

Биологические мембраны: Журнал мембранной и клеточной биологии, 2023, T. 40, № 2, стр. 112-121
Теоретическое объяснение вариабельности активации тромбоцитов через рецептор GPVI
А. А. Мартьянов a, b, М. Г. Степанян a, А. Н. Свешникова a, b, c, *
a Центр теоретических проблем физико-химической фармакологии РАН
109029 Москва, Россия
b Национальный медицинский исследовательский центр детской гематологии,
онкологии и иммунологии им. Д. Рогачева
117997 Москва, Россия
c Московский государственный университет им. М.В. Ломоносова,
факультет фундаментальной физико-химической инженерии
119991 Москва, Россия
* E-mail: a.sveshnikova@physics.msu.ru
Поступила в редакцию 28.11.2022
После доработки 09.12.2022
Принята к публикации 11.12.2022
- EDN: KZDAFN
- DOI: 10.31857/S0233475523020044
Аннотация
Одним из ключевых рецепторов на поверхности тромбоцитов является гликопротеин VI (GPVI) – рецептор к белку межклеточного матрикса коллагену. GPVI запускает каскад тирозинкиназной сигнализации в тромбоцитах, инициируя кальциевую сигнализацию через фосфолипазу Сγ2 (PLCγ2), а также активацию фосфоинозитид-3-киназ (PI3K) и генерацию PIP3. Ранее нашей группой было продемонстрировано, что среди здоровых доноров наблюдается более чем двукратная вариабельность по ответам тромбоцитов на активацию через рецептор GPVI. В представленной работе предлагается компьютерная модель активации тромбоцитов через рецептор GPVI для объяснения данного явления. Настоящая модель представляет собой систему обыкновенных дифференциальных уравнений, интегрируемых методом LSODA. Уравнения модели были выведены на основе ранее опубликованной модели активации тромбоцитов через рецептор CLEC-2. С помощью разработанной модели была предсказана монотонная зависимость степени активации тромбоцита от количества рецепторов GPVI. Анализ чувствительности модели к ее параметрам показал, что ответ тромбоцита на активацию через GPVI лимитируется количеством рецепторов GPVI, а также каталитическими параметрами тирозинкиназ, при этом двукрат ного изменения количества рецепторов достаточно для объяснения наблюдаемого феномена. Таким образом, теоретически было предсказано, что вариабельность кальциевых ответов тромбоцитов на их стимуляцию через рецептор GPVI может определяться вариабельностью количества рецепторов GPVI на поверхности тромбоцитов здоровых доноров.
ВВЕДЕНИЕ
Тромбоциты – безъядерные клетки крови размером 2–4 мкм, производящиеся в костном мозге и циркулирующие в кровотоке в течение 7–10 дней до момента утилизации в селезенке или печени [1]. Основная задача тромбоцитов – предотвращение кровопотери при нарушении целостности кровеносной системы [2, 3]. Для выполнения данной задачи тромбоциты способны переходить в так называемое активированное состояние при контакте с повреждением [4, 5]. Активированный тромбоцит может адгезировать к стенкам сосуда и агрегировать с другими клетками за счет активных интегринов, секретировать гранулы и молекулы-активаторы тромбоцитов, стимулировать свертывание плазмы крови [2, 6, 7].
При повреждении кровеносных сосудов происходит активация и гибель клеток сосудистого эндотелия, приводящая к появлению растворимых активаторов тромбоцитов – ADP и тромбина, а также обнажение белков межклеточного матрикса, одним из которых является коллаген [1]. Тромбоциты адгезируют к коллагену не напрямую, а через молекулы фактора фон Виллебранда (ФВ) – мультимерного белка плазмы крови, который также секретируется эндотелиоцитами при их активации – одним концом связывающиеся с коллагеном, а другим – с тромбоцитами [8]. При этом тромбоциты оказываются в непосредственной близости от коллагена, который активирует тромбоцитарный рецептор гликопротеин VI (GPVI) [9, 10]. Активация тромбоцитов коллагеном значительно замедлена по сравнению с другими известными агонистами, такими как ADP и тромбин [11].
Связывание рецепторов GPVI с коллагеном инициирует кластеризацию этих рецепторов на поверхности тромбоцитов [9, 12, 13]. Ассоциированные с GPVI тирозинкиназы семейства Src (Fyn/Lyn) при этом оказываются в непосредственной близости от других рецепторов GPVI и потому могут фосфорилировать их цитоплазматические домены [14]. С фосфорилированными GPVI связываются неактивные Syk, что приводит к их активации [15]. Для активации одной Syk-киназы достаточно одной молекулы GPVI. Активные Syk запускают формирование “LAT-сигналосомы”, в основе которой лежит линкерный адаптерный Т-клеточный белок (LAT) [14]. В составе LAT-сигналосомы в результате ассоциации с фосфотирозином и фосфорилирования со стороны тирозинкиназ происходит активация фосфолипазы Cγ2 (PLCγ2). PLCγ2 гидролизует фосфоинозитол-4,5-бисфосфат (PIP2), находящийся в мембране тромбоцитов, производя растворимый инозитол-3,4,5-трифосфат (IP3) – вторичный мессенджер, который активирует рецептор к IP3 (IP3R) на поверхности производной эндоплазматического ретикулума плотной тубулярной системы (ЭР), являющейся хранилищем для ионов кальция в тромбоците [14]. Активация IP3R инициирует выход из ЭР в цитозоль свободных ионов кальция, что приводит к резкому повышению его концентрации и активации кальций-чувствительных белков в цитозоле тромбоцита [4, 16]. Повышение концентрации кальция в цитозоле приводит как к закрытию IP3R, так и к активации кальциевых ATP-аз плазматической мембраны (plasma membrane calcium ATPase, PMCA) и мембраны ЭР (sarcoplasmic/endoplasmic reticulum calcium ATPase, SERCA) [17], эффективно удаляющих кальций из цитозоля. Благодаря названным обратным связям происходит развитие кальциевых “осцилляций” в цитозоле активированных тромбоцитов [17, 18].
Кальциевая сигнализация определяет все функциональные ответы тромбоцитов на активацию: изменение формы, необходимое для увеличения площади поверхности закрытия повреждения, активацию мембранных интегринов αIIbβ3, необходимых для формирования тромбоцитарных агрегатов, секрецию тромбоцитарных α-гранул и плотных гранул, необходимых для активации близлежащих клеток, прокоагулянтный ответ – митохондриально-зависимый некроз тромбоцитов, при котором происходит выставление фосфатидилсерина, основы для активации плазменного каскада свертывания крови [1, 17, 19]. Определение ключевых механизмов инициации тирозинкиназной сигнализации в тромбоцитах является ключом для тонкой регуляции активации тромбоцитов через данный путь [20].
Ранее [10] нами было сделано наблюдение, что при активации тромбоцитов здоровых доноров коллаген-подобными пептидами (collagen-related peptide, CRP) – общепризнанными агонистами GPVI [21], наблюдается значительная вариабельность по максимально достижимым кальциевым ответам тромбоцитов, однако ни один из исследованных доноров не имел склонностей к кровоточивостям или тромбозам. Одной из возможных причин подобного разброса может являться значительная вариабельность по экспрессии GPVI на поверхности тромбоцитов среди здоровых доноров. Так, в работе [22] было показано, что экспрессия GPVI у здоровых доноров может различаться более чем в 5 раз. С другой стороны, в той же работе не было обнаружено значительной разницы по агрегации тромбоцитов доноров в ответ на стимуляцию их конвульксином (экзогенный агонист GPVI), независимо от количества GPVI на поверхности [22]. Вариабельность по количествам GPVI среди здоровых доноров была показана и в другой работе, однако авторами был получен разброс только в полтора раза среди 102 здоровых доноров [23]. Авторы [23] также показали, что у пациентов с миелопролиферативными заболеваниями, такими как эссенциальная тромбоцитопатия (ЭТП) и истинная полицетемия (ИПМ), наблюдается значительное снижение экспрессии GPVI по сравнению со здоровыми донорами, а также наблюдается более чем двукратный разброс по экспрессии рецепторов на мембране тромбоцитов. Причинами столь значительного разброса по количеству рецепторов на тромбоцитах может являться как наследственность, так и потенциальные патологические факторы.
Таким образом, возникает закономерный вопрос о причинах и значимости наблюдаемой вариабельности ответа тромбоцитов на активацию рецептора GPVI. В настоящей работе мы используем компьютерное моделирование для описания кальциевой сигнализации, возникающей в тромбоцитах при активации через рецептор GPVI. В результате исследования разработанной модели мы делаем вывод, что двукратный разброс между донорами в уровне экспрессии рецепторов достаточен для объяснения наблюдаемой вариабельности ответа.
МАТЕРИАЛЫ И МЕТОДЫ
Разрабатываемая модель представляет собой систему обыкновенных дифференциальных уравнений (ОДУ), описывающих кинетику концентраций веществ, участвующих в реакциях в соответствующих компартментах. Каждое из ОДУ описывает скорости изменения концентраций веществ. Для описания реакций ассоциации–диссоциации соединений, а также для описания неферментативных реакций был использован закон действующих масс. Ферментативные реакции описывались с помощью кинетики Михаэлиса–Ментен. В некоторых случаях для описания реакций были использованы эмпирические законы, полученные исходя из механистических представлений о реакциях.
Численное интегрирование и анализ модели проводился в COPASI.org [24], интегрирование проводилось методом LSODA [25] с относительной чувствительностью 10–6. Начальные условия для моделей были взяты из доступных данных по протеомному составу тромбоцитов [26]. Подбор параметров модели по экспериментальным данным проводился методом эволюционного программирования с размером популяции 200 [27].
Анализ чувствительности [28] модели к значениям параметров проводился в подходе OAT (“one-at-a-time”) с относительным отклонением 0.001, при этом в качестве выходного значения рассматривалась максимальная концентрация активных PLCγ2 при стимуляции 10 мкг/мл CRP.
РЕЗУЛЬТАТЫ
Разработка компьютерной модели активации тромбоцита через рецептор GPVI
Модель активации тромбоцитов через рецептор GPVI была построена на основе ранее опубликованной [20, 29] модели активации тромбоцитов через рецептор CLEC-2. Несмотря на общую схожесть CLEC-2- и GPVI-индуцированной сигнализации в тромбоцитах, на начальных стадиях инициируемой ими сигнализации присутствуют следующие значимые отличия [10] (рис. 1):
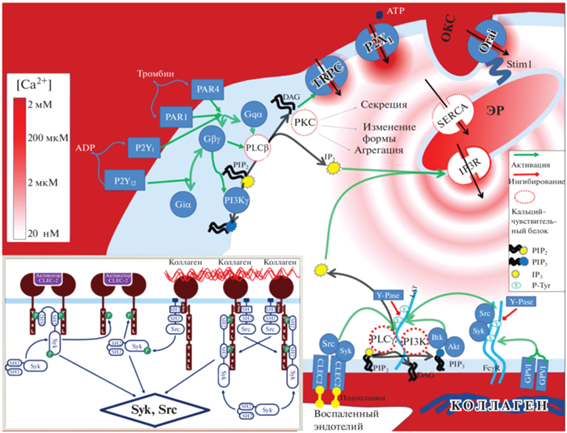
Рис. 1.
Упрощенная схема кальциевой и фосфоинозитидной внутриклеточной сигнализации при активации тромбоцита. Активация тромбоцита управляется концентрациями вторичных мессенджеров – ионов кальция (концентрация обозначена оттенками фона) и молекул фосфоинозитол-3,4,5-трифосфата (PIP3). Концентрация ионов кальция в цитозоле появляется в результате активности фосфолипаз (PLC). PLC гидролизуют фосфоинозитол-4,5-дифосфат (PIP2) до диацилглицерола (DAG) и инозитол-1,4,5-трифосфата (IP3). IP3 приводит к открытию каналов-рецепторов IP3R в мембране ЭР тромбоцитов, что запускает кальциевую сигнализацию. Ионы кальция возвращаются в ЭР с помощью кальциевой ATP-азы SERCA. Концентрация PIP3 повышается в результате активности фосфоинозитид-3-киназы (PI3K), которая фосфорилирует мембранные фосфоинозитиды, в первую очередь PIP2 до PIP3. Появление PIP3 приводит к локализации в окрестности сигналосомы тирозинкиназы Btk, которая фосфорилирует ассоциированные с адаптером LAT PLCγ2, переводя их в активное состояние. На вставке показано, как рецепторы CLEC-2 и GPVI передают сигнал через одну и ту же сеть тирозинкиназ, однако внутриклеточные домены CLEC-2 фосфорилируются тирозининазами Syk, а внутриклеточные домены GPVI тирозинкиназами SFK. Кроме того, фосфорилирование CLEC-2 замедленно по сравнению с фосфорилированием GPVI из-за отсутствия ассоциации между Syk и CLEC-2 в покоящихся тромбоцитах. Активные Syk-киназы фосфорилируют адаптеры LAT, к которым через фосфотирозины присоединяются PI3K и PLCγ2. PI3K при этом переходят в активное состояние. Обозначения: G – субъединицы G-белка; PAR1, PAR4 – активируемые протеазами рецепторы; P2Y1, P2Y12 – пуринэргические рецепторы; TRPC, P2X1, Orai1 – катионные каналы; PKC – протеинкиназа С; Y-Pase – тирозинфосфотаза; ОКС – открытая канальцевая система тромбоцита.
1. количество GPVI на тромбоцитах в среднем составляет 5000 молекул [30], в то время как молекул CLEC-2 только 2000 [31];
2. молекулы рецепторов GPVI через полипролиновый домен ассоциированы с тирозинкиназами SFK [32], что обеспечивает их большую доступность для более быстрого фосфорилирования, в то время как CLEC-2 в покоящемся состоянии клетки не ассоциированы ни с одними из киназ;
3. при активации тромбоцита цитоплазматический домен рецептора GPVI фосфорилируется тирозинкиназами SFK (в первую очередь Fyn и Lyn) [14], в то время как цитоплазматический домен рецептора CLEC-2 фосфорилируется только тирозинкиназами Syk [33], которых в тромбоцитах значительно меньше [34];
4. кластеризация рецепторов GPVI в области липидных рафтов может быть дополнительно ускорена благодаря их ассоциации с SFK-киназами, которые пальмитоилированы, и за счет этого имеют значительное сродство к мембранным микродоменам, обогащенным холестерином [32, 35];
5. в ассоциированной с GPVI FcγR-цепью находится ITAM-домен, содержащий две YxxL последовательности, которые могут быть фосфорилированы, в то время как в цитоплазматическом домене CLEC-2 находится домен hemITAM, содержащий одну YxxL последовательность, доступную для фосфорилирования соответствующими тирозинкиназами; таким образом, одна молекула GPVI может активировать одну тирозинкиназу Syk, в то время как CLEC-2 для этого необходимо сформировать димер.
Так как разные лиганды значительным образом могут влиять на динамику и паттерны кластеризации рецепторов GPVI тромбоцитов [9], в качестве основного исследуемого лиганда был выбран CRP [36]. Параметры модуля кластеризации были получены нами ранее [13] на основе экспериментальных данных по кластеризации рецепторов GPVI в ответ на появление лиганда [9]. В настоящей работе по данным активации тромбоцитов в суспензии в ответ на CRP [10, 30] проводился дополнительный подбор параметров: аффинность Syk к фосфорилированным рецепторам GPVI, константа реакции дефосфорилирования GPVI, а также константы аффинности тирозинкиназ SFK к рецептору GPVI (табл. 1). Результаты подбора параметров модели приведены на рис. 2. В результате был сделан вывод, что модель может качественно описывать получаемые экспериментальные данные.
Таблица 1.
Уравнения и параметры модели, специфичные для GPVI
| Реакция | Уравнение | Параметры | Источн. |
|---|---|---|---|
| $D \leftrightarrows D{\kern 1pt} *$ | ${{S}_{{PM}}}\left( {\frac{{\left( {\frac{{F{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}{{kcat_{{cat}}^{{Src}}} \mathord{\left/ {\vphantom {{kcat_{{cat}}^{{Src}}} 3}} \right. \kern-0em} 3} + \frac{{{{F}^{P}}{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}{{2kcat_{{cat}}^{{Src}}} \mathord{\left/ {\vphantom {{2kcat_{{cat}}^{{Src}}} {3 + \frac{{F{\kern 1pt} *{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}kcat_{{cat}}^{{Src}}}}} \right. \kern-0em} {3 + \frac{{F{\kern 1pt} *{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}kcat_{{cat}}^{{Src}}}}} \right)}}{{K{{m}^{{Src}}} + \frac{{D{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}}}} \right)D - {{S}_{{PM}}}K{{r}^{{CD148}}}D{\kern 1pt} *$ | $k_{{cat}}^{{Src}}$ = 2.1 с–1 | [37, 38] |
| KmSrc = 3 мкМ | |||
| KrCD148 = 90.8 с–1 | [20] | ||
| $Cs \leftrightarrows Cs{\kern 1pt} *$ | ${{S}_{{PM}}}\left( {\frac{{\left( {\frac{{F{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}{{kcat_{{cat}}^{{Src}}} \mathord{\left/ {\vphantom {{kcat_{{cat}}^{{Src}}} 3}} \right. \kern-0em} 3} + \frac{{{{F}^{P}}{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}{{2kcat_{{cat}}^{{Src}}} \mathord{\left/ {\vphantom {{2kcat_{{cat}}^{{Src}}} {3 + \frac{{F{\kern 1pt} *{\kern 1pt} {{S}_{{PM}}}}}{{{{V}_{{JM}}}}}kcat_{{cat}}^{{Src}}}}} \right. \kern-0em} {3 + \frac{{F{\kern 1pt} *{\kern 1pt} {{S}_{{PM}}}}}{{{{V}_{{JM}}}}}kcat_{{cat}}^{{Src}}}}} \right)}}{{K{{m}^{{Src}}} + \frac{{Cs{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}}}} \right)Cs - {{S}_{{PM}}}K{{r}^{{CD148}}}Cs{\kern 1pt} *$ | $k_{{cat}}^{{Src}}$ = 2.1 с–1 | [37, 38] |
| KmSrc = 3 мкМ | [20] | ||
| KrCsk = 1.0 с–1 | [20] | ||
| ${{F}_{P}} \leftrightarrows F$ | ${{S}_{{PM}}}\left( {\frac{{\frac{{D{\kern 1pt} *{\kern 1pt} {{S}_{{PM}}}}}{{{{V}_{{JM}}}}}k_{{cat}}^{{C{{D}_{{148}}}}}}}{{K{{m}^{{CD148}}} + \frac{{{{F}_{P}}{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}}}} \right){{F}_{P}} - {{S}_{{PM}}}\left( {\frac{{\frac{{Cs{\kern 1pt} *{\kern 1pt} {{S}_{{PM}}}}}{{{{V}_{{JM}}}}}k_{{cat}}^{{Csk}}}}{{K{{m}^{{Csk}}} + \frac{{F{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}}}} \right)F$ | $k_{{cat}}^{{CD148}}$ = 9.7 с–1 | [39] |
| KmCD148 = 9.1 мМ | |||
| $k_{{cat}}^{{Csk}}$ = 1.9 с–1 | [40] | ||
| KmCsk = 10 мкМ | |||
| $F \leftrightarrows {{F}^{P}}$ | ${{S}_{{PM}}}\left( {\frac{{\left( {\frac{{F{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}{{kcat_{{cat}}^{{Src}}} \mathord{\left/ {\vphantom {{kcat_{{cat}}^{{Src}}} 3}} \right. \kern-0em} 3} + \frac{{{{F}^{P}}{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}{{2kcat_{{cat}}^{{Src}}} \mathord{\left/ {\vphantom {{2kcat_{{cat}}^{{Src}}} 3}} \right. \kern-0em} 3} + \frac{{F{\kern 1pt} *{\kern 1pt} {{S}_{{PM}}}}}{{{{V}_{{JM}}}}}kcat_{{cat}}^{{Src}}} \right)}}{{K{{m}^{{Src}}} + \frac{{F{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}}}} \right)$$F - {{S}_{{PM}}}\left( {\frac{{\frac{{D{\kern 1pt} *{\kern 1pt} {{S}_{{PM}}}}}{{{{V}_{{JM}}}}}k_{{cat}}^{{C{{D}_{{148}}}}}}}{{K{{m}^{{CD148}}} + \frac{{{{F}^{P}}{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}}}} \right)$FP | $k_{{cat}}^{{Src}}$ = 2.1 с–1 | [37, 38] |
| KmSrc = 3 мкМ | |||
| $k_{{cat}}^{{CD148}}$ = 9.7 с–1 | [39] | ||
| KmCD148 = 9.1 мМ | |||
| ${{F}^{P}} \leftrightarrows F{\kern 1pt} *$ | ${{S}_{{PM}}}sk_{{SF1}}^{{SH2}}2R_{P}^{*}{{F}^{P}} - {{S}_{{PM}}}kD_{{SFK}}^{{SH2}}k_{{SF1}}^{{SH2}}F{\kern 1pt} *$ | $S$ – средний размер кластера рецепторов GPVI | [20] |
| $k_{{SF1}}^{{SH2}} = 0.6\,\frac{{{\text{мк}}{{{\text{м}}}^{2}}}}{{{\text{с}}\,\,{\text{мкмоль}}}}$ | |||
| $kD_{{SFK}}^{{SH2}} = {{10}^{6}}\,\frac{{{\text{мкмоль}}}}{{{\text{мк}}{{{\text{м}}}^{2}}\,\,{\text{с}}}}$ | [41], подбор | ||
| $T \leftrightarrows T{\kern 1pt} *$ | ${{V}_{{Cyt}}}Kf_{{Syk}}^{{TULA{\kern 1pt} - {\kern 1pt} 2}}S{\kern 1pt} *{\kern 1pt} T - {{V}_{{Cyt}}}Kr_{{Syk}}^{{TULA{\kern 1pt} - {\kern 1pt} 2}}T{\kern 1pt} *$ | $Kf_{{Syk}}^{{TULA2}} = 0.1\,\frac{1}{{{\text{с}}\,\,{\text{мкМ}}}}$ | [20] |
| $Kr_{{Syk}}^{{TULA2}}$ = 0.007 с–1 | |||
| $R_{C}^{*} \leftrightarrows R_{P}^{*}$ | ${{S}_{{PM}}}\left( {\frac{{S{\kern 1pt} *k_{{cat}}^{{Syk}}}}{{K{{m}^{{Syk}}} + \frac{{R_{C}^{*}{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}}}} \right)R_{C}^{*} - {{S}_{{PM}}}K{{r}^{{{\text{Phosph}}}}}R_{P}^{*}$ | $k_{{cat}}^{{Syk}}$ = 11.85 с–1 | [42] |
| KmSyk = 9.1 мкМ | |||
| KrPhosph = 10–4 с–1 | [20] | ||
| $S \leftrightarrows S{\kern 1pt} *$ | ${{V}_{{Cyt}}}\frac{{sk_{{S1}}^{{SH2}}{{S}_{{PM}}}R_{P}^{*}}}{{{{V}_{{JM}}}}}\left( {\frac{{s{\kern 1pt} *{\kern 1pt} k_{{cat}}^{{Syk}}}}{{K{{m}^{{Syk}}} + \frac{{S{{S}_{{PM}}}}}{{{{V}_{{JM}}}}}}}} \right)S - {{V}_{{cyt}}}(kD_{{Syk}}^{{SH2}}k_{{S1}}^{{SH2}} + K{{r}^{{Syk}}} + Kr_{{TULA2}}^{{Syk}}T{\kern 1pt} *)S{\kern 1pt} *$ | $k_{{S1}}^{{SH2}} = 0.026\,\frac{1}{{{\text{с}}\,\,{\text{мкМ}}}}$ | [20] |
| $k_{{cat}}^{{Syk}}$ = 11.85 с–1 | [42] | ||
| KmSyk = 9.1 мкМ | |||
| $kD_{{Syk}}^{{SH2}}$ = 0.176 мкМ | [43] | ||
| KrSyk = 10 c–1 $Kr_{{TULA2}}^{{Syk}} = 3.05\,\frac{1}{{{\text{с}}\,\,{\text{мкМ}}}}$ |
Подбор, [20] |
Примечание. RC – концентрация кластеров GPVI; RP – концентрация фосфорилированных кластеров GPVI; F – концентрация SFK (Fp – неактивные, F – 1/3 активные, Fp – 2/3 активные, F* – полностью активные); D – концентрация CD148; Cs – концентрация Csk; T – концентрация TULA-2; * означает активную форму фермента; SPM – площадь мембраны, Vcyt – объем цитозоля.
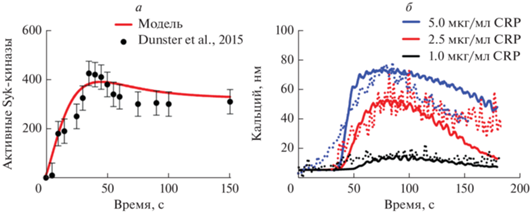
Рис. 2.
Подбор неизвестных параметров компьютерной модели по экспериментальным данным. а – Данные о зависимости активности тирозинкиназ от времени. Красная кривая показывает зависимость от времени количества активированных Syk-киназ при стимуляции CRP в концентрации 10 мкг/мл. Кривая получена при следующих параметрах модели: KM(Syk–GPVI) = 0.176 мкM, k–1 (GPVI–P) = 10–4 с–1, KD(SFK–GPVI) = 10 мкM. Черные точки показывают экспериментально определенное среднее количество активированных Syk-киназ в суспензии тромбоцитов при стимуляции CRP в концентрации 10 мкг/мл [26]. б – данные о зависимости мобилизации кальция от времени и концентрации активатора. Средняя концентрация кальция в тромбоцитах в ответ на активацию их 5 (синие кривые), 2.5 (красные кривые) и 1 (черные кривые) мкг/мл CRP: точки – эксперимент [10], кривые – теоретический расчет. Приведен результат для типичного взрослого здорового донора из n = 5 здоровых доноров.
Определение управляющих параметров модели
Анализ чувствительности разработанной модели показал, что в ответ на изменение следующих параметров модели максимум мобилизации кальция значительно меняется (относительная чувствительность более 1). Наиболее значимыми для сигнализации параметрами являются начальное количество рецепторов GPVI на поверхности тромбоцитов (относительная чувствительность – 1.2), параметры Syk-киназ – каталитическая константа (относительная чувствительность – 1.67), начальное количество (1.58) и константа Михаэлиса (относительная чувствительность – 1.19).
Гипотеза о вкладе вариабельности количества рецепторов GPVI на тромбоцитах в ответ на активацию подтверждается высокой, хоть и не максимальной, чувствительностью модели к их количеству. Следует иметь ввиду, что нелинейная зависимость концентрации кальция от концентрации IP3 [44] могла нивелировать влияние количества копий рецептора на кальциевые ответы. Для дополнительного исследования этого вопроса, мы рассчитали зависимость максимальной концентрации кальция в ответ на CRP от количества GPVI на тромбоцитах (рис. 3). При рассчете было получено, что изменение количества рецепторов GPVI с 5000 до 3000 копий на тромбоцит может привести к двукратному уменьшению максимальной концентрации кальция в ответ на стимуляцию. Таким образом, можно сделать вывод, что при варьировании количеств рецепторов в физиологических пределах можно наблюдать кальциевые ответы, получаемые в экспериментах [10, 23]. Важно подчеркнуть, что вариабельность по кальциевым ответам наблюдается только при активации насыщающими концентрациям CRP, что подразумевает задействование всех рецепторов GPVI. Это также косвенно подтверждает вывод о том, что одной из причин вариабельности ответов тромбоцитов на активацию их через GPVI является вариабельность количества самих по себе рецепторов.
Рис. 3.
Расчетная зависимость максимума мобилизации кальция в тромбоците от количества рецепторов GPVI. Предсказываемая моделью зависимость максимально достижимой концентрации кальция от начального количества рецепторов GPVI на поверхности тромбоцитов. Расчеты проводились при тех же параметрах модели, что и на рис. 2б для концентрации CRP 10 мкг/мл. Выделен диапазон количеств GPVI на поверхности тромбоцитов здоровых доноров, ранее полученный в эксперименте [22].
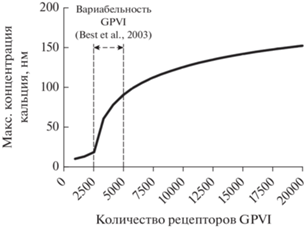
Важно также отметить, что в работе [30] теоретически была предсказана двукратная вариабельность активации тромбоцитов через GPVI только при уменьшении количества рецепторов более чем в 4 раза. Одной из причин подобных различий между моделью из [30] и разработанной здесь моделью может быть отсутствие в модели из [30] полноценной стадии кластеризации рецепторов.
ОБСУЖДЕНИЕ
Значимость рецептора GPVI для нормального функционирования тромбоцитов человека была многократно продемонстрирована. Показано, что у пациентов с наследственными дефицитами данного рецептора наблюдаются серьезные кровотечения. Также было продемонстрировано, что введение антител к рецептору GPVI приводит к нестабильности тромбов у мышей [45]. Таким образом, у полученной нами вариабельности активации тромбоцитов агонистами рецепторов GPVI может быть патологическая значимость, что подчеркивает необходимость исследования данного феномена.
Одной из интересных особенностей GPVI является то, что при активации тромбоцитов различными физиологическими лигандами происходит активация металлопротеина ADAM10, осуществляющего гидролиз GPVI на поверхности тромбоцитов [46, 47]. Показано, что даже при кратковременном приложении напряжения сдвига (в течение минуты) в плазме здоровых доноров происходит повышение концентрации растворимого эктодомена GPVI массой от 50 до 55 кДа, называемого sGPVI [48], что является одним из характерных признаков активности металлопротеиназ на тромбоцитах. Предположительно, подобные сдвиговые стрессы могут возникать в ранах [49] и при заборе крови. В то время как врожденный и/или приобретенный дефицит GPVI у людей встречается достаточно редко [46], снижение количества GPVI на тромбоцитах в результате активности металлопротеиназ может приводить к аналогичному фенотипу и кровоточивости [47]. Здесь важно отметить, что у пациентов со значительной кровопотерей, произошедшей в результате травмы, наблюдается значительное снижение количества рецепторов GPVI на поверхности тромбоцитов и соответствующее повышение количества sGPVI в плазме крови [46].
Таким образом, полученные ранее в литературе данные о значительном разбросе по количеству рецепторов GPVI на поверхности тромбоцитов здоровых доноров может быть одной из причин значимых разбросов ответов тромбоцитов. С помощью теоретической модели нами было получено, что количество рецепторов на поверхности тромбоцитов является одним из наиболее влияющих на степень кальциевой активации тромбоцитов параметров. Более того, разброс по количеству рецепторов на поверхности тромбоцитов здоровых доноров находится в области наибольшей зависимости концентрации свободных ионов кальция от количества GPVI на тромбоцитах. Исходя из этого, можно предположить, что даже незначительное отклонение количеств рецептора от нормы, например, из-за стресса у донора при взятии крови, может привести к значительному изменению ответа тромбоцита на активацию через GPVI.
Помимо инициации кальциевой сигнализации, GPVI индуцирует PI3K-Akt сигнализацию в тромбоцитах [14]. Исходя из этого, можно также предположить, что вариабельность кальциевых ответов может коррелировать с вариабельностью активности PI3K-Akt сигнализации в тромбоцитах. Однако, экспериментальное исследование изменений концентрации фосфоинозитидов в мембранах тромбоцитов является чрезвычайно сложным с экспериментальной точки зрения. Наравне с экспериментальным исследованием корреляции количества рецепторов GPVI и кальциевых ответов тромбоцитов в ответ на активацию их через GPVI, данные исследования должны быть проведены в дальнейшем.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Источники финансирования. Работа выполнена при поддержке Российского фонда фундаментальных исследований и Лондонского Королевского Общества (проект № 21-51-10 005).
Соответствие принципам этики. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
Пантелеев М.А., Свешникова А.Н. 2014. Тромбоциты и гемостаз. Онкогематология. 9 (2), 65–73.
Sveshnikova A., Stepanyan M., Panteleev M., Sveshnikova A., Stepanyan M., Panteleev M. 2021. Platelet functional responses and signalling: the molecular relationship. Part 1: Responses. Syst. Biol. Physiol. Reports. 1 (1), 20.
Bergmeier W., Stefanini L. 2009. Novel molecules in calcium signaling in platelets. J. Thromb. Haemost. 7, 187–190.
Martyanov A., Panteleev M. 2021. Platelet functional responses and signalling: The molecular relationship. Part 2: Receptors. Syst. Biol. Physiol. Reports. 1 (3), 13–30.
Gear A.R. 1994. Platelet adhesion, shape change, and aggregation: rapid initiation and signal transduction events. Can. J. Physiol. Pharmacol. 72 (3), 285–294.
Канева В.Н., Мартьянов А.А., Морозова Д.С., Пантелеев М.А., Свешникова А.Н. 2019. Тромбоцитарные интегрины αIIBβ3. Биол. мембраны. 36 (1), 15–31.
Podoplelova N.A., Sveshnikova A.N., Kotova Y.N., Eckly A., Receveur N., Nechipurenko D.Yu., Obydennyi S.I., Kireev I.I., Gachet C., Ataullakhanov F.I., Mangin P.H., Panteleev M.A. 2016. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood. 128 (13), 1745–1755.
Bryckaert M., Rosa J.-P., Denis C.V., Lenting P.J. 2015. Of von Willebrand factor and platelets. Cell Mol. Life Sci. 72 (2), 307–326.
Poulter N.S., Pollitt A.Y., Owen D.M., Gardiner E.E., Andrews R.K., Shimizu H., Ishikawa D., Bihan D., Farndale R.W., Moroi M., Watson S.P., Jung S.M. 2017. Clustering of glycoprotein VI (GPVI) dimers upon adhesion to collagen as a mechanism to regulate GPVI signaling in platelets. J. Thromb. Haemost. 15 (3), 549–564.
Степанян М.Г., Филькова А.А., Гарсон Дасгупта А.К., Мартьянов А.А., Свешникова А.Н. 2020. Активация тромбоцитов через рецептор GPVI: вариабельность ответа. Биол. мембраны. 37 (6), 442–452.
Johnson E.N., Brass L.F., Funk C.D. 1998. Increased platelet sensitivity to ADP in mice lacking platelet-type 12-lipoxygenase. Proc. Natl. Acad. Sci. USA. 95 (6), 3100–3105.
Pollitt A.Y., Poulter N.S., Gitz E., Navarro-Nuñez L., Wang Y.-J., Hughes C.E., Thomas S.G., Nieswandt B., Douglas M.R., Owen D.M., Jackson D.G., Dustin M.L., Watson S.P. 2014. Syk and Src family kinases regulate C-type lectin receptor 2 (CLEC-2)-mediated clustering of podoplanin and platelet adhesion to lymphatic endothelial cells. J. Biol. Chem. 289 (52), 35 695–35 710.
Garzon Dasgupta A.K., Martyanov A.A., Filkova A.A., Panteleev M.A., Sveshnikova A.N. 2020. Development of a simple kinetic mathematical model of aggregation of particles or clustering of receptors. Life. 10 (6), 97.
Watson S.P., Herbert J.M.J., Pollitt A.Y. 2010. GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 8 (7), 1456–1467.
Rayes J., Watson S.P., Nieswandt B. 2019. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Invest. 129 (1), 12–23.
Balabin F.A., Sveshnikova A.N. 2016. Computational biology analysis of platelet signaling reveals roles of feedbacks through phospholipase C and inositol 1,4,5-trisphosphate 3-kinase in controlling amplitude and duration of calcium oscillations. Math Biosci. 276, 67–74.
Sveshnikova A.N., Balatskiy A.V., Demianova A.S., Shepelyuk T.O., Shakhidzhanov S.S., Balatskaya M.N., Pichugin A.V., Ataullakhanov F.I., Panteleev M.A. 2016. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J. Thromb. Haemost. 14 (10), 2045–2057.
Jackson S.P., Schoenwaelder S.M. 2010. Procoagulant platelets: Are they necrotic? Blood. 116 (12), 2011–2018.
Podoplelova N.A., Nechipurenko D.Y., Ignatova A.A., Sveshnikova A.N., Panteleev M.A. 2021. Procoagulant platelets: Mechanisms of generation and action. Hämostaseologie. 41 (02), 146–153.
Martyanov A.A., Balabin F.A., Dunster J.L., Panteleev M.A., Gibbins J.M., Sveshnikova A.N. 2020. Control of platelet CLEC-2-mediated activation by receptor clustering and tyrosine kinase signaling. Biophys. J. 118 (11), 2641–2655.
Moroi M., Jung S.M. 2004. Platelet glycoprotein VI: Its structure and function. Thromb. Res. 114 (4), 221–233.
Furihata K., Clemetson K.J., Deguchi H., Kunicki T.J. 2001. Variation in human platelet glycoprotein VI content modulates glycoprotein VI-specific prothrombinase activity. Arterioscler. Thromb. Vasc. Biol. 21 (11), 1857–1863.
Best D., Senis Y.A., Jarvis G.E., Eagleton H.J., Roberts D.J., Saito T., Jung S.M., Moroi M., Harrison P., Green F.R., Watson S.P. 2003. GPVI levels in platelets: Relationship to platelet function at high shear. Blood. 102 (8), 2811–2818.
Hoops S., Sahle S., Gauges R., Lee C., Pahle J., Simus N., Singhal M., Xu L., Mendes P., Kummer U. 2006. COPASI – a COmplex PAthway SImulator. Bioinformatics. 22 (24), 3067–3074.
Petzold L., Hindmarsh A. 1997. LSODA (Livermore solver of ordinary differential equations). Computing and Mathematics Research Division, Lawrence Livermore National Laboratory, Livermore, CA, 24.
Burkhart J.M., Vaudel M., Gambaryan S., Radau S., Walter U., Martens L., Geiger J., Sickmann A., Zahedi R.P. 2012. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 120 (15), e73–82.
Back T. 1996. Evolutionary algorithms in theory and practice: Evolution strategies, evolutionary programming, genetic algorithms. Oxford: Oxford university press.
Saltelli A., Ratto M., Tarantola S., Campolongo F. 2005. Sensitivity analysis for chemical models. Chem. Rev. 105 (7), 2811–2828.
Мартьянов А.А., Балабин Ф.А., Майоров А.С., Шамова Е.В., Пантелеев М.А., Свешникова А.Н. 2018. Компьютерное моделирование внутриклеточной сигнализации при активации тромбоцитов крови фукоиданом. Биол. мембраны. 35 (5), 364–375.
Dunster J.L., Mazet F., Fry M.J., Gibbins J.M., Tindall M.J. 2015. Regulation of early steps of GPVI signal transduction by phosphatases: A systems biology approach. PLoS Comput. Biol. 11 (11), e1004589.
Rayes J., Watson S.P., Nieswandt B. 2019. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Invest. 129 (1), 12–23.
Senis Y.A., Mazharian A., Mori J. 2014. Src family kinases: At the forefront of platelet activation. Blood. 124 (13), 2013–2024.
Séverin S., Pollitt A.Y., Navarro-Nuñez L., Nash C.A., Mourão-Sá D., Eble J.A., Senis Y.A., Watson S.P. 2011. Syk-dependent phosphorylation of CLEC-2. J. Biol. Chem. 286 (6), 4107–4116.
Burkhart J.M., Vaudel M., Gambaryan S., Radau S., Walter U., Martens L., Geiger J., Sickmann A., Zahedi R.P. 2012. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 120 (15), e73–82.
Quinter P.G., Quinton T.M., Dangelmaier C.A., Kunapuli S.P., Daniel J.L. 2005. Role of lipid rafts in GPVI agonist-induced platelet signaling. Blood. 106 (11), 3576.
Miura Y., Takahashi T., Jung S.M., Moroi M. 2002. Analysis of the interaction of platelet collagen receptor glycoprotein VI (GPVI) with collagen. A dimeric form of GPVI, but not the monomeric form, shows affinity to fibrous collagen. J. Biol. Chem. 277 (48), 46 197–46 204.
Kemble D.J., Wang Y.H., Sun G. 2006. Bacterial expression and characterization of catalytic loop mutants of Src protein tyrosine kinase. Biochemistry. 45 (49), 14 749–14 754.
Bradshaw J.M. 2010. The Src, Syk, and Tec family kinases: Distinct types of molecular switches. Cell. Signal. 22 (8), 1175–1184.
Ren L., Chen X., Luechapanichkul R., Selner N.G., Meyer T.M., Wavreille A.-S., Chan R., Iorio C., Zhou X., Neel B.G., Pei D. 2011. Substrate specificity of protein tyrosine phosphatases 1B, RPTPα, SHP-1, and SHP-2. Biochemistry. 50 (12), 2339–2356.
Lin X., Lee S., Sun G. 2003. Functions of the activation loop in Csk protein-tyrosine kinase. J. Biol. Chem. 278 (26), 24 072–24 077.
Park M.J., Sheng R., Silkov A., Jung D.J., Wang Z.G., Xin Y., Kim H., Thiagarajan-Rosenkranz P., Song S., Yoon Y., Nam W., Kim I., Kim E., Lee D.G., Chen Y., Singaram I., Wang L., Jang M.H., Hwang C.S., Honig B., Ryu S., Lorieau J., Kim Y.M., Cho W. 2016. SH2 domains serve as lipid-binding modules for pTyr-signaling proteins. Mol. Cell. 62 (1), 7–20.
Tsang E., Giannetti A.M., Shaw D., Dinh M., Tse J.K.Y., Gandhi S., Ho A., Wang S., Papp E., Bradshaw J.M. 2008. Molecular mechanism of the Syk activation switch. J. Biol. Chem. 283 (47), 32650–32659.
Hughes C.E., Sinha U., Pandey A., Eble J.A., O’Callaghan C.A., Watson S.P. 2013. Critical role for an acidic amino acid region in platelet signaling by the HemITAM (hemi-immunoreceptor tyrosine-based activation motif) containing receptor CLEC-2 (C-type lectin receptor-2). J. Biol. Chem. 288 (7), 5127–5135.
Sveshnikova A.N., Balatskiy A.V., Demianova A.S., Shepelyuk T.O., Shakhidzhanov S.S., Balatskaya M.N., Pichugin A.V., Ataullakhanov F.I., Panteleev M.A. 2016. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J. Thromb. Haemost. 14 (10), 2045–2057.
Ahmed M.U., Kaneva V., Loyau S., Nechipurenko D., Receveur N., Le Bris M., Janus-Bell E., Didelot M., Rauch A., Susen S., Chakfé N., Lanza F., Gardiner E.E., Andrews R.K., Panteleev M., Gachet C., Jandrot-Perrus M., Mangin P.H. 2020. Pharmacological blockade of glycoprotein VI promotes thrombus disaggregation in the absence of thrombin. Arterioscler. Thromb. Vasc. Biol. 40 (9), 2127–2142.
Montague S.J., Delierneux C., Lecut C., Layios N., Dinsdale R.J., Lee C.S.-M., Poulter N.S., Andrews R.K., Hampson P., Wearn C.M., Maes N., Bishop J., Bamford A., Gardiner C., Lee W.M., Iqbal T., Moiemen N., Watson S.P., Oury C., Harrison P., Gardiner E.E. 2018. Soluble GPVI is elevated in injured patients: Shedding is mediated by fibrin activation of GPVI. Blood Adv. 2 (3), 240–251.
Montague S.J., Andrews R.K., Gardiner E.E. 2018. Mechanisms of receptor shedding in platelets. Blood. 132 (24), 2535–2545.
Al-Tamimi M., Tan C.W., Qiao J., Pennings G.J., Javadzadegan A., Yong A.S.C., Arthur J.F., Davis A.K., Jing J., Mu F.-T., Hamilton J.R., Jackson S.P., Ludwig A., Berndt M.C., Ward C.M., Kritharides L., Andrews R.K., Gardiner E.E. 2012. Pathologic shear triggers shedding of vascular receptors: a novel mechanism for down-regulation of platelet glycoprotein VI in stenosed coronary vessels. Blood. 119 (18), 4311–4320.
Yakusheva A.A., Butov K.R., Bykov G.A., Závodszky G., Eckly A., Ataullakhanov F.I., Gachet C., Panteleev M.A., Mangin P.H. 2022. Traumatic vessel injuries initiating hemostasis generate high shear conditions. Blood Adv. 6 (16), 4834–4846.
Дополнительные материалы отсутствуют.
Инструменты
Биологические мембраны: Журнал мембранной и клеточной биологии