

Биологические мембраны: Журнал мембранной и клеточной биологии, 2022, T. 39, № 6, стр. 457-473
Миелоидная дифференцировка повышает устойчивость лейкозных клеток к TRAIL-индуцированной гибели путем снижения экспрессии рецепторов DR4 и DR5
Я. В. Ломовская a, *, М. И. Кобякова a, А. С. Сенотов a, И. С. Фадеева a, b, А. И. Ломовский a, К. С. Краснов a, b, Д. Ю. Штатнова a, b, В. С. Акатов a, b, Р. С. Фадеев a, b
a Институт теоретической и экспериментальной биофизики РАН
142290 Пущино, Московская обл., Россия
b Пущинский государственный естественно-научный институт
142290 Пущино, Московская обл., Россия
* E-mail: yannalomovskaya@gmail.com
Поступила в редакцию 18.04.2022
После доработки 20.06.2022
Принята к публикации 22.06.2022
- EDN: JIZHWG
- DOI: 10.31857/S023347552206010X
Аннотация
Изучение механизмов устойчивости опухолевых клеток к TRAIL-индуцированной гибели остается актуальной задачей, так как данный цитокин является важным высокоизбирательным молекулярным эффектором противоопухолевого иммунитета. В нашей работе показано, что у клеток лейкоза человека THP-1, HL-60 и K562 индукция экзогенными факторами миелоидной дифференцировки in vitro во всех направлениях миелопоэза, кроме эритроидоподобного, повышает устойчивость к TRAIL-индуцированной гибели, снижая экспрессию рецепторов DR4 и DR5 на клеточной поверхности. Было установлено также, что ONC201, туникамицин и SAHA (субероиланилид гидроксамовой кислоты), способные вызывать повышение экспрессии DR5 у лейкозных клеток, подавляли их TRAIL-резистентность, индуцированную факторами дифференцировки. Полученные результаты представляют интерес для разработки препаратов и стратегий для повышения эффективности лечения миелоидных лейкозов.
ВВЕДЕНИЕ
Цитокин TRAIL/Apo2L является важным молекулярным эффектором противоопухолевого иммунитета благодаря его высокоизбирательной способности индуцировать рецептор-опосредованную апоптотическую гибель опухолевых, но не здоровых клеток организма [1–6]. Однако возникновение TRAIL резистентности у части опухолевых клеток ограничивает потенциал противоопухолевого иммунитета и возможность использования этого цитокина в терапии злокачественных заболеваний [7, 8]. Формирование устойчивости у лейкозных клеток к противоопухолевому иммунитету, опосредованному действием TRAIL, может являться одной из основных причин прогрессии миелоидных лейкозов [8–11]. Поэтому на сегодняшний день актуальным является изучение механизмов, опосредующих снижение чувствительности лейкозных клеток к TRAIL-индуцированной гибели.
Ранее нами было показано, что лейкозные клетки в высокоплотных культурах, которые могут отражать условия клеточного микроокружения в нишах костного мозга, способны приобретать резистентность к TRAIL-индуцированной гибели на фоне формирования у них признаков, характерных для воспалительной активации [12] или макрофагоподобного фенотипа [13]. В то же время появление признаков дифференцировки у лейкозных клеток под воздействием экзогенных стимулов соотносится с формированием у них резистентности к TRAIL-опосредованному апоптозу [14, 15]. Это важно учитывать при использовании стратегий дифференцирующей терапии, так как обнаружено участие дифференцированных лейкозных клеток в рецидиве заболевания [16–18]. Кроме того, показана возможность возникновения устойчивости и рецидива миелоидного лейкоза, обусловленная формированием злокачественных высокодифференцированных опухолевых клонов, в том числе под воздействием используемых в клинике химиопрепаратов [19–21]. Исходя из вышесказанного, можно предположить, что в условиях in vivo стрессовое микроокружение клеток в опухолях и/или действие на них противоопухолевых агентов различной природы может способствовать приобретению лейкозными клетками признаков дифференцировки, что сопряжено с усилением защиты лейкозных клеток от иммунной системы и с опухолевой прогрессией.
В данной работе мы исследовали влияние индукции дифференцировки лейкозных клеток линий THP-1, HL-60 и K562 в разных направлениях миелопоэза на их чувствительность к TRAIL-индуцированной гибели. В работе показано, что индукция моноцитарной, макрофагальной, гранулоцитарной, мегакариоцитарной, но не эритроидоподобной дифференцировки лейкозных клеток in vitro повышает их устойчивость к цитотоксическому действию лиганда TRAIL. Представлены данные об изменении экспрессии рецепторов DR4 и DR5 у лейкозных клеток в процессе миелоидной дифференцировки. Показана возможность подавления TRAIL-резистентности дифференцированных лейкозных клеток с помощью индукторов стресса эндоплазматического ретикулума (ЭПР-стресса).
МАТЕРИАЛЫ И МЕТОДЫ
Клеточные культуры. В работе использовали клетки миелоидного лейкоза человека ТНР-1, HL-60, K562, полученные из ATCC (США). Клетки культивировали в среде RPMI 1640/F12 (Sigma, США) с добавлением 10% эмбриональной телячьей сыворотки (FBS) (Gibco, США), 80 мкг/мл гентамицина сульфата (Sigma), 20 мкг/мл дифлюкана (Pfizer, США) при 37°С, в условиях 5% содержания СО2 в воздухе. Для индукции моноцитарной дифференцировки клетки ТНР-1 инкубировали с 1 мкМ транс-ретиноевой кислоты (ATRA) (Sigma) в течение 96 ч [22]. Для индукции макрофагальной дифференцировки клетки ТНР-1 инкубировали с 200 нM форболового эфира (PMA) (Sigma) в течение 96 ч [23, 24]. Для индукции гранулоцитарной дифференцировки клетки HL-60 инкубировали с 1 мкМ ATRA в течение 96 ч, а также с 1.25% диметилсульфоксида (DMSO) (Sigma) в культуральной среде в течение 96 ч [25–27]. Для индукции мегакариоцитарной дифференцировки клетки K562 инкубировали с 10 нМ PMA в течение 96 ч [28–31]. Для индукции эритроидоподобной дифференцировки клетки К562 инкубировали с 1 мМ бутирата натрия (SB) в течение 168 ч, а также с 50 мкМ гемина (Hem) (Sigma) в течение 96 ч [32–35].
Тестирование клеточных культур на заражение микоплазмой проводили с помощью набора MycoFluor Mycoplasma Detection Kit (ThermoFisher Scientific, США). Заражение культур клеток микоплазмой не выявлено.
Моноциты были получены из мононуклеарной фракции периферической крови здоровых доноров с использованием набора MojoSort Human Pan Monocyte Isolation Kit (BioLegend, США) в соответствии с рекомендациями производителя. Макрофаги получали из моноцитов периферической крови человека. Для этого моноциты культивировали в среде DMEM/F12 с добавлением 10% FBS, 40 мкг/мл гентамицина при 37°С, в условиях 5% содержания СО2 в увлажненном воздухе. Культуральную среду заменяли на свежую через 3 дня после посева клеток с добавлением 2% FBS. Через 14 дней культивирования моноциты поляризовались в макрофаги, и их использовали в экспериментах. Коктейль ферментов Accutase (Sigma) применяли для отделения макрофагов от поверхности культурального пластика.
Гранулоциты были получены путем центрифугирования гепаринизированной периферической крови человека в градиенте плотности с использованием Ficoll-Paque PREMIUM 1.084 (Sigma), 1600g в течение 20 мин. После центрифугирования для выделения гранулоцитов из смеси с эритроцитами применялся буфер хлорида аммония для лизиса эритроцитов (0.155 мМ хлорид аммония; 10 мM гидрокарбонат калия, 0.1 мM EDTA с рН 7.4). Гранулоциты культивировали в полной ростовой среде RPMI/F12 c 10% FBS с добавлением антибиотиков при 37°С в условиях 5% содержания СО2 в увлажненном воздухе.
Эритроциты и тромбоциты получали путем центрифугирования гепаринизированной периферической крови человека при 250g в течение 15 мин. Эритроциты отбирали из слоя осажденных эритроцитов, добавляли полную ростовую среду RPMI/F12 c 10% FBS и использовали в последующих экспериментах. Тромбоциты выделяли из слоя плазмы, образовавшегося после центрифугирования. Плазму разбавляли в соотношении 1 : 8 с фосфатно-солевым буфером (PBS) и центрифугировали 10 мин при 1200g [36]. Осадок тромбоцитов собирали в среду RPMI/F12 c 10% FBS и использовали в дальнейших экспериментах.
Протокол получения белка izTRAIL. Для получения растворимой тримерной формы белка izTRAIL был синтезирован мотив изолейциновой “молнии” и ген izTRAIL, которые были клонированы в плазмидный вектор pET101 (Novagen, США). Полученным вектором трансформировали клетки E. сoli BL21 (DE3) и методом микробного синтеза с последующей очисткой металл-аффинной хроматографией получили тримерную форму izTRAIL с молекулярной массой около 80 кДа [6].
Иммунофенотипирование и анализ экспрессии рецепторов к TRAIL. Для изучения экспрессии кластеров дифференцировки (CD) клетки собирали из культуральных флаконов, отмывали в буфере для окрашивания клеток (BioLegend) путем центрифугирования при 300g 5 мин. Окрашивание проводили с использованием панели моноклональных антител (BioLegend): APC anti-human CD11b, FITC anti-human CD11c, anti-human CD14, PE anti-human CD36, FITC anti-human CD45, FITC anti-human CD163, FITC anti-human CD68, FITC anti-human CD66, PE anti-human CD284, PE anti-human CD68, PE anti-human CD41, PE anti-human CD61, PE anti-human CD235a, APC anti-human CD71, PE anti-human HLA-DR. Для анализа экспрессии TRAIL-рецепторов использовали: PE anti-human TRAIL-R1 (CD261) (BD Bioscience, США), APC anti-human CD262 (TRAIL-R2) (BioLegend), FITC Anti-human CD263 (TRAIL-R3) (BD Bioscience), PE anti-human CD264 (TRAIL-R4) (BioLegend). Для определения неспецифического связывания клетки окрашивали контрольными антителами изотипа: APC Mouse IgG1 k isotype Ctrl, FITC Mouse IgG1 k isotype Ctrl, PE Mouse IgG1 k isotype, PE Mouse IgG2a k isotype, APC Mouse IgG2a k isotype, полученными от BioLegend. Окрашивание проводили при комнатной температуре в темноте в течение 30 мин. После окрашивания клетки фиксировали 2% раствором параформальдегида. Анализ экспрессии CD проводили с помощью проточного цитометра BD Accuri C6 (BD Biosciences). Гистограммы экспрессии поверхностных маркеров формировали с помощью программы FlowJo v10 (BD Biosciences).
Анализ фагоцитарной активности и ДНК цитометрия. Фагоцитарную активность оценивали через 2 ч инкубации клеток в ростовой среде с добавлением 1 мг/мл pHrodo Green Escherichia coli (ThermoFisher Scientific). Для контроля неспецифического окрашивания клетки инкубировали с 10 мкг/мл цитохалазина D (Sigma) в течение 30 мин в СО2-инкубаторе, затем добавляли 1 мкг/мл pHrodo Green E. coli и продолжали инкубацию еще 2 ч. Флуоресценцию измеряли с помощью проточного цитометра BD Accuri C6. Распределение по фазам клеточного цикла оценивали с помощью анализа содержания ДНК на проточном цитометре. Для оценки содержания клеточной ДНК клетки суспендировали в PBS, фиксировали 70% этанолом и окрашивали 1 мкг/мл йодида пропидия (Sigma). Клеточный цикл анализировали с использованием программного обеспечения ModFit LT 4.1 (Verity Software House, США).
Анализ окислительной активности и продукции гемоглобина. Индуцируемую и конститутивную внутриклеточную окислительную активность оценивали с использованием зонда DCFH-DA (возбуждение: 485 нм, эмиссия: 530 нм). В начале клетки нагружали флуоресцентным зондом путем инкубирования в ростовой среде с 10 мкМ DCFH-DA в течение 15 мин. Далее для оценки индуцибельной окислительной активности часть клеток после нагрузки отмывали в холодном PBS и инкубировали в свежей среде с 500 нМ PMA в течение 30 мин. Параллельно для оценки конститутивной продукции АФК часть клеток после нагрузки отмывали в холодном PBS и инкубировали в отсутствие PMA в течение 30 мин. Затем клетки повторно отмывали в PBS и анализировали флуоресценцию с помощью проточного цитометра BD Accuri C6. Измерение гемоглобина в клетках К562 проводили по методике, описанной в статье [32]. Равные количества клеток промывали холодным PBS и лизировали в течение 20 мин в лизирующем буфере (0.2% Triton X-100 (Sigma) в PBS). Лизаты центрифугировали 15 мин при 1500g, 10 мкл супернатанта инкубировали с 2 мл раствора ТМБ (3,3',5,5'-тетраметилбензидин (Sigma-Aldrich, США); 5 мг/мл в ледяной уксусной кислоте) и c 2 мл 30% H2O2 в течение 10 мин при комнатной температуре. Реакцию останавливали добавлением раствора 0.16 М серной кислоты. Адсорбцию измеряли при 450 нм с использованием планшетного спектрофлюориметра Infinite F200 (Tecan, Австрия). Содержание гемоглобина в пг на клетку рассчитывали по стандартной кривой гемоглобина и данные отображали как кратное увеличение по сравнению с уровнем гемоглобина в контрольных клетках К562.
Анализ цитотоксичности izTRAIL. Клетки высевали в 96-луночные планшеты в количестве 5×103 клеток в 0.1 мл питательной среды на лунку. Препараты (izTRAIL, ONC201, SAHA (субероиланилидом гидроксамовой кислоты) и туникамицин) добавляли в культуры через 24 ч после посева клеток. Цитотоксичность препаратов и их сочетаний оценивали по соотношению количества живых клеток в опытной и контрольной (без препаратов) культурах через 24 ч после добавления препаратов. Количество живых клеток оценивали по восстановлению резазурина. Клеточные культуры инкубировали с резазурином (30 мкг/мл) в течение 4 ч при 37°С и 5% СО2, а затем измеряли интенсивность флуоресценции инкубационной среды при длине волны возбуждения 532 нм и поглощения 590 нм, используя планшетный спектрофлуориметр Infinity F200.
Статистическая обработка данных. Результаты представляли в виде среднего ± стандартное отклонение (M ± SD). Опыты проводили не менее чем в трех повторах (n ≥ 3). Статистическую значимость отличия определяли с помощью одностороннего ANOVA с последующим множественным сравнением Холма–Сидака, р < 0.05.
РЕЗУЛЬТАТЫ
Результаты иммунофенотипирования лейкозных клеток линий ТНР-1, HL-60, K562, дифференцированных экзогенными факторами
Миелопоэз заключается в дифференцировке миелоидных клеток в моноцитарном, макрофагальном, гранулоцитарном, эритроидном и мегакариоцитарном направлениях. Ряд экзогенных факторов могут индуцировать формирование в лейкозных клетках признаков дифференцировки в указанных направлениях. Каждое направление дифференцировки характеризуется определенным набором поверхностных маркеров и формированием специфических функций у клеток. Важнейшим показателем дифференцировки является изменение иммунофенотипа, т.е. приобретение специфических поверхностных маркеров – кластеров дифференцировки (CD). Иммунофенотипирование клеток THP-1ATRA и ТНР-1PMA проводили по 10 основным маркерам моноцитарно-макрофагальной дифференцировки в сравнении с контрольными клетками ТНР-1, моноцитами и макрофагами (табл. 1). Клетки THP-1ATRA являлись положительными по основным моноцитарным маркерам CD11b, CD11c, CD14, CD284, которые отсутствовали у контрольных клеток THP-1. Процент CD68+ популяции у клеток THP-1ATRA не отличался от моноцитов. Также процент HLA-DR+ клеток THP-1ATRA увеличился до 53 ± 5% относительно 21 ± 1% у контрольных клеток. Все клетки THP-1ATRA, так же как и моноциты, являлись СD68+ в отличие от контрольных клеток ТНР-1, у которых только 51 ± 3% клеток несли данный маркер. Нужно отметить, что у клеток ТНР-1ATRA также возрастал уровень экспрессии CD64 в пересчете на клетку, что выражалось в увеличении средней интенсивности флуоресценции (СИФ) с 1465 ± 43 произвольных единиц (далее пр. ед.) у ТНР-1 до 30 264 ± ± 2113 пр. ед. у ТНР-1ATRA. Также наряду с увеличением числа клеток THP-1ATRA, несущих СD68 и HLA-DR, возрастал уровень экспрессии этих маркеров в пересчете на одну клетку с 2700 ± ± 172 пр. ед. до 20546 ± 3420 пр. ед. и с 814 ± 54 пр. ед. до 3756 ± 213 пр.ед. соответственно по сравнению с контрольными клетками ТНР-1. В отличие от контрольных клеток ТНР-1 клетки THP-1PMA имели маркеры СD11c, CD14, CD36, CD284, что также характерно для макрофагов (табл. 1). Процент CD11c+, а также CD14+ клеток в популяции ТНР-1РМА был больше, чем у макрофагов. Экспрессия СD36 отсутствовала у контрольных клеток ТНР-1 и появлялась у ТНР-1РМА, однако процент СD36+ клеток в популяции ТНР-1PMA был ниже, чем у макрофагов. В свою очередь, процент CD284+ клеток у ТНР-1PMA был небольшим и близок к тому, что выявлялся у макрофагов. Таким образом, клетки ТНР-1ATRA и THP-1PMA имели различный иммунофенотип, но у обоих клеточных вариантов наблюдалось появление дифференцировочных маркеров, характерных для моноцитарной и макрофагальной дифференцировки соответственно.
Таблица 1.
Процент клеток, несущих CD-маркеры моноцитарно-макрофагальной дифференцировки, в культурах клеток THP-1 ATRA, THP-1PMA, THP-1, у моноцитов и макрофагов
| CD | THP-1 | THP-1ATRA | THP-1PMA | Моноциты | Макрофаги |
|---|---|---|---|---|---|
| Интегрин αM (CD11b) | – | 88 ± 1 | – | 93 ± 1 | 90 ± 3 |
| Интегрин αX (CD11c) | – | 48 ± 3 | 90 ± 1 | 90 ± 2 | 66 ± 1 |
| Корецептор LPS (СD14) | – | 26 ± 2 | 95 ± 1 | 81 ± 2 | 47 ± 2 |
| SCARB3 (CD36) | – | – | 20 ± 1 | 95 ± 1 | 97 ± 1 |
| PTPRC (CD45) | 96 ± 8 | 99 ± 1 | 99 ± 1 | 99 ± 1 | 99 ± 1 |
| Fc-γ рецептор 1 (СD64) | 95 ± 1 | 99 ± 1 | 28 ± 4 | 85 ± 1 | 55 ± 4 |
| Макросиалин (CD68) | 51 ± 3 | 94 ± 4 | – | 99 ± 1 | 95 ± 4 |
| TLR4 (CD284) | – | 72 ± 9 | 15 ± 1 | 49 ± 6 | 9 ± 1 |
| Рецептор для гемоглобин- гаптоглобинового комплекса (СD163) | – | – | – | 75 ± 1 | – |
| MHC II (HLA-DR) | 21 ± 1 | 53 ± 5 | – | 73 ± 1 | 59 ± 2 |
Иммунофенотип клеток HL-60DMSO и HL-60ATRA был характерным для гранулоцитоподобных клеток (табл. 2). У этих клеток появлялись маркеры CD11b, CD11c и увеличивалась субпопуляция клеток, несущих CD66, в сравнении с контрольными клетками HL-60. Сильнее всего менялся иммунофенотип клеток, обработанных ATRA. Процент клеток CD11b+ и CD66+ у них достоверно не отличался от соответствующих значений для гранулоцитов и включал почти всю популяцию клеток. Количество положительных по CD11c клеток HL-60ATRA было почти в 4 раза больше в сравнении с клетками HL-60DMSO, 43 ± 4% и 12 ± 1% соответственно. На клетках HL-60DMSO выявлялась более высокая представленность исследуемых маркеров в сравнении с контрольными клетками HL-60, но меньшая, чем на клетках HL-60ATRA и гранулоцитах (данные не представлены). Таким образом, хотя клетки HL-60DMSO и HL-60ATRA имели одинаковый гранулоцитоподобный иммунофенотип, степень представленности СD11b, CD66 и СD11 на клетках и процент клеток, положительных по этим маркерам, были выше у клеток HL-60ATRA, что может указывать на более выраженный дифференцированный иммунофенотип этих клеток.
Таблица 2.
Процент клеток, несущих CD-маркеры гранулоцитарной дифференцировки, в культурах клеток HL-60DMSO и HL-60ATRA, HL-60 и у гранулоцитов
| CD | HL-60 | HL-60DMSO | HL-60ATRA | Гранулоциты |
|---|---|---|---|---|
| Интегрин αM (CD11b) | – | 78 ± 3 | 95 ± 1 | 99 ± 1 |
| Интегрин αX (CD11c) | – | 12 ± 1 | 43 ± 4 | 94 ± 1 |
| CEACAM3 (CD66) | 41 ± 1 | 74 ± 10 | 90 ± 5 | 99 ± 1 |
Мегакариоцитоподобная дифференцировка сопровождалась появлением у клеток K562PMA двух главных маркеров мегакариоцитов и зрелых тромбоцитов – CD41 и CD61 [37], которые полностью отсутствовали у контрольных клеток (табл. 3).
Таблица 3.
Процент клеток, несущих CD-маркеры мегакариоцитарной дифференцировки в культурах клеток K562PMA, K562 и у тромбоцитов
| CD | K562 | K562 PMA | Тромбоциты |
|---|---|---|---|
| Интегрин αIIb (CD41) | – | 20 ± 1 | 99 ± 1 |
| Интегрин β3 (CD61) | – | 86 ± 2 | 98 ± 2 |
Экспрессия эритроидного маркера CD71 присуща всем клеткам эритроидного ростка, кроме самой зрелой формы – эритроцитов, а наличие CD235a характерно для всех эритроидных клеток, начиная с проэритробластов [38]. Мы показали, что популяция как дифференцированных клеток K562Hem и К562SB, так и контрольных клеток К562 несли данные маркеры (табл. 4). Однако представленность этих CD маркеров, выраженная в пр. ед. СИФ, на дифференцированных клетках была выше и отличалась от того, что было у контрольных клеток. Наибольшее значение СИФ как для CD71, так и для CD235a среди исследуемых вариантов имели клетки К562SB (рис. 1а). Таким образом, иммунофенотип дифференцированных клеток К562SB и К562Hem выражал маркеры эритроподобной дифференцировки.
Таблица 4.
Процент клеток, несущих CD-маркеры эритроидной дифференцировки в культурах клеток K562Hem, K562SB, K562 и у эритроцитов
| CD | K562 | K562Hem | K562SB | Эритроциты |
|---|---|---|---|---|
| Рецептор трансферрина (CD71) | 88 ± 9 | 74 ± 4 | 97 ± 1 | – |
| Гликофорин А (CD235a) | 99 ± 1 | 93 ± 3 | 97 ± 1 | 100 |
Рис. 1.
а – Гистограммы экспрессии CD71 и СD235a на клетках К562, К562Hem и K562SB, окрашенных антителами, конъюгированными с флуорохромами. б – Гистограммы указанных на рисунке клеток после фагоцитирования ими флуоресцентных комплексов pHrodo Green E. сoli, представленные гистограммы являются репрезентативными для трех независимых экспериментов. в – Конститутивная и РМА-индуцибельная окислительная активность указанных клеток. СИФ – средняя интенсивность флуоресценции клеток, окращенных зондом DCFH-DA, выраженная в произвольных единицах (пр. ед.). г – Процент популяции клеток К562 и К562РМА в G0 и G1 (G0/G1), G2 и М (G2/М) фазах клеточного цикла, а также содержащих количество ДНК большее, чем в G2/М фазах (полиплоидные клетки). Окраска иодидом пропидия; д – Продукция гемоглобина клетками K562Hem и K562SB, выраженная как кратная таковой для контрольных клеток К562. * – p <0.05 по сравнению с клетками К562.
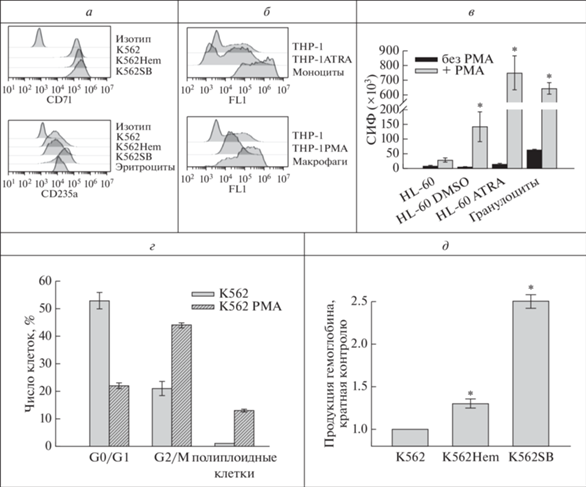
Было также обнаружено, что лейкозные клетки в процессе миелоидной дифференцировки приобретают специфические функциональные маркеры. Фагоцитоз является одной из основных функций моноцитов и макрофагов, а наличие в клетках THP-1ATRA и THP-1PMA мембранных рецепторов CD14 и CD284, входящих в состав мультимолекулярного рецепторного комплекса, распознающего липополисахарид клеточной стенки бактерий, указывает на потенциальную способность данных клеток осуществлять фагоцитоз. Для клеток THP-1ATRA и THP-1PMA была обнаружена высокая фагоцитарная активность по отношению к комплексам pHrodo Green E. coli, сопоставимая с моноцитами и макрофагами, что отражено на гистограммах (рис. 1б). Для гранулоцитов была показана высокая индуцируемая окислительная активность, что также характерно для клеток HL-60DMSO и HL-60ATRA, в отличие от недифференцированных клеток HL-60 (рис. 1в). Помимо многоядерности, для мегакариоцитоподобных клеток K562РМА была характерна полиплоидность, что отражается в увеличении количества ДНК в клетках и является маркером мегакариоцитов (рис. 1г). Основным функциональным маркером эритроидных клеток является продукция гемоглобина [38], и у дифференцированных клеток К562Hem, а также у K562SB она была в несколько раз выше, чем у контрольных клеток (рис. 2д). У K562Hem продукция Hb была выше в 1.36 ± 0.03 раза, чем у клеток K562, а у клеток К562SB – в 2.45 ± 0.04 раза, что указывает на более выраженный эритроидоподобный фенотип клеток K562SB.
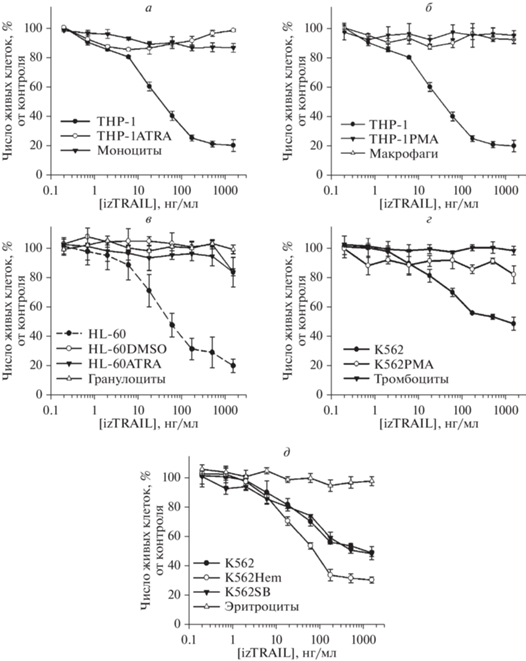
Дифференцировка лейкозных клеток экзогенными факторами во всех миелоидных направлениях, кроме эритроидного, сопровождается повышением их устойчивости к TRAIL-индуцированной гибели
Было установлено, что приобретение моноцитарно-макрофагальных черт у клеток ТНР-1 сопровождается снижением их чувствительности к TRAIL-индуцированной гибели. Не было обнаружено токсического действия белка izTRAIL даже в высоких концентрациях (до 1.5 мкг/мл) на клетки ТНР-1ATRA и моноциты, что свидетельствует об их TRAIL-резистентности (рис. 2а). Аналогично, дифференцировка в макрофагоподобном направлении сопровождалась формированием TRAIL-резистентности в клетках THP-1PMA. Такая же полная TRAIL-резистентность была характерна для макрофагов (рис. 2б). Чувствительность клеток HL-60 к действию izTRAIL также изменилась после их гранулоцитоподобной дифференцировки. Клетки HL-60DMSO и HL-60ATRA приобрели резистентность к TRAIL-индуцированной гибели (рис. 2в). При максимальной используемой концентрации izTRAIL (1.5 мкг/мл) 84 ± 10% клеток HL-60DMSO и 83 ± ± 2% клеток HL-60ATRA являлись нечувствительными к izTRAIL, что примерно в 4 раза больше в сравнении с клеточными культурами HL-60, в которых было только 20 ± 1% TRAIL-нечувствительных клеток.
Клетки K562 обладали повышенной в сравнении c ТHP-1 и HL-60 устойчивостью к TRAIL-индуцированной гибели. Для них концентрация полумаксимального ингибирования (IC50) белка izTRAIL составила 1.5 мкг/мл, т.е. максимальную используемую дозу (рис. 2г). Дифференцированные клетки K562PMA, как и тромбоциты, были TRAIL-резистентным (рис. 2г). В то же время дифференцировка в эритроподобном направлении клеток K562 не способствовала повышению их TRAIL-устойчивости (рис. 2д). Клетки К562, обработанные гемином, стали более чувствительны к izTRAIL. При концентрации белка izTRAIL 1.5 мкг/мл процент живых клеток в культурах К562Hem был равен 30 ± 2%, а для контрольных культур К562 и для K562SB он равнялся 52 ± 4% (рис. 2д).
Таким образом, наши результаты показывают повышение TRAIL-устойчивости лейкозных клеток при их дифференцировке во всех миелоидных направлениях, кроме эритроидоподобного.
Дифференцировка лейкозных клеток по всем миелоидным направлениям, кроме эритроидного, сопровождается снижением доли клеток, несущих DR4 и DR5
Известно, что изменение экспрессии рецепторов к TRAIL на клетках влияет на их чувствительность к цитотоксическому действию данного цитокина [1, 8]. Поэтому мы изучили, как дифференцировка в миелоидном направлении лейкозных клеток влияет на экспрессию TRAIL-рецепторов. Было обнаружено, что изменение TRAIL-чувствительности у дифференцированных клеток согласуется с представленностью на них проапоптотических TRAIL-рецепторов DR4 и DR5. Экспрессия антиапоптотических рецепторов TRAIL (DcR1 и DcR2) отсутствовала во всех исследованных типах клеток. Мы проанализировали процент субпопуляций, несущих TRAIL-рецепторы DR4 и DR5 (DR4+/DR5+), только DR4 (DR4+/DR5–) или только DR5 (DR4–/DR5+), а также не несущих ни один из этих рецепторов (DR4–/DR5–), во всех дифференцированных линиях клеток и сравнили их с контрольными (недифференцированными) клетками, а также с соответствующими дифференцированными клетками организма. Для этого проводили окрашивание клеток совместно антителами к DR4 и DR5 и анализировали сдвиг цитограммы флуоресценции этой популяции относительно клеток, окрашенных двумя контрольными изотипами. Было обнаружено, что в популяции THP-1ATRA по сравнению с THP-1 наблюдалось значительное (p < 0.05), более чем в 3 раза, снижение количества дважды позитивных клеток DR4+/ DR5+ (с 67 ± 3% до 15 ± 6% соответственно), а также уменьшение популяции DR4+/DR5– с 22 ± 2% до 11 ± 1% (рис. 3а). У моноцитов полностью отсутствовали рецепторы DR5. Наиболее значимо было увеличение дважды негативной популяции DR4–/DR5– у клеток ТНР-1ATRA (более чем в 10 раз) в сравнении с ТНР-1, у которых только 6 ± 1% клеток не имели ни DR4, ни DR5. У моноцитов процент популяции DR4–/DR5– также был намного больше (>17 раз), чем у ТНР-1 клеток. У макрофагоподобных клеток ТНР-1РМА и макрофагов было выявлено полное отсутствие популяции DR4+/DR5+. У клеток ТНР-1РМА полностью отсутствовали рецепторы DR4. Большинство клеток ТНР-1РМА (85 ± 1%), а также макрофагов (98 ± 1%) не имели проапоптотических рецепторов (рис. 3б).
Рис. 3.
Процент клеток, которые экспрессируют TRAIL-рецепторы DR4 и DR5 (DR4+/DR5+), только DR4 (DR4+/DR5–) или только DR5 (DR4–/DR5+) или ни DR4, ни DR5 (DR4–/DR5–): а – в популяциях THP-1, THP-1ATRA и моноцитов; б – в популяциях THP-1PMA в сравнении с THP-1 и макрофагами; в – в популяциях HL-60DMSO и HL-60ATRA в сравнении с Hl-60 и гранулоцитами; г – в популяциях клеток К562РМА и K562 в сравнении с тромбоцитами; д – в клетках K562Hem, K562SB в сравнении с К562 и эритроцитами; * – p < 0.05 по сравнению с соответствующими контрольными клетками.
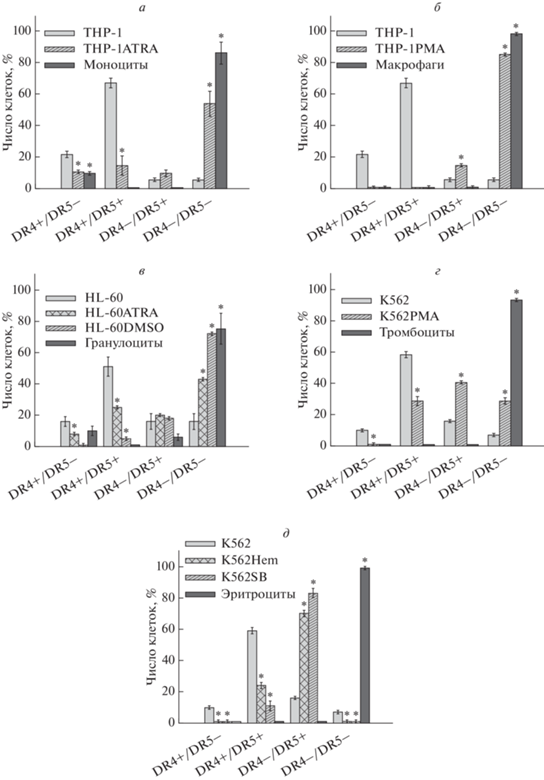
При гранулоцитоподобной дифференцировке мы также обнаружили потерю рецепторов DR4 и DR5 (рис. 3в). Было установлено, что для 75 ± ± 10% гранулоцитов характерно одновременное отсутствие обоих рецепторов на клетках. Тенденция к увеличению дважды негативной DR4–/DR5– популяции наблюдалась у HL-60DMSO (72 ± 1%) и у HL-60ATRA (43 ± 1%). У контрольных клеток HL-60 данная популяция составляла только 16 ± 5%, а большинство клеток (51 ± 6%) несли оба рецептора DR4 и DR5 на своей мембране. У клеток HL-60ATRA популяция DR4+/DR5+ была вдвое меньше, чем у контрольных клеток, а у HL-60DMSO она составляла всего 5 ± 1% относительно общего числа.
У мегакариоцитоподобных клеток K562PMA также наблюдалось изменение в экспрессии DR4 и DR5. Мы выявили, что в клеточной культуре K562PMA был снижен процент DR4+/DR5+ и повышен процент DR4–/DR5– клеток в сравнении с контрольными K562. Так, субпопуляция клеток DR4+/DR5+ у К562РМА составила 29 ± ± 3%, а у контрольных К562 – в 2 раза больше, 59 ± 2%. Вместе с тем, процент дважды негативных клеток DR4–/DR5– составил 29 ± 2% в популяции К562РМА и 7 ± 1% у клеток К562 (рис. 3г). Литературные данные показывают наличие у тромбоцитов экспрессии DR4 и DR5 [39, 40], однако мы не обнаружили популяций, несущих данные рецепторы, что согласуется с полной резистентностью тромбоцитов к izTRAIL.
Дифференцировка клеток К562 в эритроидоподобном направлении также сопровождалась изменением в экспрессии DR4 и DR5, что отражено на рис. 3д. Обнаружено достоверное снижение количества клеток популяции DR4+/DR5+ в 2.5 раза у К562Hem и в 5 раз у К562SB в сравнении с контрольными клетками. Вместе с тем, выявлено увеличение процента клеток, несущих только DR5 у дифференцированных клеток. Так, популяция DR4–/DR5+ составила 70 ± 2% у клеток K562Hem, 83 ± 3% у клеток К562SB и достоверно меньшую величину 16 ± 1% у контрольных клеток К562 (рис. 3д). Популяция клеток, не несущих ни один из апоптотических рецепторов к TRAIL, полностью отсутствовала у обоих вариантов дифференцированных клеток, а у контрольных клеток К562 составила всего 7 ± 1%.
Таким образом, повышение устойчивости к TRAIL-индуцированной гибели у лейкозных клеток, дифференцированных во всех миелоидных направлениях, кроме эритроидоподобного, сопровождалось снижением экспрессии проапоптотических TRAIL-рецепторов на клеточной поверхности.
Индукторы ЭПР-стресса повышают чувствительность дифференцированных лейкозных клеток к TRAIL-индуцированной гибели
Мы показали, что миелоидная дифференцировка лейкозных клеток в моноцитоподобном, макрофагоподобном, гранулоцитоподобном и мегакариоцитоподобном, но не в эритроидоподобном, направлениях сопровождалась приобретением устойчивости к TRAIL-индуцированной гибели и снижением экспрессии рецепторов DR4 и DR5. Следовательно, сама представленность проапоптотических TRAIL-рецепторов может опосредовать устойчивость к TRAIL у лейкозных клеток при миелоидной дифференцировке. Мы оценили возможность применения препаратов, вызывающих ЭПР-стресс (ONC201, туникамицин и SAHA), для сенситизации дифференцированных лейкозных клеток к цитотоксическому действию TRAIL, так как известна их способность индуцировать экспрессию DR5 [41–43]. Были подобраны такие концентрации данных веществ, которые наряду с минимальной токсичностью, оказываемой на лейкозные клетки, вызывали наиболее выраженный цитотоксический эффект при совместном применении с izTRAIL. Было установлено, что все три препарата достоверно повышали чувствительность моноцитоподобных клеток THP-1ATRA к действию izTRAIL (рис. 4а). При совместном применении izTRAIL и ONC201, 69 ± 5% этих клеток были чувствительны к TRAIL-индуцированной гибели. Аналогично, совместное применение izTRAIL c туникамицином или с SAHA индуцировало гибель 57 ± 3% и 64 ± 1% клеток ТНР-1ATRA соответственно (рис. 4а). Также применение этих индукторов ЭПР-стресса повышало чувствительность макрофагоподобных клеток ТНР-1РМА к TRAIL-опосредованной гибели (рис. 4б). Наибольший эффект сенситизации этих макрофагоподобных клеток к izTRAIL оказал туникамицин. Его применение более чем в 5 раз увеличило цитотоксичность izTRAIL, уменьшив количество живых клеток с 96 ± 4% до 18 ± 6%. Наименьший эффект оказал ОNC201, который уменьшил TRAIL-устойчивость клеток ТНР-1РМА в 1.5 раза. Применение SAHA в 8 раз повысило чувствительность клеток ТНР-1РМА к izTRAIL, однако для этого потребовалось добавить SAHA в слаботоксичной концентрации. В отношении мегакариоцитоподобных клеток К562 SAHA оказал наиболее значимый эффект сенситизации к TRAIL-индуцированной гибели среди применяемых препаратов. SAHA снизил количество TRAIL-резистентных клеток К562РМА с 92 ± 5% до 62 ± 2% в популяции (рис. 4в). Эффект ONC201 и туникамицина был менее значимым. Количество TRAIL-устойчивых клеток К562РМА при применении ONC201 снизилось до 75 ± 2%, а при воздействии туникамицина – до 76 ± 5% клеток. Гранулоцитоподобные HL-60ATRA и HL-60DMSO клетки отвечали на сенситизацию к izTRAIL различными индукторами ЭПР-стресса по-разному. SAHA оказал выраженный сенсибилизирующий эффект к TRAIL у клеток HL-60ATRA. Применение этого агента увеличило число чувствительных к TRAIL клеток до 63 ± 2%, а использование ONC201 и туникамицина повысило чувствительность только у 38 ± 2% и 21 ± 3% клеток HL-60ATRA соответственно (рис. 4г). SAHA не оказал какого-либо эффекта на чувствительность клеток HL-60DMSO (рис. 4д). На данные гранулоцитоподобные клетки близкий сенсибилизирующий эффект оказали ONC201 и туникамицин. После их применения 31 ± 2% и 23 ± 1% клеток соответственно приобрели чувствительность к TRAIL-индуцированной гибели.
Рис. 4.
Сенсибилизация дифференцированных лейкозных клеток к цитотоксическому действию izTRAIL индукторами ЭПР-стресса. a – THP-1ATRA совместно со 100 мкМ ONC201, 3.3 мкМ туникамицина (Tm) и 100 мкМ SAHA; б – THP-1PMA совместно со 100 мкМ ONC201, 1 мкМ туникамицина (Tm) и 33 мкМ SAHA; в – K562PMA совместно со 100 мкМ ONC201, 3.3 мкМ туникамицина (Tm) и 33 мкМ SAHA; г – HL-60ATRA совместно с 10 мкМ ONC201, 3.3 мкМ туникамицина (Tm) и 100 SAHA; д – HL-60DMSO совместно с 10 мкМ ONC201, 1.1 мкМ туникамицина (Tm) и 11 мкМ SAHA; * – p <0.05 по сравнению с эффектом только индуктора ЭПР-стресса.
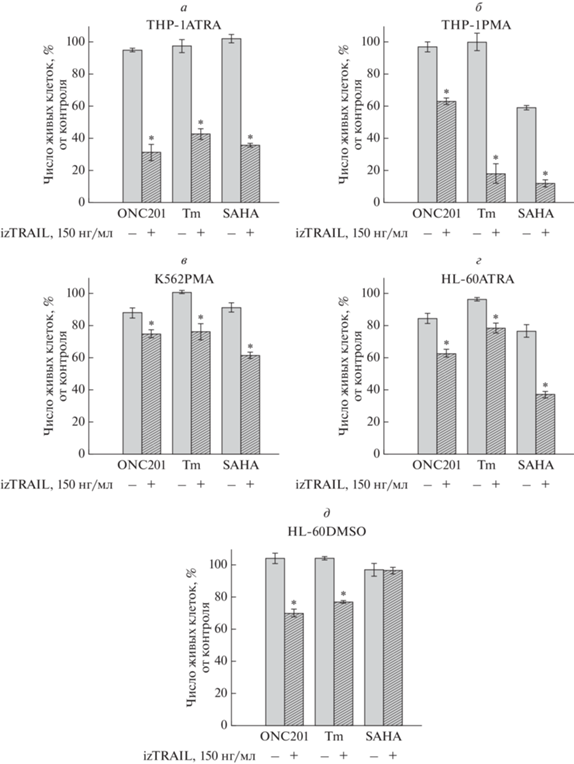
Таким образом, мы выявили, что использование индукторов ЭПР-стресса, модулирующих экспрессию TRAIL рецепторов, способствовало повышению чувствительности клеток, дифференцированных в миелоидном направлении. Это указывает на то, что приобретение устойчивости к TRAIL-индуцированной гибели данных клеток может быть связано с изменением экспрессии TRAIL-рецепторов.
Индукторы ЭПР-стресса повышают экспрессию DR5 у лейкозных клеток, дифференцированных в миелоидном направлении
Для оценки механизма сенситизации дифференцированных лейкозных клеток к TRAIL-индуцированной гибели было выполнено исследование влияния препаратов ONC201, туникамицина и SAHA на представленность рецепторов DR4 и DR5 на этих клетках. Оказалось, что эффект сенситизации к TRAIL-индуцированной гибели указанными агентами сочетался с повышением экспрессии рецептора DR5 на всех исследованных лейкозных клетках, дифференцированных в миелоидном направлении (рис. 5). Причем у всех клеток наблюдалась тенденция увеличения числа популяции DR4–/DR5+ и снижения количества дважды негативных DR4–/DR5– клеток. У моноцитоподобных клеток ТНР-1ATRA туникамицин вызвал достоверное повышение процента DR4–/DR5+ клеток (с 10 ± 2% до 45 ± 1%) и уменьшение популяции DR4–/DR5– клеток (с 54 ± 8% до 30 ± 5% клеток) (рис. 5а). Эффекты, оказываемые ONC201 и SAHA на экспрессию DR4 и DR5 в клетках THP-1ATRA, были менее выражены, чем у туникамицина. Хорошо выраженный и схожий эффект повышения экспрессии DR4 и DR5 на макрофагоподобных клетках THP-1PMA оказали туникамицин и SAHA, но не ONC201 (рис. 5б). Туникамицин и SAHA повышали процент DR4–/DR5+ клеток THP-1PMA с 40 ± 5% в контроле (без ингибиторов) до 70 ± 1% и 63 ± 3% (с ингибиторами) соответственно и уменьшали процент DR4–/DR5– клеток с 50 ± 4% в контроле до 28 ± 2% и 35 ± 2% соответственно. Аналогично, достоверно значимый эффект на экспрессию DR4 и DR5 у мегакариоцитоподобных клеток К562РМА оказали туникамицин и SAHA в противоположность ONC201 (рис. 5в). Туникамицин и SAHA повысили в 2 раза количество клеток K562PMA, несущих только DR5, c 41 ± 1% в контроле до 81 ± 2% и 82 ± 2% соответственно. В то же время данные препараты уменьшили процент DR4–/ DR5– клеток K562PMA более чем в 2 раза, с 29 ± 2% до 12 ± 2% и 11 ± 1% соответственно. Применение индукторов ЭПР-стресса также отразилось в изменении экспрессии DR5 у гранулоцитоподобных клеток HL-60ATRA и HL-60DMSO. У клеток HL-60ATRA наибольший эффект на экспрессию рецепторов оказал туникамицин (рис 5г). Туникамицин индуцировал в культуре клеток HL-60ATRA увеличение популяции DR4–/DR5+ почти в 4 раза, с 20 ± 1% до 71 ± 1% клеток, а также уменьшение популяции дважды негативных клеток с 43 ± 1% до 18 ± 1%. В клетках HL-60DMSO все три препарата оказали одинаковый эффект на экспрессию DR5, а именно повысили количество клеток DR4–/DR5+ почти в 3 раза и уменьшили число DR4–/DR5– клеток более чем в 1.5 раза (рис. 5д).
Рис. 5.
Влияние индукторов ЭПР-стресса на процент субпопуляций клеток, которые экспрессируют TRAIL-рецепторы DR4 и DR5 (DR4+/DR5+), только DR4 (DR4+/DR5–) или только DR5 (DR4–/DR5+) или ни DR4, ни DR5 (DR4–/DR5–): a –ТНР-1ATRA, 100 мкМ ONC201, 3.3 мкМ туникамицина (Tm) и 100 мкМ SAHA; б –ТНР-1PMA, 100 мкМ ONC201, 1 мкМ Tm и 33 мкМ SAHA; в – K562PMA, 100 мкМ ONC201, 3.3 мкМ Tm и 33 мкМ SAHA; г – HL-60ATRA, 10 мкМ ONC201, 3.3 мкМ Tm и 100 мкМ SAHA; д – HL-60ATRA, 10 мкМ ONC201, 1.1 мкМ Tm и 11 мкМ SAHA. * – p < 0.05 в сравнении с контролем (без индуктора).
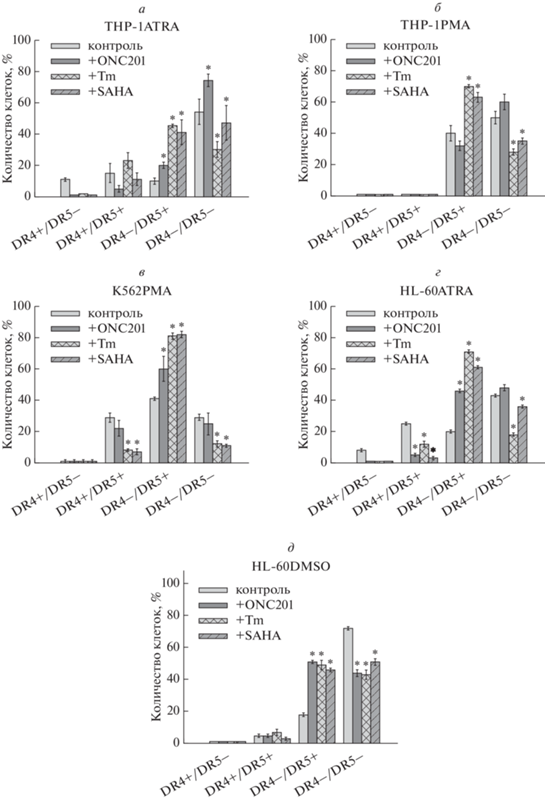
Полученные результаты указывают на то, что механизм повышения TRAIL-устойчивости лейкозных клеток, дифференцированных в миелоидном направлении, связан с уменьшением представленности на них рецепторов DR4 и DR5. В свою очередь, препараты ONC201, туникамицин, а также SAHA способствуют подавлению TRAIL-резистентности дифференцированных в миелоидном направлении лейкозных клеток, повышая экспрессию проапоптотического TRAIL-рецептора DR5 на клеточной поверхности.
ОБСУЖДЕНИЕ
Литературные данные о способности лейкозных клеток in vivo дифференцироваться с приобретением более “злокачественного” фенотипа [16, 18–21], а также о подавлении TRAIL-чувствительности дифференцированных лейкозных клеток [13–15] предполагают участие механизма дифференцировки лейкозных клеток в повышении их резистентности к TRAIL-индуцированной гибели.
На различных моделях дифференцировки лейкозных клеток in vitro в работе установлено, что повышение устойчивости к TRAIL-индуцированной гибели характерно для лейкозных клеток, дифференцированных во всех исследуемых направлениях миелопоэза, кроме эритроидоподобного, независимо от индуцирующего агента. Помимо этого мы обнаружили тенденцию к уменьшению экспрессии рецепторов DR4 и DR5, а также увеличение числа клеток, не несущих ни один из проапоптотических TRAIL-рецепторов, во всех дифференцированных лейкозных клетках, кроме эритроидоподобных. Можно сказать, что дифференцировка в эритроидоподобном направлении сопровождалась перераспределением экспрессии TRAIL-рецепторов внутри популяции DR4/DR5-позитивных клеток, так как число клеток в популяции DR4+/DR5+ снижалось наряду с увеличением числа клеток, обладающих только DR5. Это согласуется с повышением чувствительности к TRAIL у клеток K562 при дифференцировке гемином, что опосредуется снижением уровня экспрессии сFLIP [44]. Отсутствие эффекта повышения TRAIL-устойчивости при эритроидоподобной дифференцировке клеток K562 может быть связано с особой ролью TRAIL как физиологического ингибитора эритропоэза. Известно, что присутствие рецептора эритропоэтина (EpoR) на клеточной мембране может повышать устойчивость эритроидных клеток к действию TRAIL независимо от экспрессии DR4 и DR5 через Epo-зависимую регуляцию внутриклеточного уровня протеинкиназы Cϵ (PKCϵ), которая влияет на экспрессию модулятора апоптоза BCL-2 [45, 46]. В то же время, экспрессия рецепторов DR4 и DR5 остается важной для негативной регуляции эритропоэза через TRAIL-опосредованную активацию ERK 1/2-зависмого сигнального пути. Так, у зрелых эритробластов совместно со снижением экспрессии EpoR уменьшается также экспрессия DR4 и DR5, что приводит к формированию TRAIL-резистентности [47, 48]. Таким образом, для эритроидной дифференцировки свойственно снижение чувствительности к TRAIL, однако эта тенденция не постоянна и не всегда коррелирует со снижением проапоптотических TRAIL-рецепторов. По-видимому, фенотип клеток K562 при обработке бутиратом натрия или гемином не обладает достаточно высокой степенью дифференцировки, нужной для формирования резистентности к TRAIL-индуцированной гибели.
В связи с полученными результатами о зависимости экспрессии DR4 и DR5 от степени дифференцировки лейкозных клеток мы оценили возможность повышения TRAIL-чувствительности лейкозных клеток посредством стимуляции на них экспрессии проапоптотических TRAIL-рецепторов. Было выявлено, что такие вещества, как ONC201, туникамицин и SAHA, достоверно повышали чувствительность к цитотоксическому действию TRAIL у дифференцированных лейкозных клеток, увеличивая процент клеток, несущих рецепторы DR5, совместно со снижением популяции клеток DR4–/DR5–, которые заведомо являются резистентными к TRAIL. При всех вариантах миелоидной дифференцировки туникамицин наиболее значительно повышал экспрессию DR5. Известны перекрестные взаимодействия между UPR (unfolded protein response) сигнальным путем, который запускает ЭПР-стресс, и DR-cигналингом [49]. Например, сенсибилизирующее к TRAIL действие туникамицина связано с вызванной ЭПР-стрессом PERK- и CHOP-опосредованной индукцией экспрессии DR5 [41, 42, 50, 51]. Повышение экспрессии DR5 при действии ONC201 и SAHA связано с активацией транскрипционных факторов ATF4, CHOP, Foxo3a, которые являются регуляторами транскрипции гена DR5 [43, 54–57], в том числе через запуск ЭПР-стресса [52, 53]. Возможность подавления TRAIL-резистентности лейкозных клеток, имеющих дифференцированный фенотип, с помощью применения индукторов ЭПР-стресса представляет интерес для разработки стратегий повышения чувствительности лейкозных клеток к TRAIL-опосредованному иммунному надзору и для создания препаратов на основе рекомбинантных белков TRAIL.
Таким образом, полученные результаты показывают, что дифференцировка лейкозных клеток в миелоидном направлении, за исключением эритроидоподобного, повышает их TRAIL-резистентность, подавляя экспрессию рецепторов DR4 и DR5 на их поверхности, а индукторы экспрессии рецепторов DR5 способны подавлять TRAIL-резистентность дифференцированных лейкозных клеток.
Благодарности. Авторы выражают благодарность А.В. Чеканову за предоставление препаратов белка izTRAIL. Работа выполнена с использованием приборов ЦКП ИТЭБ РАН.
Конфликт интересов. Авторы заявляют об отсутствии конфликтов интересов.
Источники финансирования. Исследование выполнено при финансовой поддержке РФФИ в рамках научных проектов №№ 20-34-90 061 и 20-34-90 062.
Соответствие принципам этики. Все процедуры, выполненные в исследовании с участием людей, соответствуют этическими стандартами институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 года и ее последующим изменениям или сопоставимым нормам этики. От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Список литературы
Buchsbaum D.J., Zhou T., Lobuglio A.F. 2006. TRAIL receptor-targeted therapy. Future Oncol. 2 (4), 493–508. https://doi.org/10.2217/14796694.2.4.493
Chamuleau M.E., Ossenkoppele G.J., van Rhenen A., van Dreunen L., Jirka S.M., Zevenbergen A. 2011. High TRAIL-R3 expression on leukemic blasts is associated with poor outcome and induces apoptosis-resistance which can be overcome by targeting TRAIL-R2. Leuk. Res. 35 (6), 741–749. https://doi.org/10.1016/j.leukres.2010.12.032
Kaufmann S.H., Steensma D.P. 2005. On the TRAIL of a new therapy for leukemia. Leukemia. 19 (12), 2195–2202. https://doi.org/10.1038/sj.leu.2403946
Thapa B., Kc R., Uludag H. 2020. TRAIL therapy and prospective developments for cancer treatment. J. Control. Release. 326, 335–349. https://doi.org/10.1016/j.jconrel.2020.07.013
Snajdauf M., Havlova K., Vachtenheim J.Jr., Ozaniak A., Lischke R., Bartunkova J., Smrz D., Strizova Z. 2021. The TRAIL in the treatment of human cancer: An update on clinical trials. Front Mol. Biosci. 8, 628332. https://doi.org/10.3389/fmolb.2021.628332
Fadeev R., Chekanov A., Solovieva M., Bezborodova O., Nemtsova E., Dolgikh N., Fadeeva I., Senotov A., Kobyakova M., Evstratova Y., Yakubovskaya R., Akatov V. 2019. Improved anticancer effect of recombinant protein izTRAIL combined with sorafenib and peptide iRGD. Int. J. Mol. Sci. 20 (3), 525. https://doi.org/10.3390/ijms20030525
Testa U. 2010. TRAIL/TRAIL-R in hematologic malignancies. J. Cell Biochem. 110 (1), 21–34. https://doi.org/10.1002/jcb.22549
Riccioni R., Pasquini L., Mariani G., Saulle E., Rossini A., Diverio D., Pelosi E., Vitale A., Chierichini A., Cedrone M., Foà R., Lo Coco F., Peschle C., Testa U. 2005. TRAIL decoy receptors mediate resistance of acute myeloid leukemia cells to TRAIL. Haematologica. 90 (5), 612–624.
Austin R., Smyth M.J., Lane S.W. 2016. Harnessing the immune system in acute myeloid leukaemia. Crit. Rev. Oncol. Hematol. 103, 62–77. https://doi.org/10.1016/j.critrevonc.2016.04.020
Sag D., Ayyildiz Z.O., Gunalp S., Wingender G. 2019. The role of TRAIL/DRs in the modulation of immune cells and responses. Cancers (Basel). 11 (10), 1469. https://doi.org/10.3390/cancers11101469
Cheng J., Hylander B.L., Baer M.R., Chen X., Repasky E.A. 2006. Multiple mechanisms underlie resistance of leukemia cells to Apo2 Ligand/TRAIL. Mol. Cancer Ther. 5 (7), 1844–1853. https://doi.org/10.1158/1535-7163.MCT-06-0050
Kobyakova M.I., Evstratova Y.V., Senotov A.S., Lomovsky A.I., Minaichev V.V., Zvyagina A.I., Solovieva M.E., Fadeeva I.S., Akatov V.S., Fadeev R.S. 2021. Appearance of signs of differentiation and pro-inflammatory phenotype in acute myeloid leukemia cells THP-1 at their Increased TRAIL resistance in cell aggregates in vitro. Biochemistry (Moscow), Supplement Series A: Membrane and Cell Biology. 15 (1), 97–105. https://doi.org/10.1134/S1990747821010050
Lomovskaya Y.V., Kobyakova M.I., Senotov A.S., Lomovsky A.I., Minaychev V.V., Fadeeva I.S., Shtatnova D.Y., Krasnov K.S., Zvyagina A.I., Akatov V.S., Fadeev R.S. 2022. Macrophage-like THP-1 cells derived from high-density cell culture are resistant to TRAIL-induced cell death via down-regulation of death-receptors DR4 and DR5. Biomolecules. 12 (2), 150. https://doi.org/10.3390/biom12020150
Evstratova Y.V., Kobyakova M.I., Novikova V.V., Senotov A.S., Akatov V.S., Fadeev R.S. 2019. Monocyte-macrophage differentiation suppresses the expression of proapoptotic receptors to Apo2L/TRAIL and increases resistance to TRAIL-induced apoptosis. Biophysics. 64, 729–731. https://doi.org/10.1134/S0006350919050038
Shiiki K., Yoshikawa H., Kinoshita H., Takeda M., Ueno A., Nakajima Y., Tasaka K. 2000. Potential mechanisms of resistance to TRAIL/Apo2L-induced apoptosis in human promyelocytic leukemia HL-60 cells during granulocytic differentiation. Cell Death Differ. 7 (10), 939–946. https://doi.org/10.1038/sj.cdd.4400727
Bamezai S., Buske C. 2019. Cutting Off Leukemogenesis: Hydra-like plasticity of mature leukemic cells. Cell Stem Cell. 25 (2), 167–168. https://doi.org/10.1016/j.stem.2019.07.005
McKenzie M.D., Ghisi M., Oxley E.P., Ngo S., Cimmino L., Esnault C., Liu R., Salmon M.J., Bell C.C., Ahmed N., Erlichster M., Witkowski T.M., Liu J.G., Chopin M., Dakic A., Simankowicz E., Pomilio G., Vu T., Krsmanovic P., Su S., Tian L., Baldwin M.T., Zalcenstein A.D., DiRago L., Wang S., Metcalf D., Johnstone W.R., Croker A.B., Lancaster I.G., Murphy J.A., Naik H.S., Nutt L.S., Pospisil V., Schroeder T., Wall M., Dawson A.M., Wei H.A., de Thé H., Ritchie E.M., Zuber J., Dickins A.R. 2019. Interconversion between tumorigenic and differentiated states in acute myeloid leukemia. Cell Stem Cell. 25 (2), 258–272. https://doi.org/10.1016/j.stem.2019.07.001
Ngo S., Oxley E.P., Ghisi M., Garwood M.M., McKenzie M.D., Mitchell H.L., Kanellakis P., Susanto O., Hickey M.J., Perkins A.C., Kile B.T., Dickins R.A. 2021. Acute myeloid leukemia maturation lineage influences residual disease and relapse following differentiation therapy. Nat. Commun. 12 (1), 6546. https://doi.org/10.1038/s41467-021-26849-w
Boyd A.L., Aslostovar L., Reid J., Ye W., Tanasijevic B., Porras D.P., Shapovalova Z., Almakadi M., Foley R., Leber B., Xenocostas A., Bhatia M. 2019. Identification of chemotherapy-induced leukemic-regenerating cells reveals a transient vulnerability of human AML recurrence. Cancer Cell. 34 (3), 483–498. https://doi.org/10.1016/j.ccell.2018.08.007
Pei S., Pollyea D.A., Gustafson A., Stevens B.M., Minhajuddin M., Fu R., Riemondy A.K., Gillen E.A., Sheridan M.R., Kim J., Costello C.J., Amaya L.M., Inguva A., Winters A., Ye H., Krug A., Jones L.C., Adane B., Khan N., Ponder J., Schowinsky J., Abbott D., Hammes A., Myers R.J., Ashton M.J., Nemkov T., D’Alessandro A., Gutman A.J., Ramsey E.H., Savona R.M., Smith A.C., Jordan T.C. 2020. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 10 (4), 536–551. https://doi.org/10.1158/2159-8290.CD-19-0710
Kuusanmaki H., Leppa A.M., Polonen P., Kontro M., Dufva O., Deb D., Yadav B., Brück O., Kumar A., Everaus H., Gjertsen T.B., Heinäniemi M., Porkka K., Mustjoki S., Heckman A.C. 2020. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica. 105 (3), 708–720. https://doi.org/10.3324/haematol.2018.214882
Drach J., Lopez-Berestein G., McQueen T., Andreeff M., Mehta K. 1993. Induction of differentiation in myeloid leukemia cell lines and acute promyelocytic leukemia cells by liposomal all-trans-retinoic acid. Cancer Research. 53 (9), 2100–2104.
Daigneault M., Preston J.A., Marriott H.M., Whyte M.K., Dockrell D.H. 2010. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One. 5 (1), e8668. https://doi.org/10.1371/journal.pone.0008668
Forrester M.A., Wassall H.J., Hall L.S., Cao H., Wilson H.M., Barker R.N., Vickers A.M. 2018. Similarities and differences in surface receptor expression by THP-1 monocytes and differentiated macrophages polarized using seven different conditioning regimens. Cell Immunol. 332, 58–76. https://doi.org/10.1016/j.cellimm.2018.07.008
Newburger P.E., Chovaniec M.E., Greenberger J.S., Cohen H.J. 1979. Functional changes in human leukemic cell line HL-60. A model for myeloid differentiation. J. Cell Biol. 82 (2), 315–322. https://doi.org/10.1083/jcb.82.2.315
Tasseff R., Jensen H.A., Congleton J., Dai D., Rogers K.V., Sagar A., Bunaciu R.P., Yen A., Varner J.D. 2017. An effective model of the retinoic acid induced HL-60 differentiation program. Sci. Rep. 7 (1), 14327. https://doi.org/10.1038/s41598-017-14523-5
Baxter S.S., Carlson L.A., Mayer A.M., Hall M.L., Fay M.J. 2009. Granulocytic differentiation of HL-60 promyelocytic leukemia cells is associated with increased expression of Cul5. In Vitro Cell Dev. Biol. Anim. 45 (5–6), 264–274. https://doi.org/10.1007/s11626-008-9163-4
Huang R., Zhao L., Chen H., Yin R.H., Li C.Y., Zhan Y.Q., Zhang J.H., Ge C., Yu M., Yang X.M. 2014. Megakaryocytic differentiation of K562 cells induced by PMA reduced the activity of respiratory chain complex IV. PLoS One. 9 (5), e96246. https://doi.org/10.1371/journal.pone.0096246
Kim K.W., Kim S.H., Lee E.Y., Kim N.D., Kang H.S., Kim H.D., Chung B.S., Kang C.D. 2001. Extracellular signal-regulated kinase/90-KDA ribosomal S6 kinase/nuclear factor-kappa B pathway mediates phorbol 12-myristate 13-acetate-induced megakaryocytic differentiation of K562 cells. J. Biol. Chem. 276 (16), 13 186–13 191. https://doi.org/10.1074/jbc.M008092200
Nurhayati R.W., Ojima Y., Nomura N., Taya M. 2014. Promoted megakaryocytic differentiation of K562 cells through oxidative stress caused by near ultraviolet irradiation. Cell Mol. Biol. Lett. 19 (4), 590–600. https://doi.org/10.2478/s11658-014-0215-3
Herrera R., Hubbell S., Decker S., Petruzzelli L. 1998. A role for the MEK/MAPK pathway in PMA-induced cell cycle arrest: modulation of megakaryocytic differentiation of K562 cells. Exp. Cell Res. 238 (2), 407–414. https://doi.org/10.1006/excr.1997.3847
Zhang D., Cho E., Wong J. 2007. A critical role for the co-repressor N-CoR in erythroid differentiation and heme synthesis. Cell Res. 17 (9), 804–814. https://doi.org/10.1038/cr.2007.72
Huo X.F., Yu J., Peng H., Du Z.W., Liu X.L., Ma Y.N., Zhang X., Zhang Y., Zhao H., Zhang J. 2006. Differential expression changes in K562 cells during the hemin-induced erythroid differentiation and the phorbol myristate acetate (PMA)-induced megakaryocytic differentiation. Mol. Cell Biochem. 292 (1–2), 155–167. https://doi.org/10.1007/s11010-006-9229-0
Shariati L., Modaress M., Khanahmad H., Hejazi Z., Tabatabaiefar M.A., Salehi M., Modarressi H.M. 2016. Comparison of different methods for erythroid differentiation in the K562 cell line. Biotechnol. Lett. 38 (8), 1243–1250. https://doi.org/10.1007/s10529-016-2101-8
Witt O., Sand K., Pekrun A. 2000. Butyrate-induced erythroid differentiation of human K562 leukemia cells involves inhibition of ERK and activation of p38 MAP kinase pathways. Blood. 95 (7), 2391–2396. https://doi.org/10.1182/blood.V95.7.2391
Burzynski L.C., Pugh N., Clarke M.C.H. 2019. Platelet isolation and activation assays. Bio Protoc. 9 (20), e3405. https://doi.org/10.21769/BioProtoc.3405
Lannutti B.J., Blake N., Gandhi M.J., Reems J.A., Drachman J.G. 2005. Induction of polyploidization in leukemic cell lines and primary bone marrow by Src kinase inhibitor SU6656. Blood. 105 (10), 3875–3878. https://doi.org/10.1182/blood-2004-10-3934
Koury M.J., Sawyer S.T., Brandt S.J. 2002. New insights into erythropoiesis. Curr. Opin. Hematol. 9 (2), 93–100. https://doi.org/10.1097/00062752-200203000-00002
Li J., Xia Y., Bertino A.M., Coburn J.P., Kuter D.J. 2000. The mechanism of apoptosis in human platelets during storage. Transfusion. 40 (11), 1320–1329. https://doi.org/10.1046/j.1537-2995.2000.40111320.x
Wu L.S., Wang X.W., He W., Ma X.T., Wang H.Y., Han M., Li B. 2019. TRAIL inhibits platelet-induced colorectal cancer cell invasion. J. Int. Med. Res. 47 (2), 962–972. https://doi.org/10.1177/0300060518820785
Martin-Perez R., Niwa M., Lopez-Rivas A. 2012. ER stress sensitizes cells to TRAIL through down-regulation of FLIP and Mcl-1 and PERK-dependent up-regulation of TRAIL-R2. Apoptosis. 17 (4), 349–363
Guo X., Meng Y., Sheng X., Guan Y., Zhang F., Han Z., Kang Y., Tai G., Zhou Y., Cheng H. 2017. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anticancer Drugs. 28 (1), 66–74. https://doi.org/10.1097/CAD.0000000000000431
Nakata S., Yoshida T., Horinaka M., Shiraishi T., Wakada M., Sakai T. 2004. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 23 (37), 6261–6271. https://doi.org/10.1038/sj.onc.1207830
Hietakangas V., Poukkula M., Heiskanen K.M., Karvinen J.T., Sistonen L., Eriksson J.E. 2003. Erythroid differentiation sensitizes K562 leukemia cells to TRAIL-induced apoptosis by downregulation of c-FLIP. Mol. Cell Biol. 23 (4), 1278–1291. https://doi.org/10.1128/MCB.23.4.1278-1291.2003
Mirandola P., Gobbi G., Ponti C., Sponzilli I., Cocco L., Vitale M. 2006. PKCepsilon controls protection against TRAIL in erythroid progenitors. Blood. 107 (2), 508–513. https://doi.org/10.1182/blood-2005-07-2676
Vitale M., Gobbi G., Mirandola P., Ponti C., Sponzilli I., Rinaldi L., F A Manzoli A.F. 2006. TNF-related apoptosis-inducing ligand (TRAIL) and erythropoiesis: A role for PKC epsilon. Eur. J. Histochem. 50 (1), 15–18.
Secchiero P., Melloni E., Heikinheimo M., Mannisto S., Di Pietro R., Iacone A., Zauli G. 2004. TRAIL regulates normal erythroid maturation through an ERK-dependent pathway. Blood. 103 (2), 517–522. https://doi.org/10.1182/blood-2003-06-2137
De Maria R., Zeuner A., Eramo A., Domenichelli C., Bonci D., Grignani F., Srinivasula S.M., Alnemri E.S., Testa U., Peschle C. 1999. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature. 401 (6752), 489–493. https://doi.org/10.1038/46809
Lafont E. 2020. Stress management: Death receptor signalling and cross-talks with the unfolded protein response in cancer. Cancers (Basel). 12 (5), 1113. https://doi.org/10.3390/cancers12051113
Jung Y.H., Lim E.J., Heo J., Kwon T.K., Kim Y.H. 2012. Tunicamycin sensitizes human prostate cells to TRAIL-induced apoptosis by upregulation of TRAIL receptors and downregulation of cIAP2. Int. J. Oncol. 40 (6), 1941–1948. https://doi.org/10.3892/ijo.2012.1402
Glab J.A., Doerflinger M., Nedeva C., Jose I., Mbogo G.W., Paton J.C., Paton W.A., Kueh J.A., Herold J.M., Huang C.D., Segal D., Brumatti G., Puthalakath H. 2017. DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 24 (5), 944–950. https://doi.org/10.1038/cdd.2017.53
Yu L., Xie R., Tian T., Zheng L., Tang L., Cai S., Ma Z., Yang T., Han B., Yang Q. 2019. Suberoylanilide hydroxamic acid upregulates histone acetylation and activates endoplasmic reticulum stress to induce apoptosis in HepG2 liver cancer cells. Oncol. Lett. 18 (4), 3537–3544. https://doi.org/10.3892/ol.2019.10705
Prabhu V.V., Morrow S., Rahman Kawakibi A., Zhou L., Ralff M., Ray J., Jhaveri A., Ferrarini I., Lee Y., Parker C., Zhang Y., Borsuk R., Chang W., Honeyman N.J., Tavora F., Carneiro B., Raufi A., Huntington K., Carlsen L., Louie A., Safran H., Seyhan A.A., Tarapore S.R., Schalop L., Stogniew M., Allen E.J., Oster W., El-Deiry S.W. 2020. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia. 22 (12), 725–744. https://doi.org/10.1016/j.neo.2020.09.005
Schuler S., Fritsche P., Diersch S., Arlt A., Schmid R.M., Saur D., Schneider G. 2010. HDAC2 attenuates TRAIL-induced apoptosis of pancreatic cancer cells. Mol. Cancer. 9, 80. https://doi.org/10.1186/1476-4598-9-80
Cao H., Cheng Y., You L., Qian J., Qian W. 2013. Homoharringtonine and SAHA synergistically enhance apoptosis in human acute myeloid leukemia cells through upregulation of TRAIL and death receptors. Mol. Med. Rep. 7 (6), 1838–1844. https://doi.org/10.3892/mmr.2013.1440
Allen J.E., Crowder R.N., El-Deiry W.S. 2015. First-in-class small molecule ONC201 induces DR5 and cell death in tumor but not normal cells to provide a wide therapeutic index as an anti-cancer agent. PLoS One. 10 (11), e0143082. https://doi.org/10.1371/journal.pone.0143082
Zhang Y., Zhou L., Safran H., Borsuk R., Lulla R., Tapinos N., Seyhan A.A., El-Deiry W.S. EZH2i EPZ-6438 and HDACi vorinostat synergize with ONC201/TIC10 to activate integrated stress response, DR5, reduce H3K27 methylation, ClpX and promote apoptosis of multiple tumor types including DIPG. Neoplasia. 23 (8), 792–810. https://doi.org/10.1016/j.neo.2021.06.007
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии