

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 5, стр. 398-403
Мониторинг агонист-индуцированной активности PI3-киназы в клетках HEK-293 с использованием генетически кодируемого сенсора
П. Д. Котова a, *, О. А. Рогачевская a, Н. В. Кабанова a, С. С. Колесников a
a Институт биофизики клетки РАН, ФИЦ ПНЦБИ РАН
142290 Пущино, Московская обл., Россия
* E-mail: p.d.kotova@gmail.com
Поступила в редакцию 04.04.2022
После доработки 23.05.2022
Принята к публикации 24.05.2022
- EDN: IQFVHD
- DOI: 10.31857/S0233475522050097
Аннотация
IP3-зависимый выброс депонированного Са2+ вносит ключевой вклад в агонист-индуцированную мобилизацию Са2+ в невозбудимых клетках. Эффективность фосфоинозитидного каскада, сопрягающего поверхностные рецепторы с мобилизацией внутриклеточного Са2+, модулируется рядом киназ, включая фосфатидилинозитол-3-киназу (PI3K), которая, фосфорилируя PIP2, продуцирует фосфолипид PIP3. Ранее нами было показано, что ингибитор PI3K вортманнин не влияет на способность клеток HEK-293 генерировать Ca2+-ответы на ацетилхолин, тогда как ингибитор PI3K другой химической природы PI828 полностью подавляет эти ответы. Разная эффективность вортманнина и PI828 могла быть связана с тем, что в клетках HEK-293 функционируют изоформы PI3K, существенно более чувствительные к PI828. Для внесения ясности в этот вопрос нами была получена моноклональная линия клеток HEK-293, экспрессирующих два генетически кодируемых сенсора, а именно сенсор цитозольного Ca2+ (R-GECO1) и сенсор PIP3 PH(Akt)-Venus. Клетки этой линии позволяли одновременно регистрировать Са2+-сигналы и проводить мониторинг активности PI3K. Характерной особенностью R-GECO1 является увеличение интенсивности флуоресценции при повышении концентрации цитозольного Ca2+, в то время как PH(Akt)-Venus при PI3K-зависимой генерации PIP3 в плазмалемме перераспределяется из цитозоля в мембрану клетки. Оказалось, что ацетилхолин инициировал кратковременное повышение внутриклеточного Са2+, но не влиял на распределение PIP3-сенсора в цитоплазме клеток. Последнее указывало на отсутствие ацетилхолин-зависимой активации PI3K. В то же время инсулин, стимулирующий PI3K при участии тирозин-киназных рецепторов, вызывал перераспределение молекул PH(Akt)-Venus из цитозоля в мембрану клеток, что демонстрировало инсулин-индуцированную активность PI3K. Этот феномен не наблюдался в присутствии вортманнина или PI828, что свидетельствовало об эффективном подавлении активности PI3K этими соединениями. Таким образом, стимулируя внутриклеточную Са2+-сигнализацию в клетках HEK-293, ацетилхолин не инициировал активацию PI3K-пути, который, следовательно, не был вовлечен в холинергическую трансдукцию. Хотя полученные данные свидетельствуют об эффективном ингибировании активности PI3K вортманнином и PI828, последний подавлял ацетилхолин-индуцируемую Ca2+-сигнализацию неспецифически, т.е. воздействуя не на PI3K, а на какую-то иную клеточную мишень.
ВВЕДЕНИЕ
Мобилизация внутриклеточного Са2+ является одной из наиболее универсальных форм реакции клеток на внешние возмущения, включая стимуляцию первичными медиаторами, обеспечивающими аутокринную и паракринную регуляции клеточных функций. Хотя в генерации Са2+-сигналов принимает участие множество внутриклеточных сигнальных и транспортных систем, ключевой вклад в агонист-индуцированную мобилизацию Са2+ в невозбудимых клетках вносит IP3-зависимый выброс депонированного Са2+ [1, 2]. Фосфоинозитидный каскад может сопрягать разнообразные поверхностные рецепторы, включая гептаспиральные трансмембранные рецепторы (G-protein coupled receptor, GPCR-рецепторы), с мобилизацией внутриклеточного Са2+. Его эффективность модулируется при участии различных сигнальных систем, включая каскад фосфатидилинозитол-3-киназы класса I (PI3К) – фермента, катализирующего образование сигнального/регуляторного фосфолипида PIP3 (фосфатидилинозитол-3,4,5-трисфосфата) [3–5]. Так, например, показано, что PI3K/Akt-путь является важным регулятором Ca2+-сигнализации и сократительной активности кардиомиоцитов [6, 7]. Участие PI3K/Akt-каскада в регуляции выброса Ca2+ из Са2+-депо было продемонстрировано для клеток MDCK [8], COS-7 [9] и RINm5F [10]. Его роль в Ca2+-сигнализации может быть обусловлена PI3K/Akt-зависимой регуляцией активности IP3-рецепторов и/или фосфолипазы С [10–12].
Ранее при исследовании роли PI3K в генерации Са2+-ответов на GPCR-агонисты, нами был использован ряд ингибиторов PI3K, включая вортманнин и PI828, различающиеся по химической структуре. Было установлено, что вортманнин не влиял на способность клеток HEK-293 генерировать Ca2+-ответы на ацетилхолин, в то время как PI828 полностью подавлял эти ответы [13]. Причины столь разной эффективности PI828 и вортманнина оставались неясными. Одна из возможных причин состояла в том, что в сравнении с вортманнином PI828 существенно более эффективно блокировал изоформы PI3K, функционирующие в клетках HEK-293. В любом случае эти данные свидетельствовали о том, что механизм действия вортманнина и PI828, которые считаются ингибиторами PI3K, в действительности не так очевиден. Следовательно, результаты, полученные при исследовании роли PI3K в механизме генерации Ca2+-сигналов с использованием данных соединений, нельзя интерпретировать однозначно, что делает целесообразным проведение одновременного мониторинга внутриклеточного Са2+ и активности PI3K. Данная работа посвящена отработке такой методологии и ее использованию для установления роли PI3K в генерации Ca2+-ответов клеток HEK-293 на ацетилхолин.
МАТЕРИАЛЫ И МЕТОДЫ
Культура клеток. Клетки линии НЕК-293 (Российская коллекция клеточных культур позвоночных) и модифицированные клетки HEK-293/R-GECO1/PH(Akt)-Venus культивировали в среде DMEM с высоким содержанием глюкозы (Invitrogen) с добавлением 10% эмбриональной бычьей сыворотки (HyClone), 100 мг/мл гентамицина (Sigma-Aldrich), 2 мМ глутамина (Sigma-Aldrich) (ростовая среда) во влажной атмосфере c 5% содержанием CO2 в воздухе при 37°C. Для культивирования моноклональных линий в ростовую среду добавляли 300 мкг/мл селективного антибиотика генецитина G418 (Invivogen).
Трансфекция клеток. Клетки НЕК-293 трансфицировали плазмидными векторами CMV-R-GECO1 (Addgene plasmid #32444) и PH(Akt)-Venus (Addgene plasmid #85223) одновременно, используя набор для трансфекции FuGENE 6 (Promega) по протоколу, оптимизированному согласно рекомендациям производителя. Накануне трансфекции клетки рассевали из расчета (2–4) × 105 клеток в 1 лунку 12-лучночного планшета в 1 мл ростовой среды. Для трансфекции в лунку к среде с растущими клеткам добавляли трансфекционную смесь, содержащую 100 мкл среды OptiMEM (Gibco), 4 мкл FuGENE 6 (Promega), 0.5 мкг CMV-R-GECO1 и 0.5 мкг PH(Akt)-Venus. Через 24 ч среду заменяли на свежую ростовую.
Получение моноклональных линий HEK-293/R-GECO1/PH(Akt)-Venus. Популяцию трансфицированных клеток селектировали в ростовой среде с добавлением 700 мкг/мл G-418 (Invivogen) в течение 2–3 недель. Из полученной после селекции популяции с помощью клеточного сортера FACSAria SORP (Beckton Dickinson) отбирали клетки, обладающие флуоресценцией, соответствующей экспрессии обоих белков интереса: флуоресценцию R-GECO1 возбуждали при длине волны 561 нм, эмиссию регистрировали в области 610 ± 10 нм, флуоресценцию PH(Akt)-Venus возбуждали при длине волны 488 нм, эмиссию регистрировали в области 515 ± 10 нм. Клетки, обладающие наиболее яркой флуоресценцией обоих сенсоров, помещали по одной в лунку 96-луночного планшета, содержащую 150 мкл ростовой среды с повышенным до 15% содержанием сыворотки, и культивировали, наращивая таким образом моноклональные линии. Клетки каждой полученной моноклональной линии тестировали физиологически, а именно, работоспособность полученных клеток оценивали по ответам сенсоров R-GECO1 и PH(Akt)-Venus на стимуляцию ацетилхолином и инсулином соответственно. В результате проведенного тестирования была отобрана лучшая моноклональная линия клеток HEK-293/R-GECO1/PH(Akt)-Venus.
Мониторинг внутриклеточных Ca2+ и PIP3. Перед экспериментом клетки HEK-293/R-GECO1/PH(Akt)-Venus культивировали в фотометрических камерах в течение 24 ч в ростовой среде в CO2-инкубаторе. В процессе эксперимента клетки находились во внеклеточном растворе, содержащем (мМ): 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4, 10 глюкозы (все соли и буфер произведены Sigma-Aldrich). Фотометрические эксперименты проводили с использованием инвертированного флуоресцентного микроскопа Axiovert 200 (Zeiss), оборудованного объективом Plan NeoFluar 20×/0.75, цифровой sCMOS камерой Zyla 4.2P (Andor Technology), металлогалогенным источником света AMH-200-F6S (Andor Technology) и спиннинг-диском для конфокальной микроскопии Revolution DSD2 (Andor Technology). Флуоресценцию R-GECO1 и PH(Akt)-Venus возбуждали поочередно при длинах волн 560 ± 20 и 500 ± 10 нм соответственно, эмиссию регистрировали в областях 630 ± 37 и 535 ± 15 нм соответственно. Количественный фотометрический анализ изображений осуществляли с использованием программы NIS Elements (Nikon).
Аппликацию всех соединений, использовавшихся в описанных ниже экспериментах, проводили путем полной замены раствора в фотометрической камере с помощью системы перфузии. В работе использовали ацетилхолин, инсулин, вортманнин и PI828 производства Tocris Bioscience.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Мониторинг активности PI3K проводили в клетках моноклональной линии HEK-293/R-GECO1/PH(Akt)-Venus, экспрессирующих сенсор цитозольного Ca2+ R-GECO1 и сенсор PIP3 PH(Akt)-Venus, принципы функционирования которых различаются. Характерной особенностью сенсора R-GECO1 является градуальное увеличение интенсивности флуоресценции при повышении концентрации цитозольного Ca2+ [14]. В покое, когда уровень PIP3 в плазмалемме низок, сенсор PH(Akt)-Venus локализован преимущественно в цитозоле, но при PI3K-зависимой генерации PIP3 он перераспределяется из цитозоля к мембране клетки [15]. Этот феномен можно наблюдать при регистрации флуоресценции PH(Akt)-Venus на оптическом срезе клетки, в связи с этим в данной работе использовали SDCM-микроскопию (spinning disk confocal microscopy).
Оказалось, что при стимуляции клеток HEK-293/R-GECO1/PH(Akt)-Venus инсулином, запускающим PI3K-сигнализацию посредством активации тирозин-киназных рецепторов [16], регулярно наблюдалось перераспределение флуоресценции сенсора PH(Akt)-Venus из цитозоля к плазматической мембране клетки, причем этот эффект отменялся в присутствии вортманнина или PI828 (рис. 1). Эти данные свидетельствовали о том, что инсулин действительно стимулировал активность PI3K в исследовавшихся клетках, а вортманнин и PI828 эффективно ингибировали PI3K. Для выявления роли PI3K в генерации агонист-индуцированных Ca2+-сигналов клетки HEK-293/R-GECO1/PH(Akt)-Venus последовательно стимулировали ацетилхолином и инсулином (схема эксперимента представлена на рис. 2а). Хотя для стимуляции Са2+-сигнализации было достаточно аппликации ацетилхолина длительностью 1 мин, PI3K-сигнализация является более медленным процессом [17], и поэтому клетки инкубировали в присутствии ацетилхолина в течение 10 мин. При таком протоколе клетки массово генерировали кратковременные Ca2+-ответы в течение первой минуты после начала стимуляции, хотя затем некоторые клетки могли генерировать повторные Ca2+-сигналы (рис. 2б, R-GECO1). При этом изменений в пространственном распределении флуоресценции сенсора PIP3 PH(Akt)-Venus, которое в контроле было равномерным, в присутствии ацетилхолина зарегистрировано не было (рис. 2б, PH(Akt)-Venus). В то же время при последующей стимуляции клеток инсулином наблюдалась иная реакция. Оказалось, что инсулин не вызывал детектируемого изменения уровня внутриклеточного Ca2+ по отношению к покою, но инициировал аккумуляцию PH(Akt)-Venus у плазматической мембраны клеток, свидетельствуя о PI3K-зависимой генерации PIP3 (рис. 2б).
Рис. 1.
Мониторинг инсулин-индуцированной активности PI3K с помощью сенсора PH(Akt)-Venus. а, б – Репрезентативные последовательные изображения клеток HEK-293/R-GECO1/PH(Akt)-Venus, полученные непосредственно перед (левые панели) и через 5 мин после (средние панели) начала стимуляции 100 нМ инсулина. Падение флуоресценции тела клетки и появление различимых полосообразных флуоресцентных зон (указаны стрелками на средних панелях) свидетельствовали об индуцированном инсулином перераспределении сенсора PH(Akt)-Venus из цитозоля к мембране клеток. На правых панелях представлены изображения клеток, полученные в присутствии 10 мкМ вортманнина (а) и 30 мкМ PI828 (б), которые были добавлены в раствор через 5 мин после инсулина. Локализованный у мембраны после стимуляции инсулином сенсор PH(Akt)-Venus в присутствии ингибиторов PI3K возвращался в цитозоль. Флуоресцентные изображения клеток (а, б) получены с помощью SDCM-микроскопии.
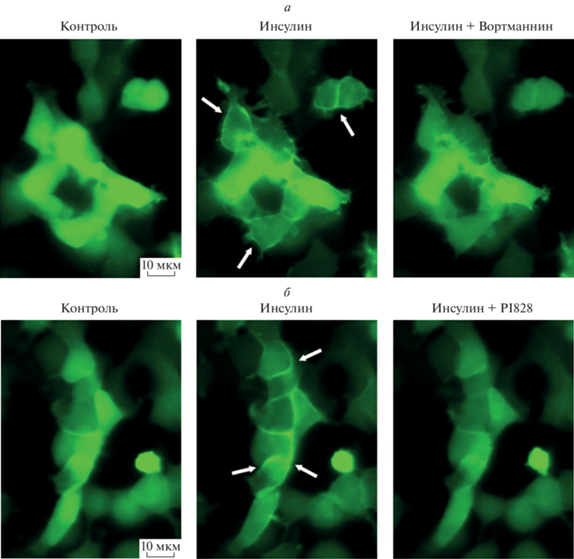
Рис. 2.
PI3K не вовлечена в сигнальные процессы, стимулируемые ацетилхолином. а – Схема протокола эксперимента; моменты и продолжительность аппликаций веществ обозначены горизонтальными линиями выше экспериментальной кривой; моменты получения изображений указаны стрелками; б – репрезентативные последовательные изображения клеток HEK-293/R-GECO1/PH(Akt)-Venus, полученные в контроле (за 30 с до стимуляции ацетилхолином) и после их стимуляции 1 мкМ ацетилхолина и 100 нМ инсулина. На верхней и нижней панелях представлены сигналы от Ca2+-сенсора R-GECO1 и PIP3-сенсора PH(Akt)-Venus соответственно. Как видно, аппликация ацетилхолина (1 мкМ) инициировала кратковременное увеличение интенсивности флуоресценции R-GECO1, что свидетельствовало о повышении концентрации цитозольного Ca2+. При этом пространственное распределение флуоресценции PH(Akt)-Venus не менялось, что указывает на отсутствие ацетилхолин-зависимой активации PI3K. Стимуляция инсулином (100 нМ) не влияла на флуоресценцию R-GECO1, т.е. не затрагивала внутриклеточный Ca2+, но приводила к аккумуляции PH(Akt)-Venus у плазмалеммы клеток (указано стрелками), свидетельствуя об активации PI3K.
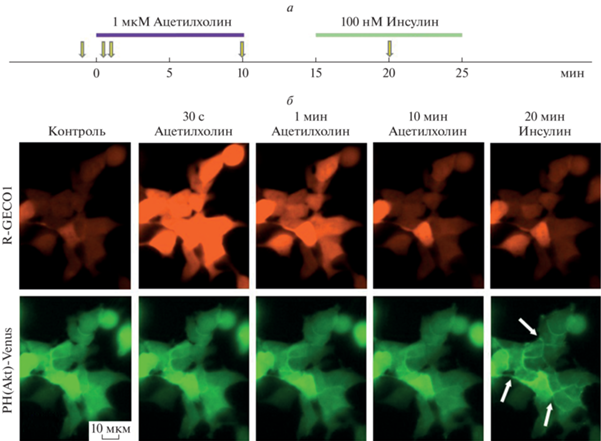
Таким образом, полученные данные наглядно демонстрируют, что, стимулируя Са2+-сигнализацию, ацетилхолин не инициирует существенную активацию PI3K-пути в клетках HEK-293. Поэтому PI3K не может быть вовлечена в трансдукцию ацетилхолина в клетках HEK-293. Как следствие, хотя результаты работы свидетельствуют об эффективном ингибировании PI3K вортманнином и PI828, последний не мог подавлять Ca2+-ответы на ацетилхолин, воздействуя на PI3K, но действовал на какую-то иную клеточную мишень. Ее идентификация представляет собой задачу для последующих исследований.
Авторы благодарят Д.М. Поташникову за помощь в проведении работ по сортировке клеток на клеточном сортере FACSAria SORP в рамках Программы развития МГУ.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа выполнена при поддержке гранта РНФ № 19-75-10068.
Соответствие принципам этики. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
Clapham D. 2007. Calcium signaling. Cell. 131, 1047–1058.
Berridge M.J. 2016. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296.
Vanhaesebroeck B., Guillermet-Guibert J., Graupera M., Bilanges B. 2010. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341.
Jean S., Kiger A.A. 2014. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 127, 923–928.
Parys J.B., Vervliet T. 2020. New insights in the IP 3 receptor and its regulation. Adv. Exp. Med. Biol. 1131, 243–270.
Graves B.M., Simerly T., Li C., Williams D.L., Wondergem R. 2012. Phosphoinositide-3-kinase/akt – dependent signaling is required for maintenance of [Ca2+]i, ICa, and Ca2+ transients in HL-1 cardiomyocytes. J. Biomed. Sci. 19, 59.
Ghigo A., Laffargue M., Li M., Hirsch E. 2017. PI3K and calcium signaling in cardiovascular disease. Circ. Res. 121, 282–292.
Santoso N.G., Cebotaru L., Guggino W.B. 2011. Polycystin-1, 2, and STIM1 interact with IP3R to modulate ER Ca2+ release through the PI3K/Akt pathway. Cell. Physiol. Biochem. 27, 715–726.
Marchi S., Marinello M., Bononi A., Bonora M., Giorgi C., Rimessi A., Pinton P. 2012. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis. 3, e304.
Fregeau M.O., Rergimbald-Dumas Y., Guillemette G. 2011. Positive regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by mammalian target of rapamycin (mTOR) in RINm5F cells. J. Cell. Biochem. 112, 723–733.
Szado T., Vanderheyden V., Parys J.B., De Smedt H., Rietdorf K., Kotelevets L., Chastre E., Khan F., Landegren U., Söderberg O., Bootman M.D., Rode-rick H.L. 2008. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA. 105, 2427–2432.
Zhang Y., Kwon S.H., Vogel W.K., Filtz T.M. 2009. PI(3,4,5)P3 potentiates phospholipase C-β activity. J. Recept. Signal Transduct. Res. 29, 52–62.
Дымова Е.А., Рогачевская О.А., Воронова Е.А., Котова П.Д. 2021. PI828 подавляет Ca2+-сигнализацию, инициируемую аминергическими агонистами, по механизму, независимому от ингибирования PI3-киназы. Биол. мембраны. 38 (5), 388–392.
Zhao Y., Araki S., Wu J., Teramoto T., Chang Y.-F., Nakano M., Abdelfattah A.S., Fujiwara M., Ishihara T., Nagai T., Campbell R.E. 2011. An expanded palette of genetically encoded Ca2+ indicators. Science. 333 (6051), 1888–1891.
O'Neill P.R., Gautam N. 2014. Subcellular optogenetic inhibition of G proteins generates signaling gradients and cell migration. Mol. Biol. Cell. 25 (15), 2305–2314.
Hopkins B.D., Goncalves M.D., Cantley L.C. 2020. Insulin-PI3K signalling: An evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 16 (5), 276–283.
Backer J.M., Schroeder G.G., Kahn C.R., Myers M.G. Jr., Wilden P.A., Cahill D.A., White M.F. 1992. Insulin stimulation of phosphatidylinositol 3-kinase activity maps to insulin receptor regions required for endogenous substrate phosphorylation. J. Biol. Chem. 267 (2), 1367–1374.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии