

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 6, стр. 446-456
Дофамин защищает нейроны от токсического действия глутамата
Е. И. Федотова a, *, А. Ю. Абрамов b, c, А. В. Бережнов a, **
a Институт биофизики клетки – ФИЦ ПНЦБИ РАН
142190 Пущино, Россия
b Институт неврологии Университетского колледжа Лондона
WC1N 3BG Лондон
c Орловский государственный университет
302026 Орел, Россия
* E-mail: delf-fenka@rambler.ru
** E-mail: g_56@rambler.ru
Поступила в редакцию 01.07.2022
После доработки 25.07.2022
Принята к публикации 26.07.2022
- EDN: DKMHHD
- DOI: 10.31857/S0233475522060056
Аннотация
Болезнь Паркинсона – нейродегенеративное заболевание, связанное с потерей нейронов в среднем мозге с последующим развитием дефицита дофамина. Мы предположили, что дофамин может действовать как антагонист глутамата, а его дефицит может усиливать вызванную глутаматом эксайтотоксичность. Используя первичную культуру гиппокампа крысы и флуоресцентную микроскопию, мы показали, что дофамин снижает амплитуду кальциевого ответа, вызванного активацией NMDA-рецепторов, не влияя при этом на кальциевый сигнал, опосредованный АМРА- и КА-рецепторами, а также активацией потенциал-зависимых кальциевые каналов. Показано, что эффект дофамина зависит не только от NMDA-рецепторов, но и от дофаминовых рецепторов D2-типа и от ГАМКА-рецепторов. Дофамин уменьшает деполяризацию митохондрий, вызванную глутаматом, и улучшает выживаемость нейронов при действии глутамата в токсических дозах. Представленные данные свидетельствуют о защитной роли дофамина при эксайтотоксичности, вызванной глутаматом.
ВВЕДЕНИЕ
Дофамин – это нейромедиатор центральной нервной системы, который участвует в регуляции различных физиологических функций. Нарушения метаболизма и сигнализации дофамина приводят к развитию таких нейродегенеративных заболеваний, как болезнь Паркинсона, болезнь Альцгеймера, шизофрения. Так, во время болезни Паркинсона наблюдается потеря дофаминергических нейронов, главным образом в черной субстанции, а также в других отделах центральной нервной системы [1].
Известно, что дофамин способен подавлять увеличение концентрации ионов кальция в цитозоле ([Ca2+]i), возникающее в ответ на глутамат, который в норме является основным возбуждающим нейромедиатором в головном мозге, а при избыточном накоплении становится токсичным и вызывает патологические состояния [2]. Во время развития болезни Паркинсона, при которой характерен дефицит дофамина, возможно, даже невысокие дозы глутамата могут приводить к нейродегенерации. Как известно, глутамат опосредует активацию группы глутаматных рецепторов, к которым относятся ионотропные рецепторы (NMDA-, AMPA- и КА-рецепторы), являющиеся лиганд-управляемыми ионными каналами и активируемые специфическими агонистами, и метаботропные рецепторы (mGlut1–mGlut8), сопряженные с G-белком [3].
Дофаминергические функции опосредованы активацией семейством дофаминовых рецепторов, состоящим из пяти типов и относящимся к двум группам: D1-подобные рецепторы (D1 и D5) и D2-подобные рецепторы (D2, D3 и D4). D1-подобные рецепторы представляют собой серпентиновые рецепторы, сопряженные с Gs-белком, которые участвуют в регуляции высвобождения нейротрансмиттеров, таких как глутамат, ГАМК и ацетилхолин. D2-подобные сопряжены с ингибирующим G-белком (Gi/0) и регулируют секрецию дофамина в нейронах в ответ на изменение внеклеточного уровня нейротрансмиттера [4].
Из литературы известно, что взаимодействие между дофаминергической и глутаматергической системами обеспечивает основу для сложных нейронных взаимодействий в головном мозге. Активация D1- и D2-подобных рецепторов может регулировать функцию и транспорт NMDA-рецепторов. Так, дофаминовые рецепторы напрямую или опосредованно через активацию протеинкиназы А взаимодействуют с NMDA-рецепторами и могут влиять на NMDA-ответы в нейронах [5].
Мы предположили, что дофамин может действовать как протектор против глутаматной нейротоксичности, поскольку добавление дофамина к культуре клеток гиппокампа ослабляет вызванные глутаматом повышение [Ca2+]i и митохондриальную деполяризацию и увеличивает количество жизнеспособных клеток после воздействия глутамата. В настоящей работе, чтобы определить мишени действия дофамина, мы исследовали его влияние на амплитуду кальциевых ответов, вызванных агонистами глутаматных и дофаминовых рецепторов.
МАТЕРИАЛЫ И МЕТОДЫ
Культура нейронов. Объектом исследования служила первичная нейроглиальная культура гиппокампа крысы. Клетки гиппокампа выделяли из головного мозга новорожденных крысят (P1–P3) породы Spraque Dawley. Согласно методике получения культуры клеток, после декапитации крысенка гиппокамп извлекали и переносили в холодный раствор Хенкса [6]. Далее ткань измельчали ножницами, инкубировали в 0.1% растворе трипсина в течение 15 мин при 37°C, отмывали 3 раза средой Neurobasal-A (Gibco, США), содержащей 2% Supplement B27 (Gibco), 1 мМ L-глутамин (Sigma, США), 7.5 мкг/мл гентамицин (Дальхимфарм, Россия), пипетируя ткань до образования суспензии клеток. После этого клетки размещали на круглых покровных стеклах диаметром 25 мм, покрытых полиэтиленимином (Fluka, США), и помещали в чашки Петри (35 мм). Через 1 ч к прикрепившимся клеткам добавляли 1.5 мл культуральной среды Neurobasal и инкубировали 5–12 дней при 37°C и 5% CО2.
Эксперименты проводили в растворе Хенкса, содержащем (в мМ): 138 NaCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 5.3 KCl, 0.45 KH2PO4, 4 NaHCO3, 0.3 Na2HPO4, 10 глюкозы, 20 HEPES (pH 7.3) при температуре 27–28°C. Эксперименты с NMDA проводили в среде без Mg2+ с последующей заменой на раствор Хенкса.
Измерение концентрации внутриклеточного кальция ([Ca2+]i). Изменения [Ca2+]i в нейронах оценивали по интенсивности флуоресценции двухволнового Са2+-чувствительного зонда Fura-2 [7]. Культуру клеток нагружали 5 мкМ Fura-2AM (Invitrogen, США) в течение 60 мин с последующей отмывкой. Для регистрации [Ca2+]i использовали систему анализа изображений на базе инвертированного моторизованного микроскопа Leica DMI6000B (Leica Microsystems, Германия), оснащенного объективом Leica HC PL APO 20×/0.7 IMM. Для возбуждения и регистрации флуоресценции Fura-2 использовали набор светофильтров FU2 (возбуждение: 340 ± 6 и 380 ± 10 нм, регистрация: 510 ± 20 нм). Также использовалась система анализа изображений Cell Observer (Carl Zeiss, Геpмания) на базе инвертированного микpоcкопа Axiovert 200M, оснащенного объективом Plan Neofluar 10×/0.3 Для возбуждения и регистрации флуоресценции Fura-2 использовали набор светофильтров Filter set 21HE (возбуждение: 340 ± 6 и 380 ± 10 нм, регистрация: 510 ± 20 нм). Изображения получали с частотой 1 кадр в 3 с. Полученные в двух различных каналах временные серии изображений обрабатывали в программе ImageJ с использованием плагина Time Series Analyzer. Амплитуду кальциевых ответов (АКО) одиночных клеток оценивали как отношение сигналов флуоресценции Fura-2 при возбуждении светом длиной волны 340 и 380 нм. В эксперименте измеряли ответ 100–200 нейронов, находящихся в поле зрения микроскопа.
Для проведения эксперимента покровное стекло с культурой клеток монтировалось в специальную измерительную ячейку и содержалось в растворе Хенкса объемом 0.5 мл. Добавки реагентов и отмывку осуществляли с помощью системы перфузии, состоящей из подающего наконечника, подключенного к резервуарам с растворами, и наконечника, подключенного к водоструйному насосу и обеспечивающего постоянный уровень среды в измерительной ячейке. Наконечники располагались на противоположных сторонах ячейки. Для осуществления добавки система перфузии включалась на 8 с, что обеспечивало десятикратную (5 мл) смену рабочего раствора в ячейке. Таким же образом производилась отмывка. Контрольные эксперименты с оптически плотным красителем (трипановый синий) показали, что в этих условиях происходит полная (более 98%) замена раствора в измерительной ячейке. Такой протокол смены растворов является результатом оптимизации и обеспечивает отсутствие реакции клеток на механическое воздействие при смене среды. Тем не менее для оценки возможной реакции клеток в начале каждого эксперимента проводилась контрольная промывка 5 мл раствора Хенкса, которая указывала на отсутствие изменений [Ca2+]i в представленных опытах.
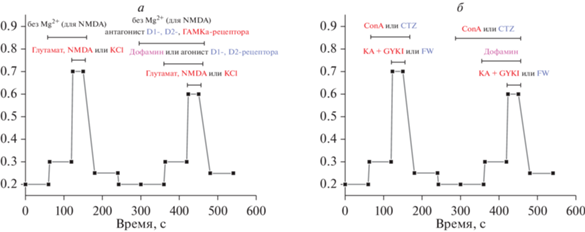
Для сравнения результатов опытов, проводимых в разные дни и на разных культурах, для нормирования сигналов в схеме опыта, а также для функционального разделения нейронов и глиальных клеток в культуре к клеткам добавляли стандартный деполяризующий раствор KCl. В работе была использована методика оценки модулирующего действия различных соединений на активность ионотропных рецепторов глутамата – NMDA-, AMPA- и KA-рецепторов [8], которая основана на том, что в контроле несколько кратковременных (20–30 с) добавок агонистов этих рецепторов с интервалом в 10–15 мин вызывают кальциевые сигналы одинаковой амплитуды. Это позволяет использовать сигнал в ответ на первую добавку в качестве контроля, а на втором сигнале тестировать эффект того или иного соединения (рис. 1а). Амплитуды сигналов измеряли относительно исходного значения до добавки. В случае активации KA-рецепторов эксперименты проводили в присутствии ингибитора AMPA-рецепторов 30 мкМ GYKI-52466 (Tocris, Великобритания), поскольку каиновая кислота (KA, Sigma) является неселективным агонистом KA- и AMPA-рецепторов, а также ингибитора десенситизации КА-рецепторов 200 мкМ конканавалина А (ConA, Sigma). Для активации AMPA-рецепторов использовали селективный агонист этих рецепторов 5-фторвиллардиин (FW, Santa Cruz Biotechnology, США) и ингибитор десенситизации AMPA-рецепторов 5 мкМ циклотиазид (CTZ, Tocris) (рис. 1б) [8].
Рис. 1.
Схемы экспериментов для определения действия дофамина на амплитуду кальциевых сигналов, вызванных агонистами глутаматных рецепторов и KCl, на основе ранее разработанной методики [8]. а – Для глутамата, NMDA и KCl; б – для KA и AMPA.
Измерение митохондриального потенциала. Для регистрации клетки нагружали флуоресцентным потенциал-чувствительным зондом 20 мкМ Rhodamine 123 (Rh123, Sigma) в течение 10 мин. После этого дважды отмывали раствором Хенкса и использовали в эксперименте. Измерения проводили с помощью системы анализа изображений Cell Observer (Carl Zeiss) на базе инвертированного микpоcкопа Axiovert 200M, оснащенного объективом Plan Neofluar 10×/0.3. Для возбуждения и регистрации флуоресценции Rh123 использовали набор светофильтров Filter set 44 (возбуждение: 490 ± 6 нм, регистрация: 550 ± 20 нм). В экспериментах использовался протокол прокраски, основанный на зависимом от концентрации красителя тушении флуоресценции в энергизованных митохондриях, при деполяризации митохондриальных мембран наблюдается увеличение флуоресценции Rh123 [9]. Изображения получали с частотой 1 кадр в 5 с, которые обрабатывали в программе ImageJ с использованием плагина Time Series Analyzer. В эксперименте измеряли ответ 100–200 нейронов, находящихся в поле зрения микроскопа. Для нормирования сигналов в схеме опыта предусмотрена добавка 2 мкМ FCCP (Sigma), выявляющая наибольшую степень деполяризации митохондрий.
Оценка жизнеспособности клеток. Для оценки количества живых клеток в смешанной нейроглиальной культуре использовали метод двойного окрашивания флуоресцентными красителями Hoechst 33342 (2 мкг/мл, Sigma) и Propidium Iodide (PI, 2 мкг/мл, Sigma) в течение 10 мин с последующей отмывкой раствором Хенкса. Флуоресценцию красителей оценивали с использованием системы анализа изображений на базе инвертированного моторизованного микроскопа Leica DMI6000B (Leica Microsystems), оснащенного объективом Leica HC PL APO 20×/0.7 IMM. Для возбуждения и регистрации флуоресценции Hoechst использовали набор светофильтров DAPI (возбуждение: 340 ± 6 нм, регистрация: 470 ± 20 нм), для PI – набор светофильтров Texas Red (возбуждение: 575 ± 10 нм, регистрация: 624 ± 10 нм). Клетки в двух каналах подсчитывали в программе ImageJ с использованием плагина Cell Counter. Краситель Hoechst проникает в клетки и связывается с ДНК, что позволяет оценить морфологию ядра. Напротив, краситель PI проникает только в поврежденные или фиксированные клетки. При колокализации сигнала от Hoechst и PI, свидетельствующей о нарушении барьерной функции плазмалеммы, клетки рассматривались нами как погибшие, а фрагментированные, ярко светящиеся в синем (Hoechst) канале ядра свидетельствовали об апоптозе. Данные представляли в виде процента нежизнеспособных и апоптотических клеток от общего числа клеток в поле зрения. В эксперименте измеряли флуоресцентный сигнал от 300–600 клеток нейроглиальной культуры.
Обработка результатов. Для построения графиков и статистической обработки использовали программы OriginPro2019 и GraphPadPrizm8 с применением параметрического анализа one-way ANOVA (post hoc Turkey test). На графиках представлены репрезентативные кривые, количество клеток в одном эксперименте N = 100–140, количество однотипных экспериментов: n = 3–4. Данные в столбцах представлены как среднее значение ± стандартное отклонение от среднего, статистически значимые различия при * p < 0.05.
РЕЗУЛЬТАТЫ
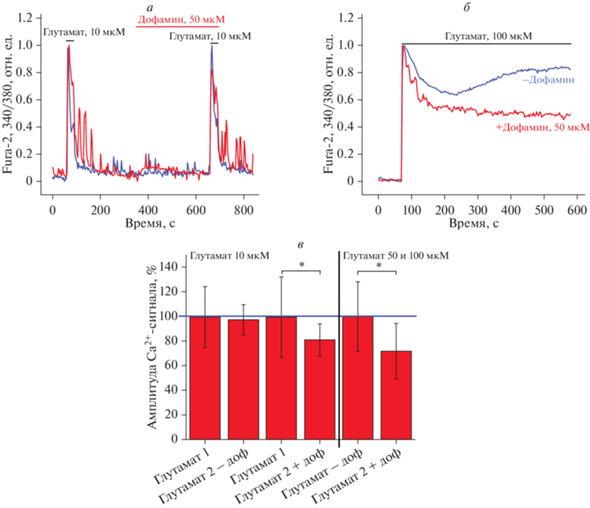
Дофамин подавляет глутамат-индуцированное увеличение [Ca2+]i в нейронах. Ранее было показано, что дофамин способен подавлять увеличение уровня цитозольного кальция ([Ca2+]i), возникающее в ответ на небольшие дозы глутамата (5 мкМ) [10]. В данной работе мы проверили, может ли дофамин защищать клетки от действия более высоких концентраций глутамата – 50 и 100 мкМ, которые являются токсичными для клеток. Мы оценили способность дофамина влиять на амплитуду кальциевого сигнала в ответ на глутамат с помощью разработанного ранее способа оценки модулирующего действия различных соединений на активность ионотропных рецепторов глутамата – NMDA-, AMPA- и KA-рецепторов [8]. Был использован протокол, при котором повторяющиеся кратковременные добавки агонистов рецепторов глутамата вызывают повторяющееся по форме и амплитуде повышение [Ca2+]i. В работе было показано, что дофамин в концентрации 50 мкМ способен снижать амплитуду кальциевого сигнала в ответ на вторую добавку 10 мкМ глутамата на 19%. Так, добавка глутамата на фоне дофамина составила 81 ± 13% от первой добавки глутамата (на рис. 2а, 2в ответ на глутамат принят за 100%). В случае действия больших доз глутамата (100 и 50 мкМ) ответ при добавлении дофамина также снижался – на 28% (72 ± 22%) (рис. 2б, 2в). Подобный эффект может приводить к предотвращению глутамат-индуцированной кальциевой перегрузки клеток и таким образом являться нейропротекторным в условиях эксайтотоксичности.
Рис. 2.
Влияние дофамина на амплитуду кальциевого сигнала в ответ на глутамат в нейронах. а – Повторяющиеся кальциевые сигналы в нейронах на кратковременные добавки 10 мкМ глутамата (синяя кривая) и влияние 50 мкМ дофамина на сигнал в клетках в ответ на добавку глутамата (красная кривая); нормировано относительно первой добавки глутамата. б – Кальциевый сигнал в ответ на 100 мкМ глутамата в присутствии (красная кривая) и в отсутствие дофамина (50 мкМ, синяя кривая); нормировано относительно ответа на глутамат. в – Амплитуда кальциевого сигнала в ответ на добавку глутамата 10 и 50–100 мкМ в отсутствие (–доф) и присутствии 50 мкМ дофамина (+доф). В случае действия 10 мкМ глутамата ответы на вторую добавку глутамата (Глутамат 2) представлены относительно ответов на первую добавку (Глутамат 1). В случае больших доз глутамата (50–100 мкМ) кальциевый ответ на глутамат совместно с дофамином (Глутамат+доф) представлен относительно ответа на глутамат без дофамина (Глутамат–доф). * p < 0.05.
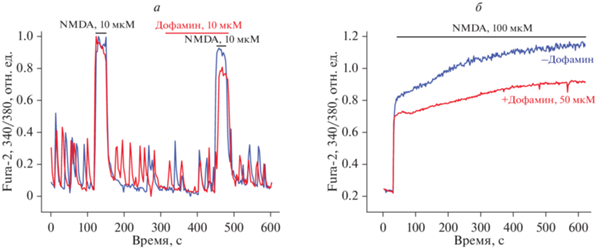
Для того чтобы выявить, какие именно рецепторы участвуют в данном эффекте, мы проверили, как дофамин влияет на амплитуду кальциевых сигналов, вызванных селективной активацией ионотропных рецепторов глутамата: NMDA-, AMPA- и KA-рецепторов. При этом NMDA-рецептор сам проницаем для ионов кальция, а активация AMPA- и KA-рецепторов в основном опосредована действием потенциал-зависимых кальциевых каналов [11]. Было показано, что дофамин подавляет активность NMDA-рецепторов, при этом не влияет на активность рецепторов каиновой кислоты (KA-рецепторов) и AMPA-рецепторов. Были получены следующие значения: дофамин в концентрации 10 мкМ подавляет кальциевый сигнал, вызванный добавлением небольших доз (5 и 10 мкМ) NMDA на 23% (77 ± 23% относительно первого сигнала агониста, рис. 3а, 5б). При действии высоких концентраций NMDA (50 и 100 мкМ) дофамин подавляет ответ на добавку этого агониста на 15% (рис. 3б).
Рис. 3.
Влияние дофамина на амплитуду кальциевого сигнала, вызванного NMDA в нейронах. а – Кальциевый сигнал в ответ на добавление 10 мкМ NMDA в отсутствие (синяя кривая) и в присутствии (красная кривая) 50 мкМ дофамина; нормировано относительно первой добавки NMDA. б – Кальциевый сигнал в ответ на добавление 100 мкМ NMDA в отсутствие (синяя кривая) и в присутствии (красная кривая) 50 мкМ дофамина.
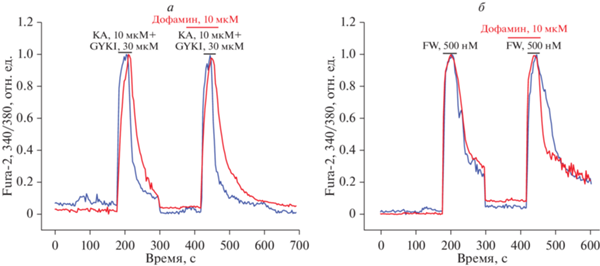
Было показано, что дофамин не уменьшает кальциевый сигнал в ответ на агонист КА-рецептора каиновую кислоту (на фоне антагониста AMPA-рецептора GYKI-52466, KA + GYKI). Кальциевый ответ на вторую добавку составил 101 ± ± 22% относительно первого сигнала (рис. 4а, 5б). Также дофамин не оказывает влияние на активность AMPA-рецепторов, поскольку добавление дофамина на фоне агониста AMPA-рецептора фторовиллардиина (FW) не изменяло кальциевый ответ (98 ± 13%) (рис. 4б, 5б). Для снятия десенситизации этих рецепторов эксперименты проводили в присутствии 200 мкг/мл конканавалина А (для KA-рецептора) или 5 мкМ циклотиазида (для АМРА-рецептора) [8].
Рис. 4.
Влияние дофамина на амплитуду кальциевого сигнала, вызванного активацией KA-и AMPA-рецепторов в нейронах. а – Кальциевый сигнал в ответ на добавление 10 мкМ каиновой кислоты (KA) совместно с 30 мкМ GYKI в отсутствие (синяя кривая) и в присутствии (красная кривая) 50 мкМ дофамина; нормировано относительно первой добавки KA+GYKI. б – Кальциевый сигнал в ответ на добавление 500 нМ фторвиллардиина (FW) в отсутствие (синяя кривая) и в присутствии (красная кривая) 50 мкМ дофамина; нормировано относительно первой добавки FW.
Также мы проверили, изменяется ли амплитуда кальциевого ответа, вызванного активацией потенциал-зависимых кальциевых каналов (ПЗКК), в ответ на дофамин. Было показано, что добавление дофамина не влияет на амплитуду и характер кальциевых сигналов, возникающих под действием 35 мМ хлорида калия, активирующего ПЗKK (рис. 5а, 5б). Так, кальциевый сигнал в ответ на вторую добавку KCl составляет 82 ± 11% относительно первой добавки, в присутствии дофамина это значение составляет 80 ± 10%.
Рис. 5.
Влияние дофамина на амплитуду кальциевого сигнала, вызванного агонистами глутаматных рецепторов и KCl. а – Кальциевый сигнал в ответ на добавление 35 мМ KCl в отсутствие (синяя кривая) и в присутствии (красная кривая) 50 мкМ дофамина; нормировано относительно первой добавки KCl. б – Амплитуда кальциевых сигналов в ответ на добавки NMDA, KA, AMPA и KCl в отсутствие (черные столбцы, –доф) и в присутствии (красные столбцы) 50 мкМ дофамина (+доф) относительно первой добавки на агонист, KCl1 – первая добавка KCl, KCl2 – вторая добавка KCl, * p < 0.05.
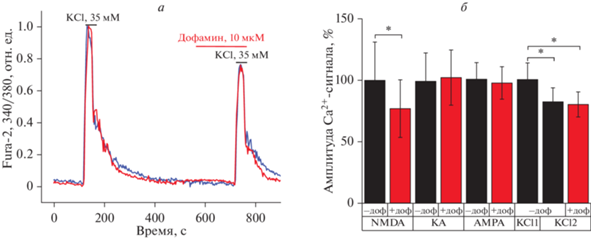
Таким образом, в работе было установлено, что при оценке амплитуд кальциевых сигналов в ответ на глутамат главной мишенью дофамина является NMDA-рецептор.
Участие дофаминовых рецепторов. Известно, что дофамин непосредственно подавляет активность NMDA-рецептора; его действие также может быть опосредовано активацией специфических дофаминовых рецепторов [4, 12].
Далее в работе мы проверили, способны ли антагонисты D1- и D2-подобных рецепторов предотвращать эффект дофамина на NMDA-индуцированный кальциевый сигнал в нейронах. На рис. 6 показано, что эффект дофамина не предотвращается в присутствии блокатора дофаминовых рецепторов типа D1 SKF-83566 (50 мкМ), о чем свидетельствует понижение амплитуды кальциевого ответа при действии NMDA совместно с дофамином на фоне SKF-83566 (76 ± 26%, рис. 6а, 6в). Добавление агониста D1-рецептора SKF-38393 (50 мкМ) не приводит к значительному изменению кальциевого сигнала в ответ на NMDA (98 ± ± 26%, рис. 6а, 6в).
Рис. 6.
Влияние агонистов и антагонистов D1- и D2-рецепторов на кальциевый ответ, вызванный NMDA. а – Кальциевый сигнал в ответ на 10 мкМ NMDA в присутствии 50 мкМ агониста D1-рецептора SKF-38393 (синяя кривая) и 50 мкМ блокатора D1-рецептора SKF-83588 совместно с дофамином (красная кривая); нормировано относительно первой добавки NMDA. б – Кальциевый сигнал в ответ на 10 мкМ NMDA в присутствии 100 нМ агониста D2-рецептора Quinpirole (красная кривая) и 1 мкМ блокатора D2-рецептора L-741,626 совместно с дофамином (синяя кривая); нормировано относительно первой добавки NMDA. в – Амплитуды кальциевых ответов, вызванных NMDA, в присутствии активаторов D1- и D2-рецепторов и их блокаторов совместно с дофамином (+доф) относительно первой добавки NMDA (100%). * p < 0.05.
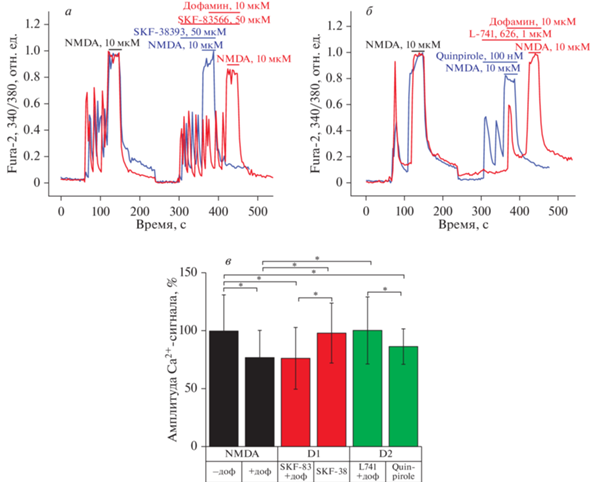
В случае действия ингибитора D2-рецептора L-741,626 (1 мкМ) не наблюдается эффект дофамина: кальциевый сигнал на вторую добавку NMDA составил 100 ± 28% (рис. 6б, 6в), а добавление к клеткам агониста D2-рецептора Quinpirole (100 нМ) уменьшает кальциевый сигнал, вызванный NMDA (86 ± 15%, рис. 6б, 6в). Это говорит о том, что действие дофамина опосредовано активацией Gi-сопряженного рецептора D2.
Таким образом, стало понятно, что дофамин оказывает свое угнетающее действие на NMDA-рецептор через активацию D2-рецептора.
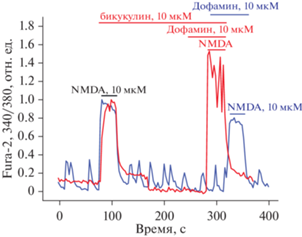
Ранее было показано, что эффект этого нейромедиатора может отменяться ингибиторами ГАМКА-рецепторов [13, 14]. В данной работе мы обнаружили, что 10 мкМ бикукулина (ингибитора ГАМКА-рецепторов) совместно с 10 мкМ дофамина не подавляют кальциевый сигнал в ответ на добавку NMDA (рис. 7).
Рис. 7.
Влияние активности ГАМКА-рецепторов на кальциевый ответ, вызванный NMDA. Кальциевый сигнал в ответ на 10 мкМ NMDA в присутствии 10 мкМ ингибитора ГАМКА-рецепторов бикукулина совместно с 10 мкМ дофамина (красная кривая) и в отсутствие бикукуллина, но с дофамином (синяя кривая); нормировано относительно первой добавки NMDA.
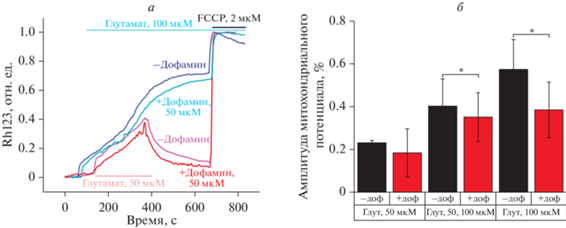
Дофамин защищает митохондрии от глутамат-индуцированной деполяризации. Известно, что в основе токсического действия глутамата лежит увеличение уровня цитозольного кальция в нейронах, для которого характерна так называемая отложенная кальциевая дисрегуляция (ОКД), возникающая из-за подавления производства ATP в митохондриях и неспособности клеток поддерживать ионный гомеостаз. При этом характерным признаком является деполяризация митохондрий [2]. Мы проверили, влияет ли дофамин на глутамат-индуцированную деполяризацию митохондрий. Было показано, что в присутствии дофамина глутамат вызывает митохондриальную деполяризацию на 15 ± 7% меньше, чем в контроле (рис. 8а, 8б). Так, в ответ на глутамат амплитуда изменения флуоресценции Rhodamine 123 (Rh123) составила 0.40 ± 0.12, тогда как при предварительной обработке дофамином это значение было 0.35 ± 0.11 (данные нормированы относительно сигнала в ответ на 2 мкМ FCCP, который вызывает полную деполяризацию митохондрий).
Рис. 8.
Влияние дофамина на глутамат-индуцированную деполяризацию митохондрий в нейронах гиппокампа крысы. а – Изменение флуоресценции родамина 123 (Rh123), вызванное 50 и 100 мкМ глутамата в присутствии (голубая и красная кривые) и в отсутствие (синяя и розовая кривые) 50 мкМ дофамина; нормировано относительно FCCP. б – Амплитуда изменения митохондриального потенциала, вызванного кратковременной добавкой 50 и 100 мкМ глутамата (Глут, 50 мкМ и 100 мкМ) в отсутствие (черные столбцы, –доф) и в присутствии (красные столбцы) 50 мкМ дофамина (+доф), представленные данные соответствуют схеме эксперимента с отмывкой глутамата (а – красная и розовая кривые). * p < 0.05.
Дофамин подавляет глутаматную эксайтотоксичность. Для того чтобы подтвердить защитную роль дофамина в условиях действия токсических доз глутамата, мы провели эксперименты по исследованию выживаемости нейронов. Было показано, что глутамат в концентрации 100 мкМ вызывает гибель 30.0 ± 1.7% нейронов в культуре при 24-часовом воздействии, что значительно превосходит данный показатель в контроле (клетки без воздействия – 18.1 ± 1.6%) (рис. 9). В случае, если в среде инкубации присутствует дофамин в концентрации 10 мкМ, количество поврежденных нейронов через 24 ч уменьшается на 53%. Так, этот показатель при совместном воздействии глутамата и дофамина составил 14.2 ± ± 1.6%. Инкубация с дофамином значимо не влияла на количество поврежденных клеток относительно контроля (17.1 ± 1.7%).
Рис. 9.
Влияние дофамина на выживаемость нейронов в нейроглиальной культуре гиппокампа крысы. Степень поврежденных клеток: мертвых (желтый) и апоптотических (голубой), относительно общего количества клеток в поле зрения (%) после 24 ч инкубации с 100 мкМ глутамата, 10 мкМ дофамина и с глутаматом и дофамином вместе. * p < 0.05 при сравнении всех нежизнеспособных клеток (мертвые + апоптотические).
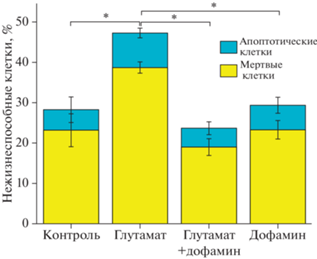
Таким образом, дофамин уменьшает глутамат-индуцированную деполяризацию митохондрий и улучшает жизнеспособность клеток в культуре, оказывая тем самым нейропротекторное действие на клетки.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В данной работе с использованием методов флуоресцентной микроскопии и ингибиторного анализа показано, что дофамин способен проявлять нейропротекторные свойства против глутаматной токсичности на примере культуры нейронов гиппокампа. В частности, дофамин в концентрации 10 мкМ снижает амплитуду кальциевого ответа на глутамат, в первую очередь, за счет подавления активности NMDA-рецептора – одного из подтипов ионотропных рецепторов глутамата. Кроме того, дофамин препятствует развитию глутамат-индуцированной деполяризации митохондрий нейронов и увеличивает выживаемость нейронов в присутствии токсических доз глутамата. Этот эффект глутамата зависит от активности D2-рецепторов, но не зависит от D1-рецепторов дофамина. Также показано, что эффект дофамина на глутамат-индуцированные изменения исследованных клеточных параметров во многом определяется активацией ГАМКергической системы, поскольку эффект дофамина также зависит и от активности ГАМКА-рецептора [14]. Было показано, что исследуемый эффект дофамина чувствителен к антагонисту ГАМКА-рецептора – бикукулину, поскольку присутствие в среде инкубации 10 мкМ бикукулина полностью подавляет действие дофамина на NMDA-индуцированный кальциевый сигнал.
Можно предположить несколько возможных механизмов, через которые дофамин реализует свой защитный эффект.
Во-первых, действие дофамина может проявляться за счет непосредственного взаимодействия с каналом NMDA-рецептора. В работах показано, что лиганды дофаминовых рецепторов, в том числе и сам дофамин, а также другие моноамины, могут взаимодействовать с NMDA-рецептором, выступая как потенциал-зависимые блокаторы открытого состояния канала и имея сайт связывания внутри поры NMDA-рецептора [15, 16].
Во-вторых, дофамин через рецептор вызывает гиперполяризацию нейронов. Еще в 1986 году на нейронах прилежащего ядра (nucleus accumbens) было показано, что дофамин способен вызывать D1-рецептор-зависимую гиперполяризацию в большинстве нейронов; кроме того, наблюдали деполяризацию, опосредованную активацией D2-рецепторов [17]. Наши эксперименты помогают расширить представления о механизмах этого взаимодействия нейротрансмиттеров. Так, одной из мишеней дофамина является именно D2-рецептор. Однако при блокировании двух типов рецепторов (D1- и D2-рецепторов) мы не видели отмену действия дофамина. Возможно, это связано с тем, что этот эффект опосредован не только этими типами рецепторов.
Также известно, что дофамин может влиять на AMPA-рецепторы, но это происходит на уровне модуляции их экстернализации. В этом случае дофамин активирует включение субъединиц GluR1 в плазматическую мембрану нейронов D1-рецептор-зависимым образом [18, 19]. Полученные нами данные о действии дофамина на кальциевые сигналы в ответ на активацию KA- и NMDA-рецепторов находятся в некотором противоречии с данными авторов [20]. В указанной работе было продемонстрировано, что хотя дофамин и защищает клетки от каинат-индуцированной нейротоксичности, но в этом эффекте не задействованы механизмы, связанные с модуляцией кальциевого гомеостаза. Более того, активность NMDA-рецептора вообще не регулировалась дофамином. В отличие от этого, мы обнаружили, что эффект дофамина связан с NMDA-рецептором, тогда как на кальциевый ответ при активации KA-рецептора дофамин не оказывал влияния.
ЗАКЛЮЧЕНИЕ
В работе показано, что дофамин обладает нейропротекторным действием против глутаматной нейротоксичности, поскольку добавление дофамина к культуре клеток гиппокампа снижает кальциевый ответ и митохондриальную деполяризацию, вызванные добавлением глутамата, и увеличивает количество жизнеспособных клеток после воздействия глутаматом. Мы также обнаружили, что дофамин, действуя через D2-подобные рецепторы, способен активировать NMDA-рецепторы и влиять на работу ГАМКергической системы.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа выполнена при поддержке Российского научного фонда (проект № 22-24-01043).
Соответствие принципам этики. Работу с лабораторными животными проводили в соответствии с положениями “Европейской конвенции о защите позвоночных животных, используемых для эксперимента и других научных целей” и “Руководством по работе с лабораторными животными ИБК РАН № 57 от 30.12.2011”.
Список литературы
Winklhofer K.F., Haass C. 2010. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta. 1802 (1), 29–44. https://doi.org/10.1016/j.bbadis.2009.08.013
Khodorov B. 2004. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Biophys. Mol. Biol. 86 (2), 279–351. https://doi.org/10.1016/j.pbiomolbio.2003.10.002
Meldrum B.S. 2000. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 130 (4S), 1007S–15S. https://doi.org/10.1093/jn/130.4.1007S
Gasiorowska A., Wydrych M., Drapich P., Zadrozny M., Steczkowska M., Niewiadomski W., Niewiadomska G. 2021. The biology and pathobiology of glutamatergic, cholinergic, and dopaminergic signaling in the aging brain. Front Aging Neurosci. 13, 654931. https://doi.org/10.3389/fnagi.2021.654931
Wang M., Wong A.H., Liu F. 2012. Interactions between NMDA and dopamine receptors: A potential therapeutic target. Brain Research. 1476, 154–163. https://doi.org/10.1016/j.brainres.2012.03.029
Беpежнов А.В., Кононов А.В., Федотова Е.И., Зинченко В.П. 2011. Способ выявления и характеристики лигандов ГАМК(А)-рецепторов с помощью кальций-чувствительных флуоресцентных зондов. Биофизика. 56 (4), 673–683.
Grynkiewicz G., Poenie M., Tsien R.Y. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260 (6), 3440–3450.
Бережнов А.В., Кононов А.В., Федотова Е.И., Зинченко В.П. 2013. Применение системы анализа изображения для характеристики лигандов ионотропных глутаматных рецепторов в культуре нейронов. Биол. мембраны. 30 (3), 179–188.
Kahlert S., Zündorf G., Reiser G. 2008. Detection of de- and hyperpolarization of mitochondria of cultured astrocytes and neurons by the cationic fluorescent dye rhodamine 123. J. Neurosci. Methods. 171 (1), 87–92. https://doi.org/10.1016/j.jneumeth.2008.02.0157
Vaarmann A., Kovac S., Holmström K.M., Gandhi S., Abramov A.Y. 2013. Dopamine protects neurons against glutamate-induced excitotoxicity. Cell Death Dis. 4 (1), e455. https://doi.org/10.1038/cddis.2012.194.9
Ferreira I.L., Duarte C.B., Carvalho A.P. 1996. Ca2+ influx through glutamate receptor-associated channels in retina cells correlates with neuronal cell death. Eur. J. Pharmacol. 302 (1–3), 153–162. https://doi.org/10.1016/0014-2999(96)00044-1
Castro N.G., de Mello M.C., de Mello F.G., Aracava Y. 1999. Direct inhibition of the N-methyl-D-aspartate receptor channel by dopamine and (+)-SKF38393. Br. J. Pharmacol. 126 (8), 1847–1855. https://doi.org/10.1038/sj.bjp.0702479
Lau C.G., Takeuchi K., Rodenas-Ruano A., Takayasu Y., Murphy J., Bennett M.V., Zukin R.S. 2009. Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem. Soc. Trans. 37 (Pt 6), 1369–1374. https://doi.org/10.1042/BST0371369
Berezhnov A.V., Fedotova E.I., Sergeev A.I., Teplov I.Y., Abramov A.Y. 2021. Dopamine controls neuronal spontaneous calcium oscillations via astrocytic signal. Cell Calcium. 94, 102359. https://doi.org/10.1016/j.ceca.2021.102359
Cui C., Xu M., Atzori M. 2006.Voltage-dependent block of N-methyl-D-aspartate receptors by dopamine D1 receptor ligands. Mol. Pharmacol. 70 (5), 1761–1770. https://doi.org/10.1124/mol.106.028332
Masuko T., Suzuki I., Kizawa Y., Kusama-Eguchi K., Watanabe K., Kashiwagi K., Igarashi K., Kusama T. 2004. Monoamines directly inhibit N-methyl-D-aspartate receptors expressed in Xenopus oocytes in a voltage-dependent manner. Neurosci Lett. 371 (1), 30–33. https://doi.org/10.1016/j.neulet.2004.08.030
Uchimura N., Higashi H., Nishi S. 1986. Hyperpolarizing and depolarizing actions of dopamine via D-1 and D-2 receptors on nucleus accumbens neurons. Brain Res. 375 (2), 368–372. https://doi.org/10.1016/0006-8993(86)90760-2
Mangiavacchi S., Wolf M.E. 2004. D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J. Neurochem. 88 (5), 1261–1271. https://doi.org/10.1046/j.1471-4159.2003.02248.x
Sun X., Zhao Y., Wolf M.E. 2005. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J. Neurosci. 25 (32), 7342–7351. https://doi.org/10.1523/JNEUROSCI.4603-04.2005
Amano T., Ujihara H. Matsubayashi H., Sasa M., Yokota T., Tamura Y., Akaike A. 1994. Dopamine-induced protection of striatal neurons against kainate receptor-mediated glutamate cytotoxicity in vitro. Brain Res. 655 (1–2), 61–69. https://doi.org/10.1016/0006-8993(94)91597-0
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии