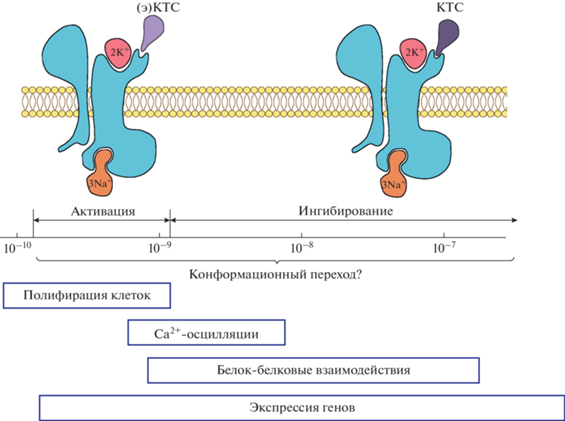
Есть ли взаимосвязь между внутриклеточным соотношением [Na+]i/[K+]i и экспрессией генов? Каковы механизм и физиологическая роль активации Na+,K+-ATP-азы кардиотоническими стероидами? Что является сенсором внутриклеточных одновалентных катионов? Эти вопросы требуют детального изучения.
ЗАКЛЮЧЕНИЕ
Итак, годы, прошедшие с момента получения Й. Скоу Нобелевской премии, показали, что Na+,K+-АТР-аза обладает не только транспортной функцией, обеспечивая перенос ионов Na+ и К+ через плазматическую мембрану. Этот фермент также является рецептором для эндогенных кардиостероидов, связывая которые он может передавать сигнал внутрь клетки за счет взаимодействия с белками-партнерами, включая таким образом сигнальные каскады, регулирующие клеточные функции. Кроме того, от транспортной функции этого фермента зависит внутриклеточная концентрация ионов Na+ и K+, изменение соотношения этих ионов в клетке прямо влияет на экспрессию многих генов [68–70]. Этот аспект влияния Na+,K+-АТР-азы на функции клетки не обсуждался в настоящей статье, но прямое действие соотношения [Na+]i/[K+]i на экспрессию генов заставляет нас задуматься о поиске сенсора этих катионов [71]. Заключая, мы можем сказать, что вопросов о роли Na+,K+-АТР-азы в функционировании клеток и о механизмах, вовлеченных в осуществление влияния фермента на клетку, пока больше, чем ответов. По этой причине Na+,K+-АТР-аза еще долго останется объектом для исследования.
Финансирование. Работа выполнена при финансовой поддержке Российского научного фонда (грант № 19-75-10 009).
Конфликт интересов. Авторы заявляют, что у них нет конфликта интересов.
Соблюдение этических норм. Все применимые международные, национальные и/или институциональные принципы ухода и использования животных были соблюдены.
Список литературы
Skou J.C. 1957. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta. 23, 394–401.
Wood E.H., Moe G.K. 1938. Studies on the effect of digitalis glycosides on potassium ion loss from the heart. Am. J. Physiol. 123, 219–220.
Schatzmann H.J. 1953. Cardiac glycosides as inhibitors of active potassium and sodium transport by erythrocyte membrane. Helv. Physiol. Pharmacol. Acta. 11, 346–354.
Skou J.C. 1960. Further investigations on a Mg++ + + Na+-activated adenosintriphosphatase, possibly related to the active, linked transport of Na+ and K+ across the nerve membrane. Biochim. Biophys. Acta. 42, 6–23.
Jørgensen L.P., Skou J.C. 1969. Preparation of highly active (Na+ + K+)-ATPase from the outer medulla of rabbit kidney. Biochem. Biophys. Res. Commun. 37, 39–46.
Lingrel J.B. 2010. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu. Rev. Physiol. 72, 395–412.
Blanco G., Mercer R.W. 1998. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. 275, F633–F650.
Geering K. 2001. The Functional role of β subunits in oligomeric P-type ATPases. J. Bioenerg. Biomembr. 33, 425–438.
Clausen M.V., Hilbers F., Poulsen H. 2017. The structure and function of the Na,K-ATPase isoforms in health and disease. Front. Physiol. 8, 371.
Forbush B., Kaplan J.H., Hoffman J.F. 1978. Characterization of a new photoaffinity derivative of ouabain: Labeling of the large polypeptide and of a proteolipid component of the Na, K-ATPase. Biochemistry. 17, 3667–3676.
Geering K. 2006. FXYD proteins: New regulators of Na-K-ATPase. Am. J. Physiol. Renal. Physiol. 290, F241-250.
Cheung J.Y., Zhang X.-Q., Song J., Gao E., Chan T.O., Rabinowitz J.E., Koch W.J., Feldman A.M., Wang J. 2013. Coordinated regulation of cardiac Na+/Ca2+ exchanger and Na+-K+-ATPase by phospholemman (FXYD1). Adv. Exp. Med. Biol. 961, 175–190.
Ahlers B.A., Zhang X.-Q., Moorman J.R., Rothblum L.I., Carl L.L., Song J., Wang J., Geddis L.M., Tucker A.L., Mounsey J.P., Cheung J.Y. 2005. Identification of an endogenous inhibitor of the cardiac Na+/Ca2+ exchanger, phospholemman. J. Biol. Chem. 280, 19 875–19 882.
Lingrel J.B., Kuntzweiler T. 1994. Na+,K+-ATPase. J. Biol. Chem. 269, 19659–19662.
Albers R.W. 1967. Biochemical aspects of active transport. Annu. Rev. Biochem. 36, 727–756.
Post R.L., Hegyvary C., Kume S. 1972. Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 247, 6530–6540.
Clarke R.J. 2009. Mechanism of allosteric effects of ATP on the kinetics of P-type ATPases. Eur. Biophys. J. 39, 3.
Dyla M., Kjærgaard M., Poulsen H., Nissen P. 2020. Structure and mechanism of P-type ATPase ion pumps. Annu. Rev. Biochem. 89, 583–603.
Burchell H.B. 1976. Coincidental bicentennials: United States and foxglove therapy. J. Hist. Med. Allied Sci. 31, 292–306.
Krickler D.M. 1985. The foxglove,“The old woman from Shropshire” and William Withering. J. Am. Coll. Cardiol. 5 (5), 3A–9A.
Patocka J., Nepovimova E., Wu W., Kuca K. 2020. Digoxin: Pharmacology and toxicology—A review. Environ. Toxicol. Pharmacol. 79, 103400.
Pavlovic D. 2019. Endogenous cardiotonic steroids and cardiovascular disease, where to next? Cell Calcium. 86, 102156.
Lopachev A.V., Abaimov D.A., Fedorova T.N., Lopacheva O.M., Akkuratova N.V., Akkuratov E.E. 2018. Cardiotonic steroids as potential endogenous regulators in the nervous system. Neurochem. J. 12, 1–8.
Hamlyn J.M., Blaustein M.P., Bova S., DuCharme D.W., Harris D.W., Mandel F., Mathews W.R., Ludens J.H. 1991. Identification and characterization of a ouabain-like compound from human plasma. Proc. Natl. Acad. Sci. USA. 88, 6259–6263.
Orlov S.N., Tverskoi A.M., Sidorenko S.V., Smolyaninova L.V., Lopina O.D., Dulin N.O., Klimanova E.A. 2020. Na,K-ATPase as a target for endogenous cardiotonic steroids: What’s the evidence? Genes Dis. 8, 259–271.
ZhuGe R., DeCrescenzo V., Sorrentino V., Lai F.A., Tuft R.A., Lifshitz L.M., Lemos J.R., Smith C., Fogarty K.E., Walsh J.V. 2006. Syntillas release Ca2+ at a site different from the microdomain where exocytosis occurs in mouse chromaffin cells. Biophys. J. 90, 2027–2037.
Kometiani P., Tian J., Nabih Z., Gick G., Xie Z. 2000. Regulation of Na/K-ATPase β1-subunit gene expression by ouabain and other hypertrophic stimuli in neonatal rat cardiac myocytes. Mol. Cell Biochem. 215, 65–72.
Hosoi R., Matsuda T., Asano S., Nakamura H., Hashimoto H., Takuma K., Baba A. 1997. Isoform-specific up-regulation by ouabain of Na+,K+-ATPase in cultured rat astrocytes. J. Neurochem. 69, 2189–2196.
Xue Z., Li B., Gu L., Hu X., Li M., Butterworth R.F., Peng L. 2010. Increased Na, K-ATPase α2 isoform gene expression by ammonia in astrocytes and in brain in vivo. Neurochem. Int. 57, 395–403.
Xie Z., Askari A. 2002. Na+/K+-ATPase as a signal transducer. Eur. J. Biochem. 269, 2434–2439.
Nie Y., Bai F., Chaudhry M.A., Pratt R., Shapiro J.I., Liu J. 2020. The Na/K-ATPase α1 and c-Src form signaling complex under native condition: A crosslinking approach. Sci. Rep. 10, 6006.
Pratt R.D., Brickman C.R., Cottrill C.L., Shapiro J.I., Liu J. 2018. The Na/K-ATPase signaling: From specific ligands to general reactive oxygen species. Int. J. Mol. Sci. 19, 2600.
Haas M., Askari A., Xie Z. 2000. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chemistry. 275, 27832–27837.
Blaustein M.P., Juhaszova M., Golovina V.A. 1998. The cellular mechanism of action of cardiotonic steroids: A new hypothesis. Clin. Exp. Hypertens. 20, 691–703.
Blaustein M.P., Hamlyn J.M. 2020. Ouabain, endogenous ouabain and ouabain-like factors: The Na+ pump/ouabain receptor, its linkage to NCX, and its myriad functions. Cell Calcium. 86, 102159.
Lichtstein D., Samuelov S., Bourrit A. 1985. Characterization of the stimulation of neuronal Na+,K+-ATPase activity by low concentrations of ouabain. Neurochem. Int. 7, 709–715.
Askari A. 2019. The sodium pump and digitalis drugs: Dogmas and fallacies. Pharmacol. Res. Perspect. 7, e00505.
Tverskoi A.M., Poluektov Y.M., Klimanova E.A., Mitkevich V.A., Makarov A.A., Orlov S.N., Petrushanko I.Y., Lopina O.D. 2021. Depth of the steroid core location determines the mode of Na,K-ATPase inhibition by cardiotonic steroids. Int. J. Mol. Sci. 22, 13268.
Rocha S.C., Pessoa M.T.C., Neves L.D.R., Alves S.L.G., Silva L.M., Santos H.L., Oliveira S.M.F., Taranto A.G., Comar M., Gomes I.V., Santos F.V., Paixão N., Quintas L.E.M., Noël F., Pereira A.F., Tessis A.C.S.C., Gomes N.L.S., Moreira O.C., Rincon-Heredia R., Varotti F.P., Blanco G., Villar J.A.F.P., Contreras R.G., Barbosa L.A. 2014. 21-Benzylidene digoxin: A proapoptotic cardenolide of cancer cells that up-regulates Na,K-ATPase and epithelial tight junctions. PLoS One. 9, e108776.
Askari A. 2019. The other functions of the sodium pump. Cell Calcium. 84, 102105.
Weigand K.M., Swarts H.G.P., Fedosova N.U., Russel F.G.M., Koenderink J.B. 2012. Na,K-ATPase activity modulates Src activation: A role for ATP/ADP ratio. Biochim. Biophys. Acta. 1818, 1269–1273.
Yuan Z., Cai T., Tian J., Ivanov A.V., Giovannucci D.R., Xie Z. 2005. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol. Biol. Cell. 16, 4034–4045.
Agalakova N.I., Kolodkin N.I., Adair C.D., Trashkov A.P., Bagrov A.Y. 2021. Preeclampsia: Cardiotonic steroids, fibrosis, Fli1 and hint to carcinogenesis. Int. J. Mol. Sci. 22, 1941.
Bagrov A.Y. 2021. High salt is a risk factor for cardiovascular and kidney diseases. What is next, fibrosis? J. Hypertens. 39, 1309–1310.
Liu L., Zhao X., Pierre S.V., Askari A. 2007. Association of PI3K-Akt signaling pathway with digitalis-induced hypertrophy of cardiac myocytes. Am. J. Physiol. Cell Physiol. 293, 1489–1497.
Wu J., Akkuratov E.E., Bai Y., Gaskill C.M., Askari A., Liu L. 2013. Cell signaling associated with Na+/K+-ATPase: Activation of phosphatidylinositide 3-kinase IA/Akt by ouabain is independent of Src. Biochemistry. 52, 9059–9067.
Aizman O., Uhlén P., Lal M., Brismar H., Aperia A. 2001. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc. Natl. Acad. Sci. USA. 98, 13420–13424.
Desfrere L., Karlsson M., Hiyoshi H., Malmersjö S., Nanou E., Estrada M., Miyakawa A., Lagercrantz H., Manira A.E., Lal M., Uhlén P. 2009. Na,K-ATPase signal transduction triggers CREB activation and dendritic growth. Proc. Natl. Acad. Sci. USA. 106, 2212–2217.
Lopina O.D., Tverskoi A.M., Klimanova E.A., Sidorenko S.V., Orlov S.N. 2020. Ouabain-induced cell death and survival. Role of α1-Na,K-ATPase-mediated signaling and [Na+]i/[K+]i-dependent gene expression. Front. Physiol. 11, 1060.
Akimova O.A., Lopina O.D., Rubtsov A.M., Gekle M., Tremblay J., Hamet P., Orlov S.N. 2009. Death of ouabain-treated renal epithelial cells: evidence for p38 MAPK-mediated ${\text{Na}}_{{\text{i}}}^{ + }$/${\text{K}}_{{\text{i}}}^{ + }$-independent signaling. Apoptosis. 14, 1266.
Triana-Martínez F., Picallos-Rabina P., Silva-Álvarez S.D., Pietrocola F., Llanos S., Rodilla V., Soprano E., Pedrosa P., Ferreirós A., Barradas M., Hernández-González F., Lalinde M., Prats N., Bernadó C., González P., Gómez M., Ikonomopoulou M.P., Fernández-Marcos P.J., García-Caballero T., Pino P. del, Arribas J., Vidal A., González-Barcia M., Serrano M., Loza M.I., Domínguez E., Collado M. 2019. Identification and characterization of cardiac glycosides as senolytic compounds. Nat. Commun. 10, 1–12.
Shatrova A., Pugovkina N., Domnina A., Nikolsky N., Marakhova I. 2022. Monovalent ions and stress-induced senescence in human mesenchymal endometrial stem cells. Research Square. https://doi.org/10.21203/rs.3.rs-1087662/v1
Jacobs M.M., Pienta R.J. 1991. In: Vitamins and minerals in the prevention and treatment of cancer. Ed Jacobs M.M. Boca Raton: CRC Press, p. 228–245.
Brown H.D. 1966. A characterization of the ouabain sensitivity of heart microsomal ATPase. Biochim. Biophys. Acta. 120, 162–165.
Palmer R.F., Lasseter K.C., Melvin S.L. 1966. Stimulation of Na+ and K+ dependent adenosine triphosphatase by ouabain. Arch. Biochem. Biophys. 113, 629–633.
Godfraind T., Ghysel-Burton J. 1977. Binding sites related to ouabain-induced stimulation or inhibition of the sodium pump. Nature. 265, 165–166.
Hougen T.J., Spicer N., Smith T.W. 1981. Stimulation of monovalent cation active transport by low concentrations of cardiac glycosides. Role of catecholamines. J. Clin. Invest. 68, 1207–1214.
Tverskoi A.M., Sidorenko S.V., Klimanova E.A., Akimova O.A., Smolyaninova L.V., Lopina O.D., Orlov S.N. 2016. Effects of ouabain on proliferation of human endothelial cells correlate with Na+,K+-ATPase activity and intracellular ratio of Na+ and K+. Biochemistry (Mosc). 81, 876–883.
Klimanova E.A., Fedorov D.A., Sidorenko S.V., Abramicheva P.A., Lopina O.D., Orlov S.N. 2020. Ouabain and marinobufagenin: physiological effects on human epithelial and endothelial cells. Biochemistry (Mosc). 85, 507–515.
Saunders R., Scheiner-Bobis G. 2004. Ouabain stimulates endothelin release and expression in human endothelial cells without inhibiting the sodium pump. Eur. J. Biochem. 271, 1054–1062.
Aydemir-Koksoy A., Abramowitz J., Allen J.C. 2001. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 276, 46 605–46 611.
Khundmiri S.J., Metzler M.A., Ameen M., Amin V., Rane M.J., Delamere N.A. 2006. Ouabain induces cell proliferation through calcium-dependent phosphorylation of Akt (protein kinase B) in opossum kidney proximal tubule cells. Am. J. Physiol. Cell Physiol. 291, C1247–C1257.
Khundmiri S.J., Salyer S.A., Farmer B., Qipshidze-Kelm N., Murray R.D., Clark B.J., Xie Z., Pressley T.A., Lederer E.D. 2014. Structural determinants for the ouabain-stimulated increase in Na–K ATPase activity. Biochim. Biophys. Acta. 1843, 1089–1102.
Holthouser K.A., Mandal A., Merchant M.L., Schelling J.R., Delamere N.A., Valdes R.R., Tyagi S.C., Lederer E.D., Khundmiri S.J. 2010. Ouabain stimulates Na-K-ATPase through a sodium/hydrogen exchanger-1 (NHE-1)-dependent mechanism in human kidney proximal tubule cells. Am. J. Physiol. Renal Physiol. 299, F77–F90.
Yoneda J.S., Scanavachi G., Sebinelli H.G., Borges J.C., Barbosa L.R.S., Ciancaglini P., Itri R. 2016. Multimeric species in equilibrium in detergent-solubilized Na,K-ATPase. Int. J. Biol. Macromol. 89, 238–245.
Petrushanko I.Y., Mitkevich V.A., Anashkina A.A., Klimanova E.A., Dergousova E.A., Lopina O.D., Makarov A.A. 2014. Critical role of γ-phosphate in structural transition of Na,K-ATPase upon ATP binding. Sci. Rep. 4, 5165.
Grimaldi S., Pascale E., Pozzi D., D’Onofrio M., Giganti M.G., Verna R. 1988. Effect of ouabain binding on the fluorescent properties of the Na+/K+-ATPase. Biochim. Biophys. Acta. 944, 13–18.
Koltsova S.V., Trushina Y., Haloui M., Akimova O.A., Tremblay J., Hamet P., Orlov S.N. 2012. Ubiquitous [Na+]i/[K+]i-sensitive transcriptome in mammalian cells: Evidence for ${\text{Ca}}_{{\text{i}}}^{{2 + }}$-independent excitation-transcription coupling. PLoS One. 7, e38032.
Klimanova E.A., Sidorenko S.V., Smolyaninova L.V., Kapilevich L.V., Gusakova S.V., Lopina O.D., Orlov S.N. 2019. Ubiquitous and cell type-specific transcriptomic changes triggered by dissipation of monovalent cation gradients in rodent cells: Physiological and pathophysiological implications. Curr. Top. Membr. 83, 107–149.
Fedorov D.A., Sidorenko S.V., Yusipovich A.I., Parshina E.Y., Tverskoi A.M., Abramicheva P.A., Maksimov G.V., Orlov S.N., Lopina O.D., Klimanova E.A. 2021. ${\text{Na}}_{{\text{i}}}^{ + }$/${\text{K}}_{{\text{i}}}^{ + }$ imbalance contributes to gene expression in endothelial cells exposed to elevated NaCl. Heliyon. 7, e08088.
Klimanova E.A., Sidorenko S.V., Tverskoi A.M., Shiyan A.A., Smolyaninova L.V., Kapilevich L.V., Gusakova S.V., Maksimov G.V., Lopina O.D., Orlov S.N. 2019. Search for intracellular sensors involved in the functioning of monovalent cations as secondary messengers. Biochemistry (Mosc). 84, 1280–1295.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии