

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 6, стр. 440-445
Изоформ-специфические ингибиторы PI3K подавляют агонист-индуцированную Ca2+-сигнализацию по PI3K-независимому механизму
Е. А. Дымова a, О. А. Рогачевская a, П. Д. Котова a, *
a Институт биофизики клетки РАН, ФИЦ ПНЦБИ РАН
142290 Пущино, Московская обл., Россия
* E-mail: p.d.kotova@gmail.com
Поступила в редакцию 14.04.2022
После доработки 10.05.2022
Принята к публикации 12.05.2022
- EDN: OKLTLL
- DOI: 10.31857/S0233475522060044
Аннотация
Ацетилхолин является первичным медиатором в аутокринных и паракринных регуляциях, контролирующих физиологические процессы в различных клетках и тканях. В данной работе с помощью микрофотометрии, Ca2+-зонда Fluo-4 и ингибиторного анализа показано, что в клетках линии HEK-293 ацетилхолин вызывает мобилизацию Са2+, стимулируя мускариновые рецепторы, сопряженные с фосфоинозитидным сигнальным каскадом. При исследовании роли PI3-киназы (PI3K) в трансдукции ацетилхолина установлено, что специфические ингибиторы PI3Kβ (AZD 6482), PI3Kγ (AS 605240) и PI3Kδ (LTURM 36) полностью подавляют агонист-индуцированные Ca2+-сигналы. При этом специфический ингибитор PI3Kα (GSK 1059615) не оказывает влияния на способность клеток HEK-293 генерировать Ca2+-ответы на ацетилхолин. Ряд фактов указывает на то, что эффекты ингибиторов PI3K на клеточные ответы обусловлены механизмами, независимыми от ингибирования PI3K, и что возможной клеточной мишенью этих соединений является мускариновый рецептор M3. Высказано предположение, что изоформ-специфические ингибиторы PI3K AZD 6482, AS 605240 и LTURM 36 являются антагонистами М3-рецепторов.
ВВЕДЕНИЕ
Ацетилхолин играет важную роль в физиологии клеток и тканей, в частности, выполняя роль нейротрансмиттера в центральной и периферической нервной системе и являясь первичным медиатором в аутокринных и паракринных регуляциях, контролирующих физиологические процессы в различных ненейрональных тканях [1, 2]. Чувствительность клеток к ацетилхолину обеспечивается поверхностными рецепторами двух классов, включая ионотропные никотиновые рецепторы, представляющие собой лиганд-активируемые Са2+-проницаемые катионные каналы, и мускариновые рецепторы, сигнальная функция которых обеспечивается сопряжением с G-белками (G-protein coupled receptor, GPCR-рецепторы) и различными внутриклеточными сигнальными путями [1]. Ряд мускариновых рецепторов (M1, M3, M5) сопряжен с фосфоинозитидным каскадом и мобилизацией внутриклеточного Са2+ [1]. Ключевыми в этом процессе являются продукция вторичного медиатора IP3, катализируемая фосфолипазой С, и IP3-зависимый выброс депонированного Са2+ из Са2+-депо [3, 4]. Регуляторная роль IP3 сводится к активации IP3-рецепторов, которые фактически являются лиганд-управляемыми Са2+-проницаемыми каналами, функционирующими в эндоплазматическом ретикулуме и некоторых других внутриклеточных органеллах [5]. Помимо IP3, коагонистом IP3-рецепторов является цитозольный Са2+, который оказывает бифазное действие на их активность через Са2+-связывающие центры двух типов [6]. Связывание Ca2+ с высокоаффинным активаторным сайтом IP3-рецептора приводит к существенному росту вероятности открытого состояния этого Ca2+-канала [7], так что ионы Ca2+, высвобождающиеся из Ca2+-депо через IP3-рецептор, потенцируют его активность. Эта положительная обратная связь лежит в основе Ca2+-индуцированного выброса депонированного Ca2+ (Ca2+-induced Ca2+ release) – регенеративного процесса, который определяет многие аспекты внутриклеточной Ca2+-сигнализации [3, 8].
На протекание процесса Ca2+-индуцированного выброса депонированного Ca2+ влияют механизмы, регулирующие IP3-рецепторы, активность которых, в частности, контролируется процессами фосфорилирования/ дефосфорилирования при участии ряда киназ, включая фосфатидилинозитол-3-киназу (PI3K) [9, 10]. Ранее нами было показано, что ингибитор PI3K вортманнин не влияет на способность клеток HEK-293 генерировать Ca2+-ответы на ацетилхолин, тогда как ингибитор PI3K другой химической природы PI828 полностью подавляет эти ответы [11]. Разная эффективность вортманнина и PI828 могла быть связана с тем, что в клетках HEK-293 функционируют изоформы PI3K, существенно более чувствительные к PI828. Для внесения ясности в этот вопрос мы исследовали влияние изоформ-специфических ингибиторов PI3K на Ca2+-сигнализацию, инициируемую ацетилхолином.
МАТЕРИАЛЫ И МЕТОДЫ
Культура клеток. Клетки линии НЕК-293 (российская коллекция клеточных культур позвоночных) культивировали в среде DMEM с высоким содержанием глюкозы (Invitrogen) с добавлением 10% эмбриональной бычьей сыворотки (HyClone), 100 мг/мл гентамицина (Sigma-Aldrich), 2 мМ глутамина (Sigma-Aldrich) (ростовая среда) во влажной атмосфере c 5% содержанием CO2 в воздухе при 37°C.
Мониторинг внутриклеточного Ca2+. Для эксперимента клетки снимали с культурального пластика 0.25% раствором трипсина (Sigma-Aldrich), а затем прикрепляли ко дну фотометрической камеры с помощью адгезивного материала Cell Tak (Corning). При дальнейших манипуляциях клетки находились во внеклеточном растворе, содержащем (мМ): 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4, 10 глюкозы, в ряде экспериментов использовали внеклеточный раствор, содержащий 260 нМ свободного Ca2+, для чего 2 мМ CaCl2 заменяли на 0.5 мМ EGTA + 0.4 мМ CaCl2 (все соли и буферы произведены Sigma-Aldrich). Для загрузки флуоресцентным Ca2+-зондом Fluo-4 клетки инкубировали в присутствии проникающего через мембрану Fluo-4 AM (4 мкМ) и детергента Pluronic (0.02%) (оба Molecular Probes) при комнатной температуре в течение 30 мин. Затем клетки отмывали внеклеточным раствором и выдерживали в нем при комнатной температуре в течение 1 ч.
Фотометрические эксперименты проводили с использованием инвертированного флуоресцентного микроскопа Axiovert 135 (Zeiss), оборудованного объективом Plan NeoFluar 20×/0.75 и цифровой EMCCD камерой LucaR (Andor Technology). Флуоресценцию клеток возбуждали на длине волны 480 ± 10 нм, эмиссию регистрировали в области 520 ± 20 нм, что соответствует спектральным характеристикам Fluo-4. Изменение уровня Са2+ в цитоплазме оценивали по относительному изменению интенсивности флуоресценции Fluo-4 ΔF/F0, где ΔF = F – F0, F и F0 – интенсивность эмиссии Са2+-индикатора в текущий момент времени и в начале регистрации соответственно. Количественный фотометрический анализ изображений осуществляли с использованием программы NIS Elements (Nikon). Полученные экспериментальные данные обрабатывали с помощью программы Sigma Plot 12.5 (Systat Software Inc).
Все химические соединения, использованные в описанных ниже экспериментах, применяли путем полной замены раствора в фотометрической камере с помощью системы перфузии. В работе использовали: ацетилхолин, ATP, AS 605240, AZD 6482, GSK 1059615, LTURM 36 (все Tocris Bioscience).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
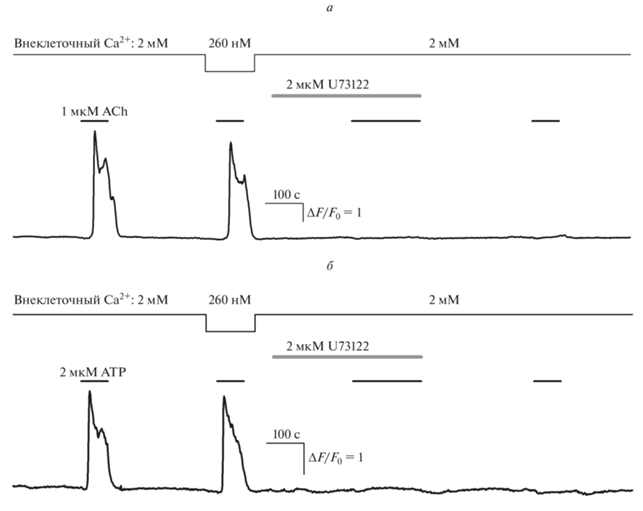
В физиологических экспериментах практически все клетки НЕК-293 генерировали Ca2+-ответы на стимуляцию ацетилхолином. Нами были проанализированы механизмы, используемые клетками HEK-293 для генерации Ca2+-ответов на ацетилхолин. Одной из решавшихся задач было выяснение вклада источников ацетилхолин-индуцируемого поступления Ca2+ в цитозоль, т.е. выброса Са2+ из внутриклеточного депо и входа наружного Са2+. Для этого, в частности, анализировали способность клеток HEK-293 генерировать Ca2+-ответы на ацетилхолин в среде с пониженным до цитоплазматического уровня содержанием Ca2+ (260 нМ). Оказалось, что все протестированные холинергические HEK-293 в среде с пониженным Ca2+ генерировали практически нормальные по амплитуде и кинетике ответы на ацетилхолин (рис. 1а). Это свидетельствует о том, что основным источником Ca2+ для исследуемых ответов служит депонированный Ca2+, а за чувствительность HEK-293 к ацетилхолину ответственны GPCR-рецепторы ацетилхолина – мускариновые рецепторы. В связи с этим было вполне ожидаемо, что Са2+-ответы на ацетилхолин подавлялись в присутствии ингибитора фосфолипазы С U73122 (рис. 1а). В совокупности эти данные свидетельствуют о том, что для генерации Ca2+-ответов на ацетилхолин клетки HEK-293 преимущественно используют мускариновые рецепторы, сопряженные с фосфоинозитидным путем трансдукции сигнала.
Рис. 1.
Вклад фосфоинозитидного каскада в Са2+-ответы HEK-293 на ацетилхолин и АТР. Репрезентативные регистрации Ca2+-ответов двух различных клеток HEK-293 при варьируемом внеклеточном Са2+ и в присутствии ингибитора фосфолипазы С U73122 (2 мкМ) при стимуляции: а – 1 мкМ ацетилхолина (ACh), б – 2 мкМ ATP. Здесь и далее моменты и продолжительность аппликаций веществ обозначены горизонтальными линиями выше экспериментальной кривой. Изменение внутриклеточного Ca2+ оценивается по относительному изменению флуоресценции Fluo-4: ΔF/F0, где ΔF = F – F0, F и F0 – текущая интенсивность эмиссии зонда и его эмиссия в начале регистрации соответственно.
При исследовании эффектов изоформ-специфических ингибиторов PI3K оказалось, что Са2+-ответы на ацетилхолин полностью подавлялись в присутствии AZD 6482, AS 605240 и LTURM 36, использовавшихся в достаточных дозах (рис. 2). Наиболее эффективным оказался ингибитор PI3Kδ LTURM 36, который в концентрации 10 мкМ блокировал ответы 48.8% клеток, чувствительных к ацетилхолину (рис. 2а). Концентрации полуэффекта для ингибиторов PI3Kβ AZD 6482 и PI3Kγ AS 605240 составляли приблизительно 30 и 40 мкМ соответственно (рис. 2б, 2в). В то же время ингибитор PI3Kα (GSK 1059615) в широком диапазоне концентраций (1–100 мкМ) не влиял на способность клеток генерировать Ca2+-ответы на ацетилхолин (рис. 2г), которые подавлялись в присутствии неспецифического ингибитора PI3K PI828 (рис. 2д) [11]. Формально полученные результаты можно рассматривать как свидетельство того, что в генерации Ca2+-ответов на ацетилхолин в клетках HEK-293 принимают участие β-, γ- и преимущественно δ-изоформа PI3K, тогда как α-изоформа не задействована в этом механизме. Однако обращал на себя внимание тот факт, что ингибирование Ca2+-ответов в присутствии LTURM 36, AZD 6482 и AS 605240 наблюдалось при аппликации этих соединений одновременно с ацетилхолином, хотя обычно ингибирование внутриклеточных мишеней в интактных клетках требует времени для проникновения ингибитора через плазматическую мембрану. В связи с этим возникал вопрос, в какой степени наблюдавшееся подавление Ca2+-ответов обусловлено воздействием перечисленных соединений именно на внутриклеточные сигнальные процессы, активируемые в HEK-293 ацетилхолином и, прежде всего, на фосфоинозитидный сигнальный путь.
Рис. 2.
Влияние специфических ингибиторов различных изоформ PI3K на Ca2+-ответы клеток HEK-293 на ацетилхолин и ATP. а–г – На левых панелях представлены репрезентативные Ca2+-ответы одиночных клеток на ацетилхолин (ACh) (1 мкМ) и ATP (2 мкМ) в контроле и присутствии ингибиторов: а – PI3Kδ LTURM 36 (10 мкМ), б – PI3Kβ AZD 6482 (30 мкМ), в – PI3Kγ AS 605240 (30 мкМ), г – PI3Kα GSK 1059615 (50 мкМ). На средних панелях приводится доля (в %) клеток, отвечающих на ацетилхолин (1 мкМ), на правых панелях – доля (в %) клеток, отвечающих на ATP (2 мкМ), в присутствии ингибиторов: а – PI3Kδ LTURM 36 (n = 341), б – PI3Kβ AZD 6482 (n = 280), в – PI3Kγ AS 605240 (n = 226), г – PI3Kα GSK 1059615 (n = 357). Данные представлены как среднее ± стандартное отклонение; усреднение по 3–5 независимым экспериментам, в каждом из которых мониторинг внутриклеточного Са2+ проводили в 80–100 индивидуальных клетках. д – Репрезентативная регистрация Ca2+-ответов одиночной клетки на ацетилхолин (1 мкМ) в контроле и присутствии ингибитора PI3Kα GSK 1059615 (50 мкМ) и неспецифического ингибитора PI3K PI828 (30 мкМ) (n = 274).
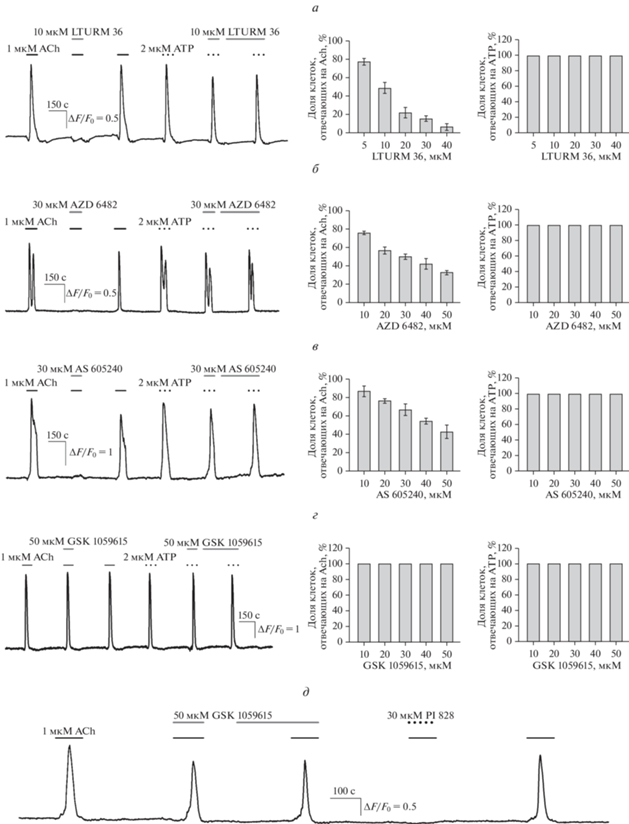
Одним из возможных вариантов проверки воздействия ингибиторов PI3K на фосфоинозитидный каскад было исследование влияния этих соединений на Са2+-сигнализацию, инициируемую не холинергическими агонистами. В эксперименте, аналогичном описанному выше, было установлено, что, действуя через P2Y-рецепторы, ATP вызывает в клетках HEK-293 Ca2+-ответы, которые также генерируются при участии фосфоинозитидного пути (рис. 1б). Оказалось, что ни один из исследовавшихся изоформ-специфических ингибиторов PI3K не влиял на способность клеток HEK-293 отвечать на ATP (рис. 2а–2г), а значит, активность PI3K вносит минимальный вклад в механизм их генерации.
Таким образом, исследовавшиеся ингибиторы PI3K по-разному влияли на мобилизацию Са2+, индуцированную различными агонистами при участии фосфоинозитидного сигнального пути. С одной стороны, AZD 6482, AS 605240 и LTURM 36 полностью подавляли Ca2+-ответы на ацетилхолин, однако когда в качестве агониста использовали ATP, то какое-либо влияние перечисленных ингибиторов PI3K на клеточные ответы отсутствовало. Полученные данные указывали на то, что подавление Ca2+-ответов на ацетилхолин соединениями AZD 6482, AS 605240, LTURM 36 связано не с ингибированием PI3K, которая модулирует фосфоинозитидный каскад, а с воздействием на другую клеточную мишень, которая участвует в генерации Ca2+-ответа на ацетилхолин, но не является частью фосфоинозитидного сигнального пути. Такой мишенью могут являться непосредственно мускариновые рецепторы ацетилхолина, что косвенно подтверждается способностью соединений AZD 6482, AS 605240, LTURM 36 блокировать Ca2+-ответы при одновременной аппликации с ацетилхолином (рис. 2а–2в). В исследованиях, предшествующих этому, нами было установлено, что за чувствительность HEK-293 к ацетилхолину отвечает М3-рецептор [12], и, следовательно, именно этот мускариновый рецептор является наиболее вероятной мишенью соединений AZD 6482, AS 605240, LTURM 36.
Ранее методами вычислительной биофизики нами была продемонстрирована возможность связывания с М3-рецепторами ингибиторов PI3K LY294002, LY303511 и Voxtalisib [12, 13]. Результаты физиологических экспериментов, описанные в данной работе, показали, что ингибитор PI3K GSK 1059615 не влиял на способность клеток отвечать на ацетилхолин, а значит, не все синтетические ингибиторы PI3-киназы обладают антагонистическими свойствами по отношению к мускариновым рецепторам. Полученные результаты по ингибированию клеточных ответов могут отражать эффективность связывания специфических ингибиторов PI3K с М3-рецепторами, которая в этом случае описывается рядом: LTURM 36 > AZD 6482 > AS 605240. Сочетание экспериментальных подходов и математического моделирования, возможно, позволит в дальнейшем установить набор молекулярных признаков, которые обеспечивают узнавание М3-рецептором ингибиторов PI3K как своих лигандов. В перспективе это может позволить создавать ингибиторы PI3K, не оказывающие неспецифического действия на активность М3-рецепторов.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа выполнена при поддержке гранта РНФ № 19-75-10 068.
Соответствие принципам этики. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
Brown D.A. 2019. Acetylcholine and cholinergic receptors. Brain Neurosci. Adv. 3, 2398212818820506.
Wessler I., Kirkpatrick C.J. 2008. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 154 (8), 1558–1571.
Berridge M.J. 2016. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296.
Thillaiappan N.B., Chakraborty P., Hasan G., Taylor C.W. 2019. IP3 receptors and Ca2+ entry. Biochim. Biophys. Acta Mol. Cell. Res. 1866 (7), 1092–1100.
Hamada K., Mikoshiba K. 2020. IP3 receptor plasticity underlying diverse functions. Annu. Rev. Physiol. 82, 151–176.
Prole D.L., Taylor C.W. 2019. Structure and function of IP3 receptors. Cold Spring. Harb. Perspect. Biol. 11 (4), a035063.
Mak D.O., Foskett J.K. 2015. Inositol 1,4,5-trisphosphate receptors in the endoplasmic reticulum: A single-channel point of view. Cell Calcium. 58, 67–78
Rios E. 2018. Calcium-induced release of calcium in muscle: 50 years of work and the emerging consensus. J. Gen. Physiol. 150, 521–537.
Szado T., Vanderheyden V., Parys J.B., De Smedt H., Rietdorf K., Kotelevets L., Chastre E., Khan F., Landegren U., Söderberg O., Bootman M.D., Rode-rick H.L. 2008. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA. 105 (7), 2427–2432.
Frégeau M.O., Régimbald-Dumas Y., Guillemette G. 2011. Positive regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by mammalian target of rapamycin (mTOR) in RINm5F cells. J. Cell Biochem. 112 (2), 723–733.
Дымова Е.А., Рогачевская О.А., Воронова Е.А., Котова П.Д. 2021. PI828 подавляет Ca2+-сигнализацию, инициируемую аминергическими агонистами, по механизму, независимому от ингибирования PI3-киназы. Биол. мембраны. 38 (5), 265–273.
Лямин О.О., Котова П.Д., Дымова Е.А., Фадеев П.Ю., Рогачевская О.А., Воронова Е.А., Колесников С.С. 2022. Ингибитор PI3K и mTOR воксталисиб нарушает сопряжение мускаринового рецептора М3 с мобилизацией Ca2+. Биол. мембраны. 33 (3), 205–214.
Kotova P.D., Kochkina E.N., Lyamin O.O., Rogachevskaja O.A., Kovalenko N.P., Ivashin D.S., Bystrova M.F., Enukashvily N.I., Kolesnikov S.S. 2020. Calcium signaling mediated by aminergic GPCRs is impaired by the PI3K inhibitor LY294002 and its analog LY303511 in a PI3K-independent manner. Eur. J. Pharmacol. 880, 173182.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии