

Биология моря, 2019, T. 45, № 1, стр. 52-60
Генетическая изменчивость и демографическая история популяции линии четных поколений горбуши Oncorhynchus gorbuscha (Walbaum, 1792) (река Бахура, остров Сахалин) на основе полиморфизма двух митохондриальных генов
А. В. Подлесных 1, *, А. Д. Кухлевский 1, 2, Вл. А. Брыков 1, 2
1 Институт биологии моря им. А.В. Жирмунского, Национальный научный центр морской биологии ДВО РАН
690041 Владивосток, Россия
2 Дальневосточный федеральный университет
690600 Владивосток, Россия
* E-mail: podlesnykh9@yandex.ru
Поступила в редакцию 20.03.2018
После доработки 04.06.2018
Принята к публикации 04.06.2018
Аннотация
Для оценки генетической изменчивости горбуши Oncorhynchus gorbuscha (Walbaum, 1792) четных поколений р. Бахура (о-в Сахалин) проанализирован полиморфизм нуклеотидных последовательностей генов ND2 и Cytb (мтДНК) общей длиной 1809 пар нуклеотидов (пн). В трех выборках горбуши, собранных в разные годы, у 211 образцов выявлено 44 гаплотипа по 39 полиморфным сайтам. С помощью тестов на нейтральность обнаружена высокая вероятность очищающего отбора в исследованных генах. Результаты анализа распределения попарных нуклеотидных замен, тесты на нейтральность и данные байесовского скайлайна поддержали модель расширения линии четных поколений горбуши. Исследование истории дивергенции гаплотипов на основе байесовской хронограммы показало, что дивергенция внутри основных филогрупп произошла в периоды 40.4–54.3 и 4.8–25 тыс. лет.
Тихоокеанский лосось горбуша Oncorhynchus gorbuscha (Walbaum, 1792) является наиболее многочисленным видом среди других представителей рода Oncorhynchus. Вследствие двухлетнего жизненного цикла и однократного размножения этот вид представлен двумя репродуктивно изолированными линиями четных и нечетных лет нереста. Популяции горбуши подвержены значительным колебаниям численности на всем ареале. На основе анализа изменчивости аллозимов митохондриальной ДНК (мтДНК) и микросателлитных маркеров были разработаны различные подходы к изучению генетического разнообразия и популяционной структуры горбуши Азии и Северной Америки (Brykov et al., 1996, 1999; Алтухов и др., 1997; Churikov, Gharrett, 2002; Beacham et al., 2012).
Одним из основных факторов, определяющих уровень внутривидового генетического полиморфизма, является периодическое колебание численности в популяциях как следствие осцилляции климата, изменения емкости среды, миграций и изоляций. Генетическая изменчивость в популяциях горбуши может быть обусловлена как естественным отбором, так и стохастическими процессами, например, генетическим дрейфом (Brykov et al., 1996, 1999; Гордеева, 2012, 2014). Результаты изучения генетической структуры и демографической истории горбуши на основе реконструкций генеалогий мтДНК подтвердили существование репродуктивной изоляции между четными и нечетными репродуктивными линиями (Brykov et al., 1999; Churikov, Gharrett, 2002; Sato, Urawa, 2017). Вместе с тем внутривидовая структура в репродуктивных линиях горбуши выражена слабо, что может определяться высоким потоком генов из-за явления стрэинга (Глубоковский, Животовский, 1986; Алтухов и др., 1997).
Митохондриальная ДНК считается удобным маркером для эволюционного и популяционного анализа благодаря наличию таких свойств, как материнское наследование, быстрая эволюция и отсутствие рекомбинаций (Brown et al., 1979). С помощью коалесцентного моделирования полиморфизма мтДНК можно определить время дивергенции основных генеалогических линий гаплотипов, реконструировать демографическую историю популяций и выявить факторы, влияющие на популяционно-генетическую структуру. Исследование отклонений от нейтральности в молекулярных маркерах, если они находятся под селективным воздействием, может быть полезно не только для анализа филогенетической структуры и демографии популяций, но и для понимания процессов локальных адаптаций видов.
В настоящей работе приведены результаты изучения генетической изменчивости в популяции горбуши р. Бахура (о-в Сахалин) в линии четных поколений на основе полиморфизма нуклеотидных последовательностей генов ND2 и Cytb (мтДНК). Выбор генов основан на проведенных ранее исследованиях с использованием полиморфизма длины рестрикционных фрагментов (ПДРФ) семи фрагментов мтДНК у трех видов тихоокеанских лососей (Churikov et al., 2001). Цель настоящей работы – реконструкция недавней демографической истории горбуши в этом регионе на основе анализа изменчивости двух фрагментов мтДНК.
МАТЕРИАЛ И МЕТОДИКА
Сбор материала. Материалом послужили сборы горбуши четных поколений в р. Бахура (юго-восточный Сахалин). Рыб отлавливали неводом в нижнем течении реки в период нерестовых миграций в 2004, 2006 и 2014 гг. Всего в трех выборках было проанализировано 211 особей (табл. 1).
Таблица 1.
Результаты анализа фрагментов генов мтДНК
| Ген | Длина (пн) | N | Vs | H | h ± SD | π ± SD | Тест | |
|---|---|---|---|---|---|---|---|---|
| Tajima D | Fu Fs | |||||||
| ND2 | 1–909 | 211 | 23 | 24 | 0.422 ± 0.044 | 0.00054 ± 0.00007 | –2.3727*** | –37.043** |
| Cytb | 910–1809 | 211 | 16 | 21 | 0.841 ± 0.011 | 0.00179 ± 0.00007 | –1.1362 | –10.962** |
| Всего | 1–1809 | 211 | 39 | 44 | 0.903 ± 0.011 | 0.00116 ± 0.00005 | –1.9977* | –44.803** |
ДНК выделяли стандартным методом из фиксированных в 96% этаноле тканей сердца и плавников (Sambrook et al., 1989). Для амплификации последовательностей генов использовали следующие пары праймеров: nd2-F 5'-CATACCCCGAAAATGTTGGT-3' и nd2-R 5'-TAGGGCTTTGAAGGCTCTTG-3' для гена ND2 и FishcytB-F/THR-Fish-R для гена Cytb (Sevilla et al., 2007) с незначительными модификациями условий полимеразной цепной реакции (ПЦР). Очищенные продукты амплификации напрямую секвенировали в обоих направлениях на ABI Prism 3500 с использованием стандартного протокола BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, США). Последовательности нуклеотидов выравнивали с помощью программы Geneious R11 (www.geneious.com). Полученные фрагменты генов ND2 и Cytb (MG911273–MG911483, MG911484–MG911694) депонировали в GenBank.
Анализ молекулярных данных. Последовательности выравнивали с использованием алгоритма MAFFT в программе Geneious R11. Длина матриц после выравнивания составила 909 пн для гена ND2 и 900 пн для гена Cytb. Комбинированный набор данных составил 1809 пн: 1770 консервативных, 39 полиморфных сайтов (из них 30 синонимичных и 9 несинонимичных) и 22 филогенетически значимых сайта.
Оценку гаплотипического (h) и нуклеотидного (π) разнообразия, распределение попарных нуклеотидных различий (mismatch distribution) между последовательностями генов ND2 и Cytb проводили в программе DnaSP v5 (Librado, Rozas, 2009). Для анализа эволюционных процессов в популяции на уровне нуклеотидных последовательностей мтДНК были использованы тесты Tajima D и Fu Fs, реализованные в программе DnaSP v5. Медианные сети гаплотипов реконструировали в программе Network 5 (http://www.fluxus-engineering.com/sharenet.htm) по алгоритму MJ.
Дифференциацию между выборками разных лет анализировали методом AMOVA в пакете программ Arlequin 3.5 (Excoffier, Lischer, 2010); для оценки различий использовали коэффициент FST, значимость оценивали на основе непараметрического подхода с использованием 17000 пермутаций.
Кроме тестов на селективную нейтральность с помощью Z-теста на основе бутстреп-метода определяли вероятности направленности отбора. Число синонимичных (dS) и несинонимичных (dN) замещений на сайт оценивали с помощью метода Нея–Годжобори (Nei, Gojobori, 1986) в программах Mega 6.0 (Tamura et al., 2013) и DnaSP v5. Отношение синонимичных и несинонимичных замещений (ω = dN/dS) использовали для определения сайтов, находящихся под действием отбора. Для этих целей применяли такие методы, как SLAC (single likelihood ancestor counting), FEL (fixed effects likelihood), REL (random effects likelihood) (Kosakovsky Pond, Frost, 2005) и FUBAR (fast unconstrained Bayesian approximation) (Murrell et al., 2013), реализованные в пакете программ HyPhy и доступные на сервере Datamonkey (www.datamonkey.org) (Kosakovsky Pond et al., 2005). Анализ сайтов проводили по NJ алгоритму на основе модели нуклеотидного замещения TN93, выбранной с помощью программы jModelTest (Posada, 2008).
Демографическую историю популяции горбуши р. Бахура изучали на основе генеалогий гаплотипов и времени их дивергенции с помощью байесовского подхода (BI) в пакете программ BEAST 1.8.4 (Drummond et al., 2012). В модуле BEAUti (BEAST 1.8.4) был задан диапазон основных условий: некоррелированные свободные часы (uncorrelated relaxed clock) с логарифмически нормальным распределением скоростей эволюции, модель нуклеотидного замещения для комбинированных данных TN93 и параметрами MCMC 50 × 106 генераций (отбор каждой тысячной генерации). Оптимальные параметры модели молекулярных часов для выравнивания демографических функций байесовских контурных диаграмм подбирали в программе Tracer 1.6.0 (Rambaut et al., 2014) на основе значений максимального правдоподобия (log Bayes Factor) (Suchard et al., 2001). Байесовские филогенетические деревья суммировали в программе TreeAnnotator (BEAST 1.8.0) и визуализировали с помощью программы FigTree (Rambaut, 2014).
РЕЗУЛЬТАТЫ
Изменчивость мтДНК. В результате анализа комбинированных гаплотипов мтДНК, что соответствовало 10.89% митохондриального генома, у 211 образцов горбуши было выявлено 44 гаплотипа по 39 полиморфным сайтам. Анализ нуклеотидных замен кодирующих участков ND2 и Cytb выявил мутации, в том числе приводящие к заменам аминокислот. Было обнаружено 30 синонимичных (S) и 9 несинонимичных (N) мутаций. Количество вариабельных сайтов (Vs) и гаплотипов (H) в последовательностях гена ND2 было больше, а гаплотипическое (h) и нуклеотидное (π) разнообразие оказалось значительно меньше, чем в последовательностях гена Cytb (табл. 1). Сходные тенденции при сравнении гаплотипического и нуклеотидного разнообразия по митохондриальным маркерам отмечены и для выборок разных лет. В целом для комбинированных данных не обнаружено существенных различий по гаплотипическому и нуклеотидному разнообразию между выборками, отобранными в разные годы (табл. 2). Результаты AMOVA показали, что 0.15% вариации изменчивости последовательностей генов ND2 и Cytb приходится на различия между выборками, а 99.85% вариабельности – на внутрипопуляционную составляющую. Дифференциация между выборками составила: FST = 0.00153 по всем локусам. Пермутационный тест не выявил значимых различий между выборками горбуши разных лет по значениям FST (р > 0.1).
Таблица 2.
Генетические характеристики выборок горбуши р. Бахура на основе комбинированных фрагментов мтДНК. Результаты анализа тестов на нейтральность и очищающий отбор
| Выборка | N | Vs | H | h ± SD | π ± SD | Тест | Значение P (метод Нея–Годжобори нейтральность/очищающий отбор) | |
|---|---|---|---|---|---|---|---|---|
| Tajima D | Fu Fs | |||||||
| bh2004 | 83 | 26 | 26 | 0.902 ± 0.020 | 0.00121 ± 0.00008 | –1.7773 | –19.663, P = 0.000** |
0.00377*/0.00181* |
| bh2006 | 81 | 24 | 27 | 0.904 ± 0.018 | 0.00110 ± 0.00008 | –1.8530* | –23.568, P = 0.000** |
0.00507*/0.00214* |
| bh2014 | 47 | 16 | 17 | 0.898 ± 0.027 | 0.00119 ± 0.0001 | –1.2685 | –9.561, P = 0.002** |
0.00307*/0.00149* |
| Всего | 211 | 39 | 44 | 0.903 ± 0.011 | 0.00116 ± 0.00005 | –1.9977* | –27.066, P = 0.000** |
0.00478*/0.00211* |
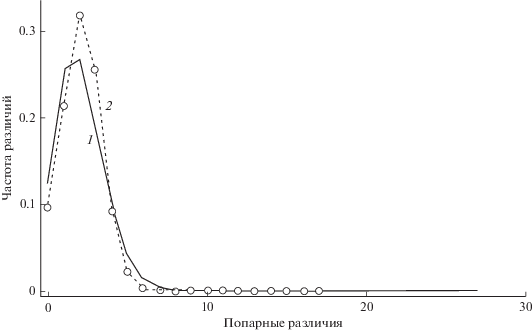
Сравнительный анализ распределений попарных нуклеотидных замен фрагментов мтДНК показал, что гаплотипы последовательностей ND2, Cytb и комбинированные данные имеют унимодальное распределение (рис. 1). Большинство попарных сравнений находится в области малых генетических дистанций и показывает хорошее совпадение с ожидаемыми распределениями.
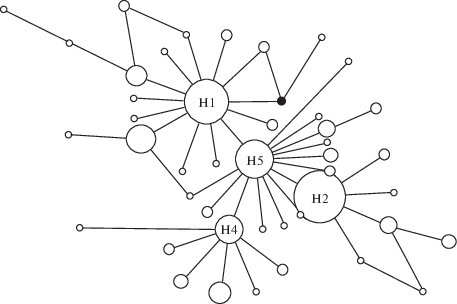
Рис. 1.
Медианная сеть гаплотипов на основе комбинированных последовательностей генов ND2 и Cytb (мтДНК) горбуши. Медианный вектор выделен темной точкой. Длина линии между гаплотипами пропорциональна количеству нуклеотидных замен. Размер окружностей пропорционален количеству особей.
Тесты на нейтральность и отбор. В результате проведенных тестов получены достоверно значимые отрицательные значения параметров Tajima D и Fu Fs для последовательностей фрагмента ND2 и комбинированных данных (табл. 1). Для фрагментов гена Cytb было отмечено отрицательное (р > 0.1) значение Tajima D. Тест на нейтральность по методу Нея–Годжобори для каждой выборки и для популяции в целом также показал достоверно отрицательные значения параметров для альтернативной гипотезы dN–dS (табл. 2). Поскольку в результате сравнительного анализа распределений попарных нуклеотидных замен и тестов на селективную нейтральность был выявлен избыток редких гаплотипов (отрицательные значения параметров Tajima D и Fu Fs) в последовательностях генов ND2 и Cytb, можно с высокой вероятностью исключить компоненту случайности для эволюции этих последовательностей. Действительно, анализ значений Z-теста для альтернативной гипотезы (dN < dS) подтверждает вероятность очищающего отбора на молекулярном уровне на 95% уровне значимости для всех выборок горбуши (табл. 2).
Средняя величина ω = dN/dS по методу SLAC составила 0.093 для 603 кодонов комбинированных данных мтДНК. Метод REL распознал 9 кодонов, находящихся под очищающим отбором. Для методов SLAC и FEL количество кодонов, находящихся под действием отрицательного отбора, составило соответственно два и 10 с двумя общими кодонами при р = 0.05. С помощью метода FUBAR было найдено четыре сайта, свидетельствующих об очищающем отборе при апостериорной вероятности PP ≥ 0.95. В целом от двух до четырех кодонов, идентифицированных методом FUBAR, также было выявлено методами SLAC, FEL и REL. С помощью использованных методов не обнаружено сайтов, находящихся под положительным отбором.
Генеалогия гаплотипов мтДНК и историческая демография. В медианной сети, иллюстрирующей филогенетические отношения между 44 гаплотипами из 211 особей горбуши (рис. 2), можно выделить четыре основных гаплотипа с множеством единичных мутаций с предполагаемым центральным гаплотипом H5. Гаплотипы H1, H2 и H4 разделены одним мутационным шагом от предположительно предкового гаплотипа H5. Сеть имеет пять петель альтернативных связей, обусловленных, вероятно, явлением гомоплазии, повторными и обратными мутациями. Дивергенция на основе нескорректированных p-дистанций между основными группами гаплотипов горбуши составила от 0.06 до 0.11%. Используя среднюю скорость эволюции комбинированных маркеров по Cytb и NADH калибровкам (p = 0.73% за 1 млн лет) (McKay et al., 1996; Crête-Lafrenière et al., 2012), можно определить начальный этап дивергенции гаплотипов в интервале от 82 до 150 тыс. лет назад.
Рис. 2.
Распределение различий между гаплотипами (mismatch distribution) комбинированных последовательностей генов ND2 и Cytb горбуши при изменяющейся численности популяции. Ожидаемая (1) и наблюдаемая (2) частота попарных различий между последовательностями при увеличении численности популяции.
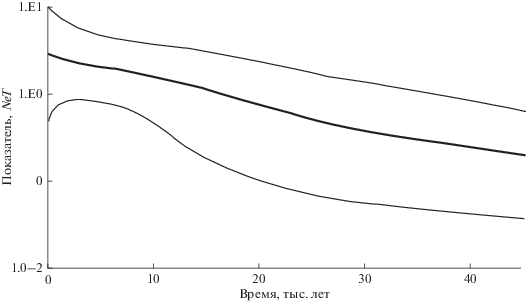
Байесовская хронограмма показала, что дивергенция основных групп гаплотипов началась около 112.8 тыс. лет назад (время дивергенции от гипотетического предкового гаплотипа, кластеры А и B по отношению к остальным кластерам) (рис. 3). На консенсусном филогенетическом дереве каждый основной гаплотип представлен двумя кластерами (обозначены буквами), которые соответствуют структуре и количеству гаплотипов, выделенных на основе медианной сети. Время дивергенции внутри основных кластеров гаплотипов на две филогруппы (А–H) в среднем составило 40.4–54.3 тыс. лет. Обнаружены также неоднократные значимые периоды дивергенции внутри основных филогрупп в интервале от 4.8 до 25 тыс. лет. Анализ эволюционной истории популяции горбуши на основе байесовского скайлайна с медианными значениями NeT показал постепенное увеличение численности популяции в 95% интервале апостериорной плотности распределения оценок, начиная c периода 45 тыс. лет назад (рис. 4).
Рис. 3.
Консенсусное BI дерево, показывающее время дивергенции основных филогрупп гаплотипов горбуши (тыс. лет) на основе комбинированных последовательностей двух фрагментов мтДНК. Значения апостериорных вероятностей PP ≥ 0.95 в узлах дерева обозначены черными точками. Справа от дерева буквами обозначены основные филогруппы гаплотипов.
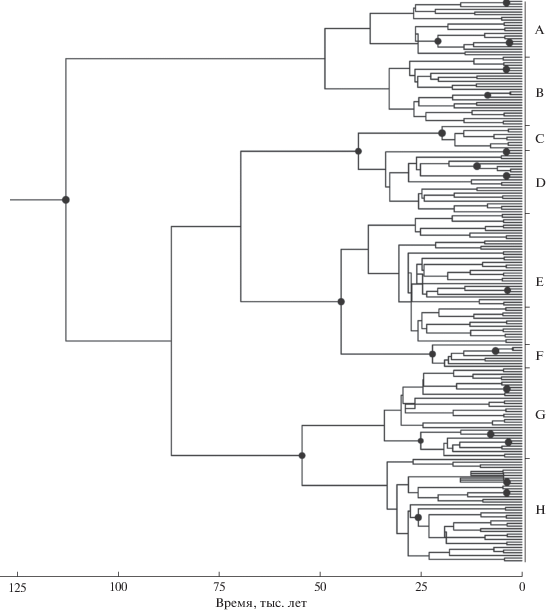
Рис. 4.
Изменение показателя NeТ (произведение эффективной численности популяции и продолжительности одного поколения) со временем (тыс. лет), рассчитанное на основе байесовского скайлайна. Жирная линия – медианные значения NeT, тонкие линии – 95% интервал апостериорной плотности распределения оценок.
ОБСУЖДЕНИЕ
В результате исследований установлено, что фрагменты гена ND2 более полиморфны по количеству вариабельных сайтов, чем последовательности гена Cytb, а гаплотипическое и нуклеотидное разнообразие оказалось существенно ниже. Нуклеотидное разнообразие фрагментов генов сопоставимо с результатами анализа ПДРФ митогеномов азиатской и аляскинской горбуши (Churikov, Gharrett, 2002). Однако последовательности гена Cytb оказались более консервативными и характеризовались меньшей разрешающей способностью для методов коалесцентного анализа, чем последовательности гена ND2.
Сравнительно низкий уровень генетического разнообразия горбуши по фрагменту ND2 может свидетельствовать как об относительно небольшом возрасте популяции, так и о прохождении ее через стадию резкого сокращения численности (“бутылочное горлышко”), возможно, неоднократно. Для последовательностей генов ND2 и Cytb было обнаружено унимодальное распределение попарных нуклеотидных замен (рис. 1), что может свидетельствовать о росте численности популяции в недавнем прошлом и о процессах расселения и увеличения численности основателя. С помощью методов коалесцентного анализа был выявлен избыток редких мутаций в генах ND2 и Cytb, а также получены достоверно отрицательные значения параметров Tajima D и Fu Fs (p < 0.01) как для фрагмента ND2, так и для комбинированных гаплотипов. В целом отрицательные значения статистик Tajima D и Fu Fs свидетельствуют об избытке редких аллелей, что можно было бы ожидать при увеличении численности горбуши после недавнего “бутылочного голышка” или в результате очищающего отбора. Данные анализа попарного распределения нуклеотидных замен и тестов на нейтральность согласуются с моделью увеличения численности для популяции четной линии горбуши р. Бахура.
Очищающий или отрицательный отбор на уровне нуклеотидных последовательностей можно охарактеризовать по соотношению числа синонимичных (dS) и несинонимичных (dN) замещений на сайт. Очищающий отбор характерен для нуклеотидных последовательностей, кодирующих функциональные гены, и наиболее часто встречается в диких популяциях. Известно, что митохондриальные гены играют ключевую роль в биоэнергетике клеточных процессов и адаптациях организма к изменению окружающей среды (Gershoni et al., 2009). Поэтому исследования несинонимичных полиморфизмов мтДНК можно использовать не только для анализа демографической экспансии популяций, но и для оценки предполагаемой роли отбора при изучении локальных адаптаций рыб.
В работах по эволюции митогеномов тихоокеанских лососей и сиговых рыб приведены доказательства наличия положительного отбора по некоторым сайтам в последовательностях гена ND5 и слабого очищающего отбора в гене ND2 (Garvin et al., 2011; Jacobsen et al., 2016). Было также идентифицировано адаптивное изменение гена Cytb у европейского анчоуса Engraulis encrasicolus, обусловленное разными температурными условиями среды обитания (Silva et al., 2014). В исследованиях Macquaria australasica (Percichthyidae) из разной среды обитания было показано, что митогеномы у этого вида развивались под очищающим отбором разной степени выраженности в зависимости от градиентов окружающей среды (Pavlova et al., 2017).
Использование Z-теста при анализе эволюции комбинированных нуклеотидных последовательностей ND2 и Cytb показало высокую вероятность (p < 0.05) очищающего отбора в исследуемых генах горбуши. При этом с помощью разных методов исследования направленности отбора по кодонам (ω = dN/dS) с высокой вероятностью было обнаружено от двух до 10 сайтов, находящихся под очищающим отбором, и не обнаружено ни одного сайта под положительным отбором. Функциональная значимость аминокислотных замен остается неясной, однако можно полагать, что такие мутации в генах ND2 и Cytb мтДНК горбуши могут быть связаны с метаболическими различиями у особей.
На медианной сети основные гаплотипы демонстрировали звездообразные генеалогии, свидетельствующие о росте популяции горбуши р. Бахура. Аналогичные структуры генеалогий были выявлены ранее для двух линий в популяциях азиатской и аляскинской горбуши (Churikov, Gharrett, 2002). Незначительная глубина генеалогии может отражать неоднократное уменьшение численности популяций из-за плейстоценовых ледниковых максимумов, а звездообразные генерации гаплотипов свидетельствуют о демографическом расширении, соответствующем началу голоценового межледникового интервала. С этим предположением согласуются и данные настоящего исследования: влияние очищающего отбора, высокий уровень гомоплазии и генетическая близость основных гаплотипов. Реконструкция демографической истории популяции горбуши на основе анализа байесовского скайлайна показала постепенное увеличение эффективной численности популяции начиная с периода около 45 тыс. лет назад (рис. 3). Время дивергенции, рассчитанное на основе байесовского консенсусного филогенетического дерева, между основными группами гаплотипов составило от 70 до 112.8 тыс. лет (начало дивергенции). Среднее время быстрого увеличения численности популяции четной линии горбуши на севере о-ва Хоккайдо, полученное на основе изменчивости контрольного региона мтДНК, оказалось близким к нашим оценкам и составило 73–128 тыс. лет назад (Sato, Urawa, 2017).
Временные оценки экспансии горбуши р. Бахура и популяций о-ва Хоккайдо по палеонтологическим данным из Охотского моря датируются межледниковыми фазами позднего плейстоцена (Nürnberg, Tiedemann, 2004; Wang et al., 2017). На байесовской хронограмме также можно выделить значимые периоды дивергенции внутри основных филогрупп в интервале 40.4–54.3 тыс. лет и неоднократные периоды дивергенции – примерно от 4.8 до 25 тыс. лет. Эти периоды образования дивергентных филогрупп могут определяться фактом существования умеренно теплых фаз в позднем плейстоцене (вюрме) и голоценового (атлантического) оптимума (Korotky et al., 1997; Микишин, Гвоздева, 2017). Таким образом, в отличие от аляскинской горбуши, для горбуши южной части ареала характерны более ранние датировки времени расширения популяций.
Исследование дифференциации горбуши на большей части ареала показало, что индексы FST по результатам AMOVA на разных уровнях иерархических и пространственных структур популяций горбуши четных поколений по микросателлитным маркерам сравнительно малы (Beacham et al., 2012; Гордеева, 2014). Полученные нами оценки дифференциации выборок горбуши в разные годы по локусам ND2 и Cytb оказались сходными с данными этих авторов. Отсутствие различий между выборками разных лет подтверждает, что на нерест приходят преимущественно поколения горбуши из одной популяции. Об этом свидетельствует и наличие отрицательного отбора в исследуемых митохондриальных генах. В целом для горбуши на большей части ареала характерны высокие значения гаплотипического и низкие показатели нуклеотидного разнообразия мтДНК, что ожидается в популяциях, подверженных историческому уменьшению численности, которое сопровождалось ростом и быстрым расширением (Churikov, Gharrett, 2002). Результаты исследования полиморфизма фрагментов мтДНК подтверждают такое состояние для популяции четной линии горбуши р. Бахура.
Работа выполнена при частичной финансовой поддержке гранта РНФ (№ 14-50-00034).
Список литературы
Алтухов Ю.П., Салменкова Е.А., Омельченко В.Т. Популяционная генетика лососевых рыб. М.: Наука. 1997. 287 с.
Глубоковский М.К., Животовский Л.А. Популяционная структура горбуши: система флуктуирующих стад // Биол. моря. 1986. № 2. С. 39–44.
Гордеева Н.В. Высокие оценки дифференциации популяций горбуши по локусу главного комплекса гистосовместимости МНС I-A1 поддерживают гипотезу “локальных стад"// Вопр. ихтиологии. 2012. Т. 52. № 1. С. 72–81.
Гордеева Н.В. Оценки дифференциации популяций по микросателлитным маркерам у горбуши Oncorhynchus gorbuscha могут быть занижены вследствие ее высокой численности // Вопр. ихтиологии. 2014. Т. 54. № 3. С. 332–343.
Микишин Ю.А., Гвоздева И.Г. Ландшафтно-климатические изменения на юге Сахалина в средне-позднеатлантическое время голоцена // Успехи соврем. естествознания. 2017. № 12. С. 207–214.
Beacham T.D., McIntosh B., MacConnachie C. et al. Population structure of pink salmon (Oncorhynchus gorbuscha) in British Columbia and Washington, determined with microsatellites // Fish. Bull. 2012. V. 110. № 2. P. 242–256.
Brown W.M., George M.Jr., Wilson A.C. Rapid evolution of animal mitochondrial DNA // Proc. Natl. Acad. Sci. U.S.A. 1979. V. 76. P. 1967–1971.
Brykov Vl.A., Polyakova N., Skurikhina L.A., Kukhlevsky A.D. Geographical and temporal mitochondrial DNA variability in populations of pink salmon // J. Fish. Biol. 1996. V. 48. P. 899–909.
Brykov Vl.A., Polyakova N., Skurikhina L.A. et al. Analysis of mtDNA indicates weak temporal genetic heterogeneity in pink salmon spawning runs in two rivers on Sakhalin Island // J. Fish. Biol. 1999. V. 55. P. 617–635.
Churikov D., Gharrett A.J. Comparative phylogeography of the two pink salmon broodlines: an analysis based on a mitochondrial DNA genealogy // Mol. Ecol. 2002. V. 11. P. 1077–1101.
Churikov D., Matsuoka M., Luan X. et al. Assessment of concordance among genealogical reconstructions from various mtDNA segments in three species of Pacific salmon (genus Oncorhynchus) // Mol. Ecol. 2001. V. 10. P. 2329–2339.
Crête-Lafrenière A., Weir L.K., Bernatchez L. Framing the Salmonidae family phylogenetic portrait: a more complete picture from increased taxon sampling // PLoS ONE. 2012. V. 7. № 10. P. e46662.
Drummond A.J., Suchard M.A., Xie D., Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7 // Mol. Biol. Evol. 2012. V. 29. № 8. P. 1969–1973.
Excoffier L., Lischer H.-E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows // Mol. Ecol. Res. 2010. V. 10. P. 564–567.
Garvin M.R., Bielawski J.P., Gharrett A.J. Positive Darwinian selection in the piston that powers proton pumps in complex I of the mitochondria of Pacific salmon // PLoS ONE. 2011. V. 6. № 9. e24127.
Gershoni M., Templeton A.R., Mishmar D. Mitochondrial bioenergetics as a major motive force of speciation // BioEssays. 2009. V. 31. P. 642–650.
Jacobsen M.W., da Fonseca R.R., Bernatchez L., Hansen M.M. Comparative analysis of complete mitochondrial genomes suggests that relaxed purifying selection is driving high nonsynonymous evolutionary rate of the NADH2 gene in whitefish (Coregonus ssp.) // Mol. Phylogenet. Evol. 2016. V. 95. P. 161–170.
Kosakovsky Pond S.L., Frost S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection // Mol. Biol. Evol. 2005. V. 22. № 5. P. 1208–1222.
Kosakovsky Pond S.L., Frost S.D.W., Muse S.V. HyPhy: hypothesis testing using phylogenies // Bioinformatics. 2005. V. 21. № 5. P. 676–679.
Korotky A., Grebennikova T., Razjigaeva N. et al. Marine terraces of Western Sakhalin Island // Catena. 1997. V. 30. № 1. P. 61–81.
Librado P., Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data // Bioinformatics. 2009. V. 25. P. 1451–1452.
McKay S.J., Devlin R.H., Smith M.J. Phylogeny of Pacific salmon and trout based on growth hormone type-2 and mitochondrial NADH dehydrogenase subunit 3 DNA sequences // Can. J. Fish. Aquat. Sci. 1996. V. 53. № 5. P. 1165–1176.
Murrell B., Moola S., Mabona A. et al. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection // Mol. Biol. Evol. 2013. V. 30. P. 1196–1205.
Nei M., Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions // Mol. Biol. Evol. 1986. V. 3. № 5. P. 418–426.
Nürnberg D., Tiedemann R. Environmental change in the Sea of Okhotsk during the last 1.1 million years // Paleoceanography. 2004. V. 19. PA4011.
Pavlova A., Gan H.M., Lee Y.P. et al. Purifying selection and genetic drift shaped Pleistocene evolution of the mitochondrial genome in an endangered Australian freshwater fish // Heredity. 2017. V. 118. № 5. P. 466–476.
Posada D. jModelTest: phylogenetic model averaging // Mol. Biol. Evol. 2008. V. 25. P. 1253–1256.
Rambaut A. FigTree 1.4.2; a graphical viewer of phylogenetic trees. 2014. http://tree.bio.ed.ac.uk/software/figtree/. Дата просмотра 20.03.2018.
Rambaut A., Suchard M.A., Xie D., Drummond A.J. Tracer v1.6. 2014. http://beast.bio.ed.ac.uk/software/tracer/. Дата просмотра 20.03.2018.
Sambrook J., Fritsch E.F., Maniatis T. Molecular cloning: A laboratory manual. New York: Cold Spring Harbor Lab. Press. 1989. 1626 p.
Sato S., Urawa S. Genetic variation of Japanese pink salmon populations inferred from nucleotide sequence analysis of the mitochondrial DNA control region // Environ. Biol. Fishes. 2017. V. 100. P. 1355–1372.
Sevilla R.G., Diez A., Norén M. et al. Primers and polymerase chain reaction conditions for DNA barcoding teleost fish based on the mitochondrial cytochrome b and nuclear rhodopsin genes // Mol. Ecol. Notes. 2007. V. 7. P. 730–734.
Silva G., Lima F.P., Martel P., Castilho R. Thermal adaptation and clinal mitochondrial DNA variation of European anchovy // Proc. Roy. Soc. В. 281. 2014. 2014.1093. http://dx.doi.org/. Дата просмотра 20.03.2018. doi 10.1098/rspb.2014.1093
Suchard M.A., Weiss R.E., Sinsheimer J.S. Bayesian selection of continuous-time Markov chain evolutionary models // Mol. Biol. Evol. 2001. V. 18. P. 1001–1013.
Tamura K., Stecher G., Peterson D. et al. MEGA 6: Molecular Evolutionary Genetics Analysis version 6.0. // Mol. Biol. Evol. 2013. V. 30. P. 2725–2729.
Wang K., Shi X., Zou J. et al. Sediment provenance variations in the southern Okhotsk Sea over the last 180 ka: Evidence from light and heavy minerals // Palaeogeogr., Palaeoclimatol., Palaeoecol. 2017. V. 479. P. 61–70.
Дополнительные материалы отсутствуют.