

Физиология человека, 2020, T. 46, № 2, стр. 94-111
Роль регуляторных белков Ерас в физиологии и патологии сердечно-сосудистой системы. Часть I. Роль белков Ерас в физиологии и патологии сосудистого русла
С. А. Крыжановский 1, *, Т. Д. Никифорова 1, А. Д. Дурнев 1
1 ФГБНУ НИИ фармакологии имени В.В. Закусова
Москва, Россия
* E-mail: sak-538@yandex.ru
Поступила в редакцию 03.07.2019
После доработки 30.09.2019
Принята к публикации 04.12.2019
Аннотация
Обзор посвящен описанию вклада регуляторных белков Ерас в физиологию и патологию сердечно-сосудистой системы. В первой части обзора приведена структура регуляторных белков Ерас, а также подробно рассмотрена роль белков Ерас в регуляции тонуса, проницаемости сосудистого русла, ангиогенезе и пролиферации эндотелиальных и гладкомышечных клеток сосудов, а также их вклад в развитие патологии сосудов.
Открытый в 1957 г. американским биохимиком E.W. Sutherland, Jr циклический 3',5'-аденозинмонофосфат (сАМР) является одним из основных вторичных мессенджеров, передающих регулирующие сигналы от нейромедиаторов и ряда эндогенных биологически активных веществ на исполнительные структуры клетки [1]. Изначально полагали, что единственным аллостерическим эффектором сАМР является открытый в 1968 г. фермент – сАМР-зависимая протеинкиназа или протеинкиназа А (РКА) [2]. Неактивная РКА представляет собой комплекс из двух димерных регуляторных (R) и двух мономерных каталитических (К) субъединиц. Активация РКА происходит в результате взаимодействия каждой R-субъединицы с двумя молекулами сАМР и последующей диссоциации от них К-субъединиц, которые и фосфорилируют остатки серина (Ser) и треонина (Thr) на многочисленных внутриклеточных белках-мишенях. К этим внутриклеточным мишеням, в частности, относятся фосфоламбан – PLN (может также фосфорилироваться кальмодулин-зависимой протеинкиназой II, CaMKII), рианодиновые рецепторы (RyR), тропонин I, миозин-связывающий белок С (cMyBRC), трансмембранные медленные потенциалзависимые Ca2+ каналы L-типа, а также фактор транскрипции сАМР (cyclic AMP response element-binding protein, CREB) [3–5], т.е. внутриклеточные мишени, ответственные за регуляцию процессов, обеспечивающих сократительный статус кардиомиоцитов и их электромеханическое сопряжение. Помимо этого, связываясь с регуляторными субъединицами РКА, сАМР контролирует процессы адгезии, миграции, пролиферации и дифференцировки клеток, апоптоз и транскрипцию генов [4]. Практически в течение 30 лет РКА рассматривали в качестве единственного эффектора сАМР (исключение составляли каналы, контролируемые циклическими нуклеотидами /CNG/ – cyclic nucleotide-gated channels, в частности управляемые циклическими нуклеотидами гиперполяризационно-активируемые каналы /HCN/ – hyperpolarization-activated cyclic nucleotide-gated channels) [6], однако в конце ХХ в. появились публикации, свидетельствующие о том, что не все клеточные эффекты сАМР могут быть опосредованы исключительно PKA и было высказано предположение, что в клетках экспрессируется “неизвестная PKA-подобная молекула” [7, 8].
В декабре 1998 г. в журналах “Nature” и “Science” были опубликованы две независимые статьи. Американские исследователи из Массачусетского технологического института сообщили о том, что ими был идентифицирован сАМФ-зависимый белок, который без участия РКА активировал малые GEFазы (cAMP-GEFs) Rap суперсемейства белков Ras (Rat sarcoma) [9]. Аналогичные данные были получены и голландскими учеными из университета Утрехта, которые помимо этого клонировали ген, кодирующий этот белок, и назвали его (белок) сАМР-регулируемый фактор обмена гуаниловых нуклеотидов (cAMP-GEF) или обменный белок, напрямую активируемый сАМР (exchange protein directly activated by cAMP, Epac) [10]. Таким образом, было показано, что в клетках существует альтернативный, не зависимый от РКА, путь активации/блокады внутриклеточных сигнальных путей. Несколько позже появились сообщения о том, что РКА и белки Ерас в одной и той же клетке могут инициировать не зависимые друг от друга, в том числе избыточные и/или противоположные эффекты [11, 12]. В настоящее временя известно, что белки Ерас, как минимум, экспрессируются в нейронах ЦНС (фронтальная кора, гиппокамп), клетках гладкой мускулатуры бронхиального дерева, иммунокомпетентных клетках, кортикальных нефронах, β-клетках поджелудочной железы, клетках гладкой мускулатуры сосудов (в том числе коронарных), клетках сосудистого эндотелия и кардиомиоцитах [13, 14].
Показано, что белки Ерас играют ключевую роль в регуляции базисных внутриклеточных сигнальных путей, ответственных за поддержание внутриклеточного гомеостаза, а их гипер/гипоэкспрессия лежит в основе патогенеза многих патологических процессов, что позволяет рассматривать их как принципиально новую биомишень для создания оригинальных, высокоэффективных лекарственных средств [13].
Следует отметить, что в последнее время помимо белков Ерас идентифицировaны и другие эффекторы cAMP – циклический нуклеотидный рецептор, участвующий в регуляции функциональной активности сперматозоидов (cyclic nucleotide receptor involved in sperm function; CRIS) [15], и экспрессирующийся в мышечной ткани белок, содержащий Popeye домен (рopeye domain containing protein; POPDC) [16].
Данный обзор посвящен роли сигнальных белков Ерас в физиологии и патофизиологии сердечно-сосудистой системы.
Белки Ерас: классификация, структура, активация
Белки Ерас по сравнению с РКА эволюционно являются более “молодой” сигнальной молекулой, поскольку показано, что РКА экспрессируется и в организме одноклеточных эукариотов, например грибков (дрожжей) из класса сахаромицетов (Saccharomyces cerevisiae), тогда как белки Ерас идентифицированы только у многоклеточных организмов, преимущественно у млекопитающих [17].
Выделяют две изоформы сигнальных белков Ерас – Ерас1 или cAMP-GEF-I (молекулярная масса около 100 kDa) и Ерас2 или cAMP-GEF-II (молекулярная масса около 110 kDa), которые кодируются различными генами. У людей EPAC1 кодируется геном RAPGEF3, содержащим 28 экзонов и расположенным на 12-ой хромосоме (12q13.11: 47,734,367–47,771,041), в то время как ген EPAC2 RAPGEF4 расположен на хромосоме 2 (2q31.1) и содержит 31 экзон [17]. Белки Eрас по своей структуре представляют единую полипептидную молекулу, тогда как РКА состоит из отдельных R и С-субъединиц, которые кодируются различными генами. Если транскрипция гена RAPGEF3 влечет за собой образование только одной изофомы Ерас1, то транскрипция гена RAPGEF4 сопровождается образованием трех изоформ белка Ерас2: Ерас2А (первоначально названный Ерас2), Ерас2В и Ерас2С. Полипептидная цепочка белка Ерас1 состоит из 923 аминокислот, белка Ерас2А – из 1011, а белков Ерас2В и Ерас2С – из 867 и 791 аминокислот соответственно.
Помимо двух изоформ сигнальных белков Ерас в 2000 г. была идентифицирована и третья изоформа белков Ерас – Repac (related to Epac, GFR, MR-GEF), которая в отличие от белков Ерас1 и Ерас2 не имеет в своей структуре регуляторного региона (рис. 1) [18]. Авторы этой статьи показали, что при определенных условиях Repac может активировать малые GТFазы Rap1 и Rap2, однако биологическая роль белков Repac до настоящего времени остается не ясной [13]. Следует отметить, что если белки Ерас1 и Ерас2А в той или иной степени экспрессируются во многих органах и тканях организма, то для белков Ерас2В и Ерас2С отмечена существенная органоспецифичность. Так, белки Ерас2В идентифицированы в β-клетках поджелудочной железы и секреторных клетках надпочечников, а Ерас2С – только в клетках печени [19, 20].
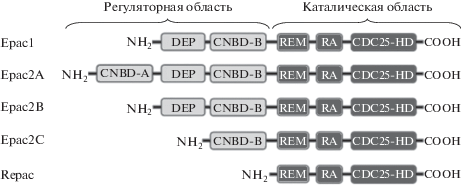
Сигнальные белки Ерас (Ерас1 и Ерас2) близки друг к другу и по своей структуре являются мультидоменными белками, содержащими NH2‑концевую регуляторную область и COOH-концевой каталитический регион (рис. 1). Полагают, что NH2-концевая регуляторная область Epac произошла из R субъединицы PKA, в то время как COOH-концевой каталитический регион по своей структуре наиболее тесно связан с представителями суперсемейства Rаs-факторами обмена гуаниловых нуклеотидов (guanine-nucleotide-exchange factors, GТFs) [22].
Каталитический регион у всех белков Ерас имеет идентичную структуру и состоит из Ras-обменного домена REM, Ras-ассоциированного домена RА и С-концевого СDC25-гомологичного домена – СDC25-HD. Домен REM необходим для поддержания стабильности каталитического региона [18]. Домен RА выполняет двоякую функцию – инициацию биологического эффекта и цитозольную локализацию белка, а в случае белка Ерас2С – его фиксацию на внутренней поверхности цитоплазматической мембраны. Домен СDC25-HD ответственен за обмен гуаниловых нуклеотидов белков Rар и локализацию белков в области ядерных поровых комплексов [23].
Регуляторный регион белка Ерас1 содержит в своем составе сАМР-связывающий домен CNBD-B (или CBD-В), который помимо взаимодействия c сАМР обеспечивает контакт белков Ерас1 с микротрубочками [24], и N-концевой домен DEP (Disheveled/Egl-10/pleckstrin domain), ответственный за локализацию белка в области внутренней поверхности клеточной мембраны [25] и обеспечивающий транслокацию Ерас1 к митохондриям [25].
Регуляторный регион белка Ерас2А также содержит CNBD-B и DEP домены, однако N-концевым доменом этого белка является не DEP домен, а присоединенный к нему дополнительный низкоаффинный CNBD-А (CBD-А) домен, который в отличие от CNBD-B домена обладает меньшим сродством к сАМР и не может индуцировать активность малых GТFаз после связывания сАМР [25, 26]. CNBD-А и DEP домены принимают участие в регуляции внутриклеточной локализации белка Ерас2А [23].
Структура регуляторного региона белка Ерас2В аналогична таковой, известной для белка Ерас1 [23]. Регуляторный регион белка Ерас2С содержит только один N-концевой домен CNBD-B.
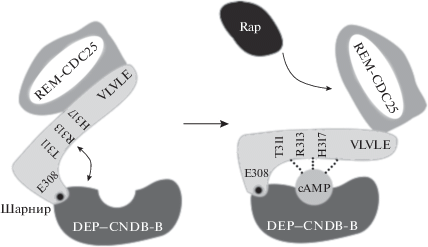
Между доменом CNBD-B регуляторного региона и REM доменом каталитической области белков Ерас располагается псевдо-β-складка, так называемый “шарнир” или “коммутатор”, содержащий в своем составе консервативную последовательность (мотив) VLVLE (321VLVLE325), состоящий из 4-х доменов – Е308, Т311, R313 и Н317 (рис. 2) [27]. Шарнир необходим для аутоингибирования белков Eрас. Домен Е308 прилежит к CNBD-B домену регуляторного региона, а консервативная последовательность VLVLE – к REM домену каталитической области (рис. 2). Мутация мотива VLVLE, в результате которой консервативная последовательность VLVLE замещается остатками аланина AAAAA, сопровождается самоактивацией модифицированного белка Ерас (АЛА)5 в отсутствии сАМР [27].
Рис. 2.
Схематическое отображение вклада последовательности VLVLE в регуляцию связи между регуляторной и каталитической областью белков Ерас (по [27]). Объяснение в тексте.
Как уже было отмечено выше, белки Repac содержат в своем составе только каталитический регион, что позволяет предположить, что либо они постоянно находятся в активном состоянии, либо их регуляторная область находится в отдельной, еще не идентифицированной, субъединице. Полагают, что белки Repac могут активировать малые GТFазы Rap1 и Rap2 посредством своего Ras-связывающего СDC25-HD домена [13].
В отсутствии сАМР белки Ерас пребывают в неактивном состоянии, поскольку регуляторный регион этих белков обладает аутоингибирующей активностью. В этих условиях CNBD-B домен регуляторного региона находится в неактивной конформации и ковалентно связан с СDC25-HD доменом каталитической области. В результате образовавшейся связи этих доменов блокируется доступ малой GТFазы Rap к каталитическому участку СDC25-HD домена (рис. 3). В основе этого феномена лежит способность консервативной последовательности VLVLE, входящей в состав “шарнира”, играть роль “покрышки”, закрывающей каталитический участок СDC25-HD домена от взаимодействия с малой GEFазой Rap [27]. При взаимодействии сАМР с CNBD-B доменом регуляторного региона белка Ерас происходят существенные конформационные изменения его структуры, напоминающие движения шарнира (рис. 2), что сопровождается переориентацией CNBD-B и DEP доменов регуляторного региона и сдвигом вверх “покрышки” (консервативной последовательности VLVLE), закрывающей каталитический участок СDC25-HD домена, что в свою очередь обеспечивает связывание этого домена с малой GТFазой Rap и последующий обмен GDP на GTP и, следовательно, активацию белков Rap (рис. 2, 3) [27].
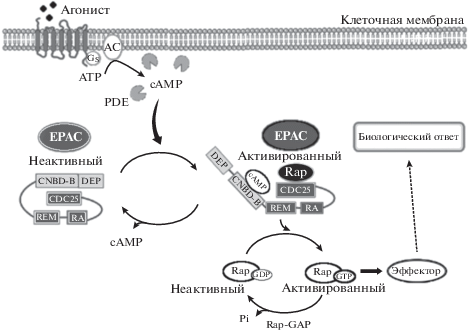
Сигнальные белки Ерас1 и Ерас2 реализуют свои внутриклеточные эффекты или самостоятельно, или в связке с РКА [23, 28]. CNBD-B домен белков Ерас имеет меньшую аффинность к сАМР, чем R субъединица РКА, т.е. для активации белков Ерас требуется более высокая концентрация сАМР [29]. Вместе с тем, физиологического повышения концентрации сАМР достаточно для активации белков Ерас и сопряженных с ними заякоривающих белков семейства АРАКs (A-kinase anchor proteins) [30, 31]. Белки АРАКs определяют специфичность действия cAMP путем компартментализации РКА и/или белков Ерас не только с фосфатазами, но и с фосфодиэстеразой, отвечающей за “выключение” сигнала.
Роль белков Ерас в физиологии и патофизиологии сосудистого русла
К настоящему времени накоплены убедительные данные, свидетельствующие о том, что белки Ерас играют существенную роль в регуляции ангиогенеза, сосудистого тонуса и проницаемости сосудистой стенки, а также в поддержании целостности эндотелиального барьера. Показано, что обе изоформы белков Ерас (Ерас1 и Ерас2) экспрессируются как в клетках гладкой мускулатуры сосудов [32, 33], так и в сосудистом эндотелии [34, 35]. Однако если роль белков Ерас1 в регуляции состояния сосудистого русла достаточно хорошо изучена, то роль белков Ерас2 на данный момент остается не ясной.
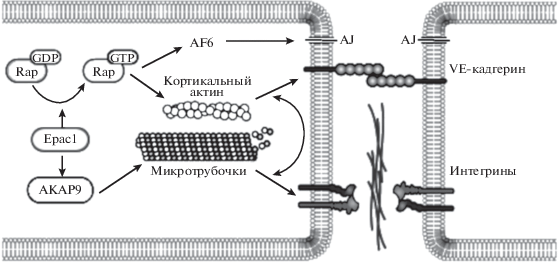
Роль белков Ерас в регуляции проницаемости сосудистого эндотелия. Сосудистый эндотелий – метаболически активный монослой клеток, выстилающий внутреннюю стенку сосудов, играет ключевую роль в регуляции их тонуса, транспорте микро- и макромолекул, тромборезистентности и т.д. Не менее важна и его барьерная функция. Контакты между клетками эндотелия, определяющие его барьерную функцию, образованы разнообразными трансмембранными адгезивными молекулами, соединенными с сетью цитоплазматических/цитоскелетных белков. Известно, что одной из основных молекул, модулирующих функцию эндотелиального барьера, является сАМР [36]. В зависимости от внутриклеточного локуса генерации сАМР его активация в эндотелиальных клетках может привести либо к сохранению, либо к дестабилизации эндотелиального барьера: аккумуляция сАМР в цитозоле клетки влечет за собой увеличение сосудистой проницаемости, а его накопление в вакуолях защищает от барьерной дисфункции [37]. Также показано, что эндотелиальные клетки сосудов экспрессируют белки Rap1, а их инактивация GTPаза-активирующим белком (GTPase-activating protein; Spa-1) оказывает негативное влияние на адгезию клеток и, следовательно, приводит к дестабилизации эндотелиального барьера [35]. В экспериментах in vitro, выполненных на культуре изолированных клеток эндотелия пупочной вены человека HUVEC, показано, что активация белков Ерас их селективным агонистом 8-CPT (8-рСРТ-2-О-Ме-сАМР) сопровождается активацией белка Rap1 с последующей активацией белка межклеточной адгезии AF6 (афадин), который связывается с другой молекулой межклеточной адгезии нектином, через которую белок AF6 связан с F-актином цитоскелета [35]. Авторы этого исследования полагают, что активация внутриклеточного каскада Epac/Rap1 приводит к повышению прочности эндотелиального барьера за счет оптимизации локализации белка AF6, который обеспечивает формирование/стабильность межклеточных слипчивых соединений за счет их связывания с интегральными белковыми комплексами межклеточных контактов AJ (adherens junctions) и TJ (tight junctions), пересекающих апикальное межклеточное пространство и регулирующих эндотелиальное прилипание клетка к клетке. Помимо того, имеются сведения о том, что активация внутриклеточного каскада Epac/Rap1 приводит к активации и транслокации белков KRIT-1 в область эндотелиальных клетка–клеточных соединений, где они стабилизируют/восстанавливают целостность соединительных волокон, ассоциированных с junctional белками [38]. Белки KRIT (ловушка для Krev1 – Krev Trapped 1, Krev – синоним белка Rap1) являются одним из эффекторов белка Rap1 и в эндотелиальных клетках, в частности, регулируют плотность межклеточных контактов и поддерживают физиологическую полярность эндотелиальных клеток [39]. В экспериментах in vitro, выполненных на культуре изолированных клеток эндотелия человека HUVEC, было показано, что активация белков Ерас1 их агонистом 8-CPT инициирует активацию якорного белка 9a-киназы (а-kinase anchor protein 9, AKAP9), который играет одну из ключевых ролей в регуляции динамики роста микротрубочек эндотелиальных клеток [40]. Активация каскада Epac/AKAP9 необходима для кортикальной реорганизации белка актина, что способствует повышению барьерной функции эндотелия сосудов, в том числе и за счет интегрин-опосредования прилипания клеток в области межклеточных щелевых контактов – gap junctions (GJ) (рис. 4). Также в этом исследовании показано, что активация AKAP9 не требует активации белков Rap.
Рис. 4.
Схематическое отображение Epac/AKAP9 опосредованного повышения барьерной функции эндотелия сосудов за счет кортикальной реорганизации белка актина (по [40]). Объяснение в тексте.
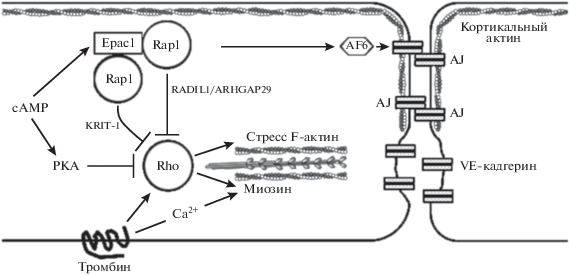
Известно, что тромбин (трипсиноподобная сериновая протеиназа) индуцирует барьерную дисфункцию эндотелия сосудов. Этот эффект реализуется за счет агонистического влияния тромбина на встроенные в клеточную мембрану эндотелиальных клеток специфичные для него сопряженные с G-белками PA-рецепторы (рrotease-activated receptors, PAR). После взаимодействия с тромбином PAR активируют сигнальный белок RhoА (GTPase-activating proteins, GAPs), относящийся к суперсемейству белков Ras, который, в свою очередь, активирует Rho-киназу (Rho-associated protein kinase; ROCK). Активация сигнального пути RhoА/RhoK влечет за собой ингибирование фосфатазы регуляторной легкой цепи миозина (мyosin light сhain рhosphatase; MLCP), фосфорилирование миозина и сборку миозиновых филаментов, что приводит к сокращению эндотелиальных клеток и, следовательно, к уменьшению плотности межклеточных щелевых контактов – увеличению проницаемости. В экспериментах in vitro, выполненных на культуре изолированных клеток эндотелия человека HUVEC, обработанных тромбином, показано, что активация белков Ерас селективным агонистом 8-CPT влечет за собой ингибирование RhoА и тем самым предохраняет эндотелий от межклеточных разрывов [35]. Полагают, что в основе подавления активности RhoА может лежать активация Ерас/Rap1/KRIT-1 сигнального каскада [39]. Известен и альтернативный путь Ерас-опосредованного ингибирования RhoА [41]. В экспериментах in vitro, выполненных на культуре изолированных клеток эндотелия человека HUVEC, показано, что в результате 8-CPT-опосредованной активации Ерас1/Rap1-сигнального каскада к клеточной мембране эндотелия перемещается белок сосудистой адгезии Ras, связывающий протеин 1 (Ras Interacting Protein 1, RASIP1) и инициирующий последующую транслокацию к мембране комплекса, включающего в себя белок, регулирующий миграцию – Rap GTPаза интерактор 1 (Rap GTPase interactor 1, RADIL1), и белок цитоскелета – Rho GTPаза, активирующий протеин 29 (Rho GTPase аctivating рrotein 29, ARHGAP29). Показано, что активация Ерас1/Rap1/RASIP1/ RADIL1/ARHGAP29 сигнального каскада приводит к транслокации к внутренней поверхности эндотелиальной мембраны комплекса RADIL1/ ARHGAP29, подавляющего активность RhoА (рис. 5).
Рис. 5.
Схематическое отображение Epac опосредованного повышения барьерной функции эндотелия сосудов за счет ингибирования Rho-киназы (по [35]). Объяснение в тексте.
Супрессор сигнализации цитокинов 3 – SOCS-3 (Suppressor of cytokine signaling 3) является одним из основных отрицательных регуляторов иммунного ответа за счет своей способности блокировать тирозинкиназную активность нерецепторной джанус-киназы 2 (JAK2), что приводит к снижению фосфорилирования преобразователя сигнала и активатора транскрипции 3 – STAT3 (signal transducer and activator of transcription 3), который теряет способность проникать в ядро и регулировать экспрессию генов, чувствительных к действию цитокинов и тем самым препятствовать дестабилизации эндотелиального барьера. Помимо этого, показано, что SOCS-3 обладает способностью блокировать рецепторы таких провоспалительных цитокинов как интерлейкины (IL-6, IL-12, IL-17 и IL-31) и интерферон γ (IFN-γ) [42]. В экспериментах in vitro и in vivo показано, что активация белков Ерас1 селективным агонистом 8-CPT приводит к SOCS-3-зависимой блокаде цитокиновой сигнализации и, как следствие этого, стабилизации барьерной функции эндотелия [42, 43].
Белок клеточной адгезии эндотелия сосудов – VE-кадгерин (VE calcium-dependent adhesion; VE-cadherin, син. CD144) рассматривают как ключевую, специфичную для эндотелия сосудов молекулу, расположенную в области контактов монослоя эндотелиальных клеток и регулирующую процессы их адгезии. В нормальных физиологических условиях VE-кадгерин связан с β-катенином и образует прочный VЕ-кадгерин/β-катениновый комплекс. β-катенин комплекса VЕ-кадгерин/β-катенин связан с α-катенином, который, в свою очередь, взаимодействует с актиновым цитоскелетом, обеспечивая стабильность клеточной адгезии. Дестабилизация VЕ-кадгерин-β/катенинового комплекса приводит к цитоплазматической аккумуляции белка p120-катенина – одного из основных регуляторов силы адгезионных межклеточных контактов, гиперэкспрессия которого сопровождается активацией белка RhoA [44]. В экспериментах in vitro, выполненных на культуре изолированных клеток эндотелия человека HUVEC, показано, что активация белков Ерас1 их селективным агонистом 8-CPT, и соответственно Ерас/Rap1 сигнального каскада, сопровождается увеличением числа и жесткости клеточно-клеточных соединений; снижением напряжения волокон актина; образованием кортикального актина и уменьшением проницаемости монослоя эндотелиальных клеток [45]. Этот эффект наблюдался только в клетках, содержащих VE-кадгерин, тогда как в клетках HUVEC, “лишенных” VE-кадгерина (VE-cadherin KN knock-out) активация Ерас/Rap1 сигнального каскада не влияла на состояние эндотелиального барьера; ре-экспрессия в этих клетках VE-кадгерина сопровождалась восстановлением Ерас-опосредованной стабилизации эндотелиального барьера [45]. Авторы этого исследования констатируют, что белки Ераc1 контролируют VE-кадгерин-связанную стабилизацию эндотелиального барьера и сопутствующую ей реорганизацию актинового цитоскелета преимущественно за счет перестройки кортикального актина. Известен еще один сигнальный путь, приводящий к Ерас-опосредованной стабилизации VE-кадгерина [46]. В экспериментах in vitro, выполненных на культуре эндотелиальных клеток артерий (HAECs) и микроартерий (HMVECs) человека, показано, что 8-CPT-опосредованая активация Ерас/Rap1 сигнального каскада влечет за собой активацию фосфодиэстеразы циклических нуклеотидов 4D (cyclic nucleotide phosphodiesterase 4D, PDE4D) которая играет важную роль в регуляции положительной адгезивной активности VE-кадгерина.
В модельных экспериментах in vitro на культуре ишемизированных изолированных клеток эндотелия человека HUVEC показано, что активация сАМР дитерпеноидным алкалоидом растения Coleus forskohlii форсколином (колеонол), который обладает способностью внерецепторно активировать сАМР, уменьшает ишемия-обусловленную гиперпроницаемость эндотелиального барьера [47]. В дальнейшем было показано, что в клетках HUVEC, “лишенных” белка Rap1 (Rap1 KN knock-out), не наблюдалось сАМР-опосредованного восстановления целостности эндотелиального барьера, а в интактных клетках целостность эндотелиального барьера поддерживается Eрас-опосредованной активацией белка Rap1 [48]. Это сообщение подтвердило ранее полученные данные о том, что активированные белки Rap1 последовательно активирует метастаз-индуцирующий белок 1 (metastasis-inducing protein 1, Tiam1) и фактор обмена гуаниловых нуклеотидов Vav2 (guanine nucleotide exchange factor Vav2) [49]. Известно, что эти белки играют важную роль в регуляции полимеризации актина в области межклеточных контактов и способствуют созреванию межклеточных контактов. Конечным этапом активации Eрас/Rap1/Tiam1/Vav2-сигнального каскада является активация малой GTPазы Rac1. Недавно было показано, что именно белки Eрас1, но не белки Ерас2, регулируют проницаемость микрососудистого барьера, поскольку в экспериментах in vivo было продемонстрировано, что у нокаутных по белку Ерас1, но не у нокаутных по белку Ерас2, мышей увеличивается проницаемость эндотелиального барьера, о чем свидетельствовало усиление трансваскулярного транспорта, который определяли при помощи динамической контрастно-усиленной магнитно-резонансной томографии (МРТ) с использованием в качестве зонда контрастного агента Gadomer-17 [48].
Роль белков Ерас в регуляции ангиогенеза и пролиферации эндотелиальных и гладкомышечных клеток сосудов. Если роль белков Ерас в регуляции проницаемости эндотелия сосудов достаточно ясна, то их функциональное значение в регуляции ангиогенеза достаточно дискутабельно. В первых посвященных этой проблеме исследованиях, выполненных на культуре изолированных клеток эндотелия человека HUVEC, было показано, что активация белков Ерас (Ерас1 и Ерас 2) селективным агонистом 8-CPT ингибирует миграцию клеток HUVEC, стимулированную фактором роста эндотелия сосудов (vascular endothelial growth factor; VEGF) [50]. Также было показано, что в отличие от белков Ерас РКА в эндотелиальных клетках не влияет на регуляцию ангиогенеза, поскольку ее активация селективным агонистом – 6-Bnz-cAMP не сопровождается подавлением VEGF-опосредованной миграции. Авторы исследования сделали вывод о том, что белки Epac и Rap1 являются центральными медиаторами, регулирующими хемотаксис эндотелиальных клеток сосудов. При изучении механизмов, лежащих в основе Ерас/Rap1-опосредованного ингибирования хемотаксиса и ангиогенеза, было показано, что активация белков Ерас селективным агонистом 8-CPT в клетках эндотелия человека HUVEC влечет за собой подавление активности ингибитора дифференциации Id1 (inhibitor of differentiation 1), подавляющего активность белка внеклеточного матрикса тромбоспондина-1 (thrombospondin 1; TSP1) – эндогенного ингибитора ангиогенеза и влечет за собой подавление активности ингибитора дифференциации Id1 (inhibitor of differentiation 1), подавляющего активность белка внеклеточного матрикса тромбоспондина-1 (thrombospondin 1; TSP1) – эндогенного ингибитора ангиогенеза, и последующее повышение экспрессии TSP1 [51]. В свою очередь, TSP1, связываясь с рецепторами мембранных белков CD36 и CD47, инициирует активацию внутриклеточных сигнальных каскадов, подавляющих ангиогенную активность VEGF.
Эти результаты представлялись достаточно спорными, поскольку еще в начале XXI века в экспериментах in vivo было показано, что активация сАМР форсколином или активация PKA 6-Bnz-cAMP влечет за собой VEGF-опосредованную активацию ангиогенеза [52].
Несколько позднее было продемонстрировано, что активация сопряженных с белком Rap1 сигнальных путей стимулирует ангиогенез [53, 54]. В основе стимуляции ангиогенеза лежит способность белков Rap1a и Rap1b активировать VEGF-рецепторы 2 типа (VEGFR2); этот эффект частично опосредуется мембранным белком интегрином αvβ3 [54]. Более того, было показано, что у нокаутных по белку Rap1 мышей интенсивность миграции и пролиферации эндотелиальных клеток в ответ на стимуляцию VEGF и/или фактором роста фибробластов (fibroblast growth factor; FGF) существенно снижается [54]. Результаты этих исследований хорошо согласуются с данными о том, что белки Ерас активируют внутриклеточные проангиогенные каскады [32, 55, 56]. Так, в экспериментах in vitro, выполненных на культуре изолированных клеток эндотелия человека HUVEC, показано, что активация белков Ерас1 селективным агонистом 8-CPT влеклa за собой фосфорилирование (активацию) внеклеточной сигнал-регулируемой киназы-1/2 (extracellular signal–regulated kinase-1/2; ERK1/2, син. MAPK3) и последующую стимуляцию ангиогенеза [55]. В этом же исследовании был проведен анализ результатов цепной реакции обратной транскрипции полимеразы, свидетельствующий о том, что белок Epac1, но не белок Epac2, вызывает фосфорилирование ERK1/2.
В экспериментах in vitro, выполненных на культуре изолированных клеток эндотелия человека HUVEC, показано, что белок Ерас реализует свои ангиогенные эффекты посредством активации сигнального пути, регулируемого фосфоинозитид-3-киназой (рhosphoinositide 3-kinas; PI3K) PI3K/Akt/eNOS/NO сигнального пути [56]. Было показано, что ангиогенез, инициированный агонистом сАМР форсколином, подавляется ингибитором PI3K – LY294002 и/или ингибитором эндотелиальной NO-синтазы (еNO-synthase; eNOS, син. NOS3) – NMA, но его интенсивность практически не изменяется при блокаде РКА ее эндогенным ингибитором PKI. Помимо этого, было показано, что форсколин-индуцированное фосфорилирование транскрипционного фактора CREB (cAMP response element-binding protein) и последующая экспрессия VEGF блокируются ингибитором РКА – PKI. Авторы этого исследования делают заключение, что в эндотелиальных клетках присутствуют два параллельных, взаимодополняющих cAMP-опосредованных проангиогенных сигнальных пути, регулируемыe РКА и белками Ерас1. При этом, если РКА-сопряженный сигнальный каскад (РКА/рCREB) инициирует экспрессию VEGF, то белки Ерас1 активируют/регулируют ангиогенез по VEGF-независимым сигнальным путям: Rap1/PI3K/Akt/ eNOS/NO и/или Rap1/MEK/ERK [56]. Однако позднее было показано, что активация в клетках HUVEC белков Ерас1 селективным агонистом 8-CPT сопровождается увеличением образования VEGFR-2 и экспрессии VEGF [32]. Также было показано, что РКА регулирует ангиогенез посредством активации сопряженного с ERK сигнального каскада [57].
Если роль белков Ерас в регуляции ангиогенеза и пролиферации эндотелиальных клеток дискутабельна, то их место в регуляции пролиферации гладкомышечных клеток сосудов представляется достаточно определенным. Еще в начале XXI века in vitro и in vivo было показано, что форсколин-опосредованная активация cAMP ингибирует пролиферацию гладкомышечных клеток сосудов посредством дестабилизации S-фазного киназа-ассоциированного белка 2 (S-phase kinase-associated protein 2; Skp2), являющегося главным компонентом Skp2/SCF-комплекса, регулирующего цикл клеточного деления [58]. Позднее эти же авторы показали, что РКА и белки Ерас являются синергистами в отношении регуляции пролиферации гладкомышечных клеток сосудов [59, 60]. Так, в экспериментах in vitro, выполненных на культуре гладкомышечных клеток сосудов, полученных от Спраг-Доули (SD) крыс, показано, что селективная активация PKA 6-Bnz-cAMP не препятствовала пролиферации гладкомышечных клеток [59]. Аналогичный результат был получен и при активации белков Ерас1 селективным агонистом 8-CPT, несмотря на активацию их эффекторов – белков Rap1а и Rap1b. Вместе с тем, совместная активация PKA 6-Bnz-cAMP и белков Ерас1 8-CPT эффективно подавляла пролиферацию гладкомышечных клеток. В основе этого эффекта лежит корпоративное подавление фосфорилирования ERK и c-Jun N-концевой киназы (c-Jun N-terminal kinase; JNK) и, следовательно, ингибирование их активности, PKA и белком Ерас1. В процессе этих исследований было подтверждено, что корпоративный эффект PKA и белков Ерас1 реализуется в гладкомышечных, но не эндотелиальных, клетках сосудов [60]. Также было показано, что подавление пролиферации гладкомышечных клеток, как минимум частично, связано со способностью PKA/Ерас1 ингибировать активность Ras-связанного субстрата ботулотоксина C3,1 – белкa Rac1 (Ras-related C3 botulinum toxin substrate 1). Ингибирование C3,1 – Rac1 приводило к быстрому ремоделированию цитоскелета, ядерному экспорту ERK1/2, дефосфорилированию фактора транскрипции Elk1 (ETS domain-containing protein) и ингибированию активности цис-регуляторного элемента SRE (участок ДНК, регулирующий экспрессию генов).
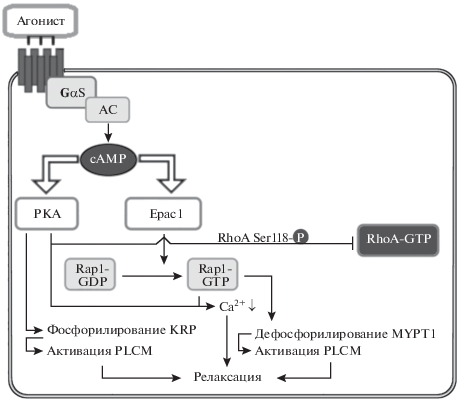
Роль белков Ерас в регуляции тонуса сосудистого русла. Регуляция тонуса сосудистого русла – сложный многофакторный процесс, в котором одну из ключевых ролей играет Rho-ассоциированная протеинкиназа А (Rho-associated protein kinase; RhoKα, син. ROCK2), активация которой инициирует вазоконстрикцию. Показано, что RhoKα-опосредованная вазоконстрикция реализуется по двум путям: Са2+-независимому пути (за счет ингибирования фосфатазы легких цепей миозина – РLCМ посредством фосфорилирования ее регуляторной субъединицы MYPT1, что приводит к снижению активности РLCМ и ее диссоциации от миозина и, следовательно, сокращению гладкой мышцы) и Са2+-зависимому пути (за счет усиления входа ионов Са2+ в цитозоль гладкомышечной клетки в результате повышения активности трансмембранных потенциалзависимых Са2+-каналов L-типа) [61]. Известно, что, в отличие от RhoKα, cAMP инициирует вазодилатацию. В ряде исследований показано, что вазодилатирующее действие реализуется по Са2+-независимому механизму посредством активации cAMP-сопряженных сигнальных каскадов, инициирующих дефосфорилирование регуляторной субъединицы MYPT1 фосфатазы LCМ, что приводит к восстановлению ее активности и расслаблению гладкомышечных клеток сосудов [62]. Этот эффект связан c активацией сAMP/РКА сигнального пути [63] и реализуется в крупных магистральных сосудах [64]. Однако позднее в экспериментах in vitro, выполненных на культуре гладкомышечных клеток (R518) аорты крысы, было показано, что дефосфорилирование регуляторной субъединицы MYPT1 фосфатазы LCМ связано с активацией Ерас/Rap1-сигнального пути селективным агонистом белков Ерас 8-CPT, тогда как активация PKA ее селективным агонистом 6-Bnz-cAMP инициирует вазодилатацию посредством фосфорилирования (активации) миозин-связывающего белка гладких мышц – белка KRP (Kinase Related Protein; син. фелокин), который, связываясь с субъединицей MYPT1 фосфатазы LCМ, восстанавливает ее функциональную активность [64] (рис. 6). В этом же исследовании показано, что Ерас-опосредованная вазодилатация связана не только с восстановлением активности фосфатазы LCМ, но и с уменьшением фосфорилирования регуляторной легкой цепи миозина – RLC20. В экспериментах in vitro, выполненных на лишенных эндотелия кольцевых сегментах аорты крыс, показано, что белки Ерас, также как и PKA, могут способствовать вазодилатации за счет истощения внутриклеточных запасов ионов Са2+, аккумулированых в саркоплазматическом ретикулуме [65]. Однако остается не ясным, насколько корректны данные результаты, поскольку кольцевые сегменты аорты находились в беcкальциевой среде.
Рис. 6.
Схематическое отображение Epac и РКА опосредованной регуляции релаксации гладкомышечных клеток сосудов (с изменениями по [64]). Объяснение в тексте.
Известно, что одним из физиологических регуляторов тонуса гладкомышечных клеток сосудов являются АТФ-чувствительные К+-каналы (KАТФ-каналы), относящиеся к классу K+-каналов внутреннего выпрямления (K+ inward rectifier; Kir) семейства Kir6. KАТФ-канал состоит из четырех Kir6.х субъединиц, формирующих пору канала, и четырех дополнительных белков – рецепторов к сульфонилмочевине SUR. Вазодилатация связана с открытием KАТФ-каналов, вызывающих гиперполяризацию клеточной мембраны и, как следствие этого, подавление входящего трансмебранного Са2+ тока.
Показано, что в гладкомышечных клетках сосудов активация KАТФ-каналов нейротрансмиттерами может происходить по Gs/cAMP/PKA-сигнальному пути [66]. Конечным этапом этого процесса является фосфорилирование PKA Kir6.1 субъединицы KАТФ-канала и активация SUR2B канального рецептора.
В экспериментах in vitro, выполненных на гомогенате тканей аорты и брыжеечной артерии крыс, показано, что белки Ерас1, но не белки Ерас2, локализуются на внутренней поверхности клеточной мембраны вблизи KАТФ-каналов [67]. В экспериментах in vitro, выполненных на культуре гладкомышечных клеток аорты крысы, было показано, что активация белков Ерас1 селективным агонистом 8-CPT сопровождалась активацией/модуляцией KАТФ-каналов [67]. Этот эффект белков Ерас1 опосредуется путем активации ими Са2+-кальмодулин-зависимой серин-треониновой фосфатаза 2b (PP2b фосфатаза; син кальцинейрин, CaN). Также в этом исследовании было показано, что активация KАТФ-каналов не связана с РКА-опосредованной сигнализацией, поскольку селективное ингибирование РКА ингибитором Rp-cAMPS (конкурентный ингибитор активации РКА cAMP) или ингибитором KT5720 (блокада РКА-опосредованного фосфорилирования), не влияла на Ерас-инициированную активацию KАТФ-каналов. В экспериментах in vitro, выполненных на кольцевых сегментах брыжеечных артерий, показано, что активация белков Ерас селективным агонистом 8-CPT сопровождается активацией активируемых ионами Са2+ калиевых каналов (ВКСа-каналы), встроенных в клеточную мембрану гладкомышечных клеток сосудов [68]. Известно, что активация (открытие) ВКСа-каналов сопровождается увеличением внутриклеточной концентрации ионов Са2+, что приводит к выходу ионов К+ из клетки, гиперполяризации клеточной мембраны и последующей вазодилатации. Также было показано, что блокада ВКСа-каналов ибериотоксином препятствует реализации Ерас-обусловленного расширения кольцевых сегментах брыжеечных артерий. При изучении механизмов, лежащих в основе Ерас-обусловленной активации ВКСа-каналов, было показано, что активация белков Epac вызывает релаксацию гладких мышц путем увеличения частоты локального высвобождения ионов Ca2+ из саркоплазматического ретикулума через встроенные в его мембрану рианодин-чувствительные каналы (рианодиновые рецепторы, ryanodine receptor RyRs). Выделившиеся ионы Ca2+ активируют так называемый спонтанный переходный выходящий ток ионов К+ (spontaneous transient outward K+currents; STOCs), что вызывает гиперполяризацию клеточной мембраны и уменьшает вход ионов Ca2+ в цитозоль клетки через потенциалзависимые трансмебранные медленные Ca2+ каналы L-типа. Позднее было показано, что Ерас-опосредованные STOCs связаны со способностью белков Ерас стимулировать ау-тофосфорилирование (активацию) цитозольной кальций/ кальмодулин-зависимой киназы 2 (Ca2+/calmodulin-dependent protein kinase II; CaMKIIδ), которая, в свою очередь, активирует RyR [69]. Данное заключение сделано на основании того, что активация белков Ерас1 селективным агонистом 8-CPT на фоне блокады CaMKIIδ ингибитором KN93 не приводила к активации STOCs. Помимо ВКСа-каналов в Ерас-обусловленной релаксации сосудов принимают участие и трасмебранные потенциалзависимые К+-каналы задержанного выпрямления подсемейства KV7,4 (KvLQT4 или KCNQ4). В экспериментах in vitro, выполненных на кольцевых сегментах брыжеечной артерии, показано, что на фоне блокады KV7,4-каналов селективным ингибитором pan-KV7 каналов – linopirdine не реализуются ни вазодилатация, инициированная неселективным β-адреномиметиком изопротеренолом, ни вазодилатация, инициированная активацией белков Ерас их агонистом 8-CPT, на основании чего авторы пришли к выводу, что Ерас-опосредованное расширение сосудов связано с активацией KV7,4-каналов и последующей гиперполяризацией клеточной мембраны [70]. Также было показано, что KV7,4-опосредованное вазодилатирующее действие белков Ерас связано с активацией Eрас/Rap1-сигнального каскада. В аналогичных экспериментах, выполненных на сегментах почечной артерии, белки Ерас не инициировали вазодилатацию. Авторы полагают, что в почечных артериях KV7,4-опосредованная вазодилатация связана с активацией сигнального пути РКА/якорный белок а-киназ (A-kinase anchor proteins, AKAP). В экспериментах in vitro, выполненных на кольцевых сегментах аорты, показано, что активация белков Ерас селективным агонистом 8-CPT значительно ослабила интенсивность сокращения как интактных, так и лишенных эндотелия колец аорты, вызванного агонистом α-адренорецепторов фенилэфрином [71]. Аналогичное действие оказывала и РКА активированная 6-Bnz-cAMP. Авторы исследования полагают, что вазодилатирующий эффект белков Ерас реализуется посредством увеличения продукции эндотелиальной NO-синтазы (NOS3), поскольку на фоне ингибирования NOS3 N-нитро-L-аргинин метилэфиром (L-NAME) релаксация под действием белков Ерас не развивалась.
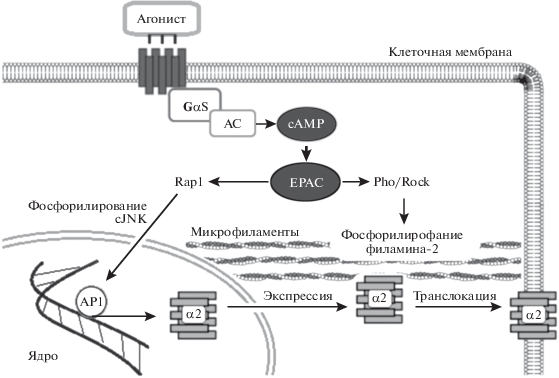
Если в крупных сосудах активация белков Ерас инициирует вазодилатацию, то в микрососудах скелетной мускулатуры белки Ерас контролируют вазоконстрикторные каскады. Изначально было показано, что холодовая стимуляция вызывает вазоконстрикцию, увеличивая экспрессию α2c-адренорецепторов в клеточной мембране гладкомышечных клеток подкожных артериол посредством активации cAMP/Rap1 сигнального пути [72]. Позднее в экспериментах in vitro, выполненных на культуре гладкомышечных клеток, полученных из кожных артериол человека, было показано, что активация белков Ерас1 селективным агонистом 8-CPT инициирует экспрессию α2c-адренорецепторов и последующее сокращение гладкомышечных клеток посредством активации белка Rap1, который фосфорилирует (активирует) с-Jun-амино-терминальную киназу (c-Jun N-Terminal Kinase; с-JNK) [73]. Фосфорилированная с-JNK активирует фактор транскрипции AP-1 (аctivator protein 1), который транслоцируется в ядро клетки, где инициирует экспрессию α2c-адренорецепторов. Позднее те же авторы в аналогичных экспериментах показали, что экспрессия α2c-адренорецепторов в гладких мышцах микрососудов кожи человека инициируется посредством активации cAMP/Ерас/Rap1А/ Rho/ROCK сигнального пути [74]. В том же исследовании было продемонстрировано, что вазоконстрикторные эффекты, опосредуемые белками Ерас, могут реализоваться не только в ответ на холодовое воздействие, но и при нормальной температуре. Полагают, что активация cAMP/ Ерас/Rap1А/Rho/ROCK сигнального пути влечет за собой фосфорилирование (активацию) актин-связывающего белка филамин-2 по Ser2113, который не только связывает отдельные микрофиламенты актина, но и регулирует экспрессию таких факторов транскрипции как SMAD1 (мothers against decapentaplegic homolog1), FOXC1 (forkhead box C1 ) и TRAF2 (TNF receptor-associated factor 2) [75]. По всей видимости, транслокацию α2c-адренорецепторов в область клеточной мембраны инициируют сопряженные с белками Ерас Rap1/с-JNK/AP-1 и Rap1А/Rho/ROCK/ филамин-2 сигнальные пути [75] (рис. 7).
Рис. 7.
Схематическое отображение Epac опосредованной инициации вазоконстрикции гладкомышечных клеток подкожных артериол. Объяснение в тексте.
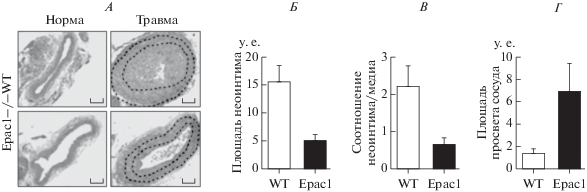
Роль белков Ерас в патологии сосудистого русла. Количество исследований, посвященных изучению роли белков Ерас в патологии сосудистого русла, незначительно, однако все они свидетельствуют о том, что их гиперэкспрессия в условиях травматического повреждения сосудов способствует формированию гиперплазии неоинтимы. Так, в модельных экспериментах in vivo, выполненных на нокаутных по белкам Ерас1 мышах обоего пола, было показано, что через 28 дней после перевязки сонной артерии у них, по сравнению с контрольными животными, интенсивность облитерации просвета лигированного сосуда значимо (р < 0.05) меньше [76] (рис. 8, А). Морфометрический анализ поврежденных сонных артерий показал, что у нокаутных по белкам Epac1 мышей, по сравнению с контрольными, толщина интимы значимо меньше (рис. 8, Б) и более чем в 3 раза уменьшено соотношение интима/медиа (рис. 8, В), что сопровождается 5-кратным увеличением площади просвета сосуда (рис. 8, Г). Помимо этого, у нокаутных по белкам Epac1 мышей было значимо (р < 0.05) снижено общее количество пролиферативных клеток. Иммуностайнинг эндотелиальных клеток показал, что у нокаутных по белкам Epac1 мышей эндотелиальный слой хорошо организован и практически интактен, в то время как эндотелий контрольных животных дезорганизован и в него из гладкомышечных клеток проникают волокна белка α-актина (аlpha-smooth muscle actin; α-SMA) – одного из индукторов фиброгенеза. Известно, что фактор роста тромбоцитов (Platelet-derived growth factor, PDGF) играет ключевую роль в переходе гладкомышечных клеток сосудов от сократительного к синтетическому фенотипу и способствует образованию неоинтимы при их ремоделировании. Этот эффект PDGF реализуется посредством активации PI3K/AKT/mTOR сигнального пути. Показано, что белки Ерас1 в условиях патологии сосудистого русла, также как и PDGF, инициируют образование неоинтимы за счет активации PI3K/AKT/mTOR сигнального пути [76]. Известно, что белки Ерас1 локализуются в том числе и на клеточной мембране митохондрий, ассоциацию с которой обеспечивает их DEP домен регуляторного региона [77]. При изучении роли белков Ерас1 в регуляции функциональной активности митохондрий в поврежденных гладкомышечных клетках сосудов было показано, что они непосредственно участвуют в регуляции скорости деления митохондрий и сопутствующей этому процессу активации митохондрии-обусловленной продукции реактивных форм кислорода (ROS) [76] и, как следствие этого, ROS-обусловленной активации PDGF. В основе этого процесса лежит Ерас1-инициированное фосфорилирование (активация) динамин-1-подобного белка (DLP-1) – ключевого регулятора скорости деления митохондрии. Показано, что при ингибировании белка Ерас1 неселективным антагонистом белков Ерас – HJC0726 не происходит фосфорилирование DLP-1 и, следовательно, блокируется митохондрии-обусловленная продукция ROS. Также в этом исследовании показано, что блокада белка Ерас1 неселективным антагонистом белков Ерас – ESI-09 подавляет процессы образования неоинтимы в поврежденном сосуде. В модельных экспериментах in vivo, в которых повреждение стенки бедренной артерии вызывали транслюминальной механической травмой, показано, что в стенке поврежденного сосуда происходит динамическое увеличение экспрессии белков Ерас1, которая достигает максимума к концу 3-ей недели после нанесения травмы [78]. При этом, если к концу 1-ой недели экспрессия белков Ерас1 отмечается только в гладкомышечных клетках сосудов, то к концу 3-ей недели белки Ерас1 экспрессируются и в гладкомышечных клетках, и в неоитиме. В этом же исследовании было показано, что РКА, в отличие от белков Ерас1, не стимулирует, а ингибирует образование неоинтимы в поврежденном сосуде. В модельных экспериментах in vivo, в которых повреждение стенки бедренной артерии вызывали транслюминальной механической травмой, показано, что у нокаутных по белкам Ерас1 мышей, в отличие от контрольных животных, не происходит избыточного формирования неоинтимы (р < 0.05) [34]. Помимо этого, у нокаутных животных длина наружных ламеллоподий (lamellipodia – выпячивания, которые клетка образует по направлению своего движения/роста) была значимо (р < 0.05) меньше. В экспериментах in vitro, выполненных на культуре гладкомышечных клеток аорты, полученных от нокаутных по белкам Ерас1 мышей, показано, что PDGF не оказывал влияния на уровень фосфорилирования (активации) актин-связывающего белка кофилин 1 (Cofilin 1, CFL1), что подавляло процесс разборки F-актина и тем самым стабилизировало цитоскелет и останавливало миграцию, тогда как в культуре гладкомышечных клеток интактных животных PDGF в присутствии белка Ерас1 инициировал дефосфорилирование (деактивацию) CFL1, что, в свою очередь, активировало процессы клеточной миграции [34].
Рис. 8.
Влияние белков Ерас на образование неоинтимы в поврежденном сосуде (по [76]). Объяснение в тексте.
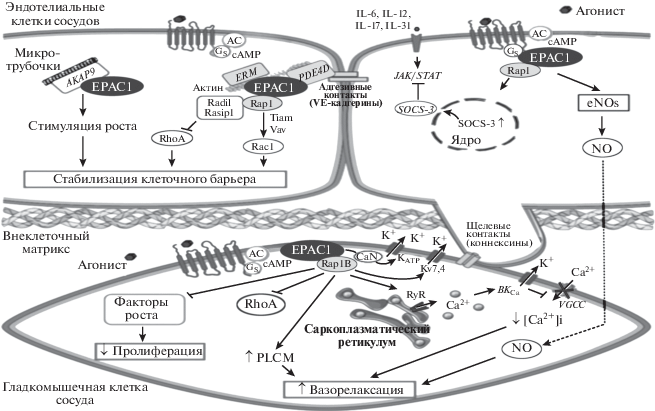
ЗАКЛЮЧЕНИЕ
Таким образом, накопленные к настоящему времени данные свидетельствуют о том, что белки Ерас1, но не белки Ерас2, в нормальных физиологических условиях играют важную, в том числе и не зависимую от РКА, многофакторную роль в регуляции функциональной активности сосудистой стенки. В результате активации катехоламинами β1-адренорецепторов и/или β2-адренорецепторов, встроенных в клеточную мембрану эндотелиальных клеток, и сопряженного с ними Gs-белка, активируется аденилатциклаза, которая инициирует синтез сАМР и последующую активацию белков Ерас с1, которые, в свою очередь, индуцируют ряд внутриклеточных сигнальных каскадов, стабилизирующих/восстанавливающих барьерную функцию эндотелия (активация Ерас1/AKAP9 и/или Eрас1/Rap1/Tiam1/ Vav2/Rac1 и/или Ерас/Rap1/PDE4D сигнальных каскадов; ингибирование RhoА посредством активации Ерас/Rap1/KRIT-1 и/или Ерас1/ Rap1/RASIP1/RADIL1/ARHGAP29 сигнальных каскадов, блокада цитокиновой сигнализации посредством Ерас-опосредованной активации SOCS-3) (рис. 9). Способность белков Ерас1 повышать барьерную функцию эндотелия сосудов универсальна и реализуется как в артериальном, так и венозном отделах сосудистого русла, что продемонстрировано в экспериментах на культуре клеток эндотелия артерий – HAECs , микроартерий – HMVECs и пупочной вены – HUVEC.
Рис. 9.
Схематическое отображение роли белков Epac в физиологической регуляции сосудистого русла (с изменениями по [23]). Объяснения в тексте.
Роль белков Ерас1 в регуляции пролиферации эндотелиальных клетках сосудов дискутабельна, однако большинство исследователей полагает, что они, активируя сопряженные с ними Rap1/PI3K/Akt/eNOS/NO и/или Rap1/MEK/ERK сигнальные каскады проявляют проангиогенную активность. Вместе с тем, остается не ясным, связаны ли эндотелиальные проангиогенные эффекты белков Ерас1 с активацией VEGF. Если роль белков Ерас1 в регуляции ангиогенеза и пролиферации эндотелиальных клеток окончательно не выяснена, то их место в регуляции пролиферации/ангиогенеза гладкомышечных клеток сосудов представляется достаточно определенным – белки Ерас1, действуя корпоративно с РКА, проявляют антиангиогенную активность. В основе этого эффекта лежит их способность ингибировать в гладкомышечных клетках сосудов ERK/JNK и/или Rac1/ERK1/Elk1 сигнальные каскады.
Не менее важную роль белки Ерас1 играют в регуляции тонической активности сосудистого русла. В крупных сосудах они проявляют выраженную комплексную вазодилатирующую активность: активируют трансмембранные ВКСа- и KV7,4-каналы, что вызывает гиперполяризацию клеточной мембраны и, следовательно, подавляет вход ионов Ca2+ в цитозоль клетки через потенциалзависимые трансмебранные медленные Ca2+-каналы L-типа; аналогичное действие оказывает Ерас1/CaN-опосредованная активация KАТФ трансмебранных каналов; ингибируют вазоконстрикторную активность Rho за счет активации Ерас/Rap1 сигнального пути и последующего дефосфорилирования регуляторной субъединицы MYPT1 фосфатазы LCМ; индуцируя эндотелий-зависимую релаксацию путем активации NOS3 и высвобождения NO (рис. 8). Напротив, в микрососудах Ерас1 инициируют вазоконстрикцию посредством активации Ерас1/Rap1А/ Rho/ROCK сигнального пути и, как следствие этого, экспрессии α2c-адренорецепторов в клеточной мембране.
В условиях патологии сосудистого русла – сосудистой травмы, белки Ерас1 активируют процессы миграции гладкомышечных клеток и рост неоинтимы, что, естественно, может вызвать стеноз/рестеноз поврежденного сосуда. Также, если принять во внимание данные о том, что белки Ерас1 могут инициировать избыточную транслокацию α2c-адренорецепторов в область клеточной мембраны в условиях нормотермии [75], т.е. все основания полагать, что они потенциально могут играть существенную роль в патогенезе болезни Рейно.
Благодарности. Авторы выражают глубокую признательность за консультативную помощь в работе над обзором литературы гл.н.с. ФГБНУ НИИ фармакологии им. В.В. Закусова, чл.-корр. РАН Ю.В. Вахитовой и за помощь в оформлении работы н.с. ФГБНУ НИИ фармакологии им. В.В. Закусова В.В. Барчукову (Москва).
Список литературы
Rall T.W., Sutherland E.W. Formation of a cyclic adenine ribonucleotide by tissue particles // J. Biol. Chem. 1958. V. 232. № 2. P. 1065.
Walsh D.A., Perkins J.P., Krebs E.G. An adenosine 3'5'-monophosphate-dependent protein kinase from rabbit skeletal muscle // J. Biol. Chem. 1968. V. 243. № 13. P. 3763.
Lymperopoulos A., Rengo G., Koch W.J. Adrenergic nervous system in heart failure: pathophysiology and therapy // Circ. Res. 2013. V. 113. № 6. P. 739.
Sands W.A., Palmer T.M. Regulating gene transcription in response to cyclic AMP elevation // Cell Signal. 2008. V. 20. № 3. P. 460.
Chin K.V., Yang W.L., Ravatn R. et al. Reinventing the wheel of cyclic AMP: novel mechanisms of cAMP signaling // Ann. N.Y. Acad. Sci. 2002. V. 968. P. 49.
Biel M., Michalakis S. Cyclic nucleotide-gated channel // Handb. Exp. Pharmacol. 2009. V. 191. P. 111.
Renström E., Eliasson L., Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cell // J. Physiol. 1997. V. 502. Pt. 1. P. 105.
Anciaux K., Van Dommelen K., Nicolai S. et al. Cyclic AMP-mediated induction of the glial fibrillary acidic protein is independent of protein kinase A activation in rat C6 glioma // J. Neurosci. Res. 1997. V. 48. № 4. P. 324.
Kawasaki H., Springett G.M., Mochizuki N. et al. A family of cAMP-binding proteins that directly activate Rap1 // Science. 1998. V. 282. № 5397. P. 2275.
de Rooij J., Zwartkruis F.J., Verheijen M.H. et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP // Nature. 1998. V. 396. № 6710. P. 474.
Aronoff D.M., Canetti C., Serezani C.H. et al. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1 // J. Immunol. 2005. V. 174. № 2. P. 595.
Cheng X., Ji Z., Tsalkova T., Mei F. Epac and PKA: a tale of two intracellular cAMP receptors // Acta Biochim. Biophys. Sin (Shanghai). 2008. V. 40. № 7. P. 651.
Muñoz-Llancao P., Henríquez D.R., Wilson C. et al. Exchange protein directly activated by cAMP (EPAC) regulates neuronal polarization through Rap1B // J. Neurosci. 2015. V. 35. № 32. P. 11315.
Breckler M., Berthouze M., Laurent A.C. et al. Rap-linked cAMP signaling Epac proteins: compartmentation, functioning and disease implications // Cell. Signal. 2011. V. 23. № 8. P. 1257.
Krähling A.M., Alvarez L., Debowski K. et al. CRIS-a novel cAMP-binding protein controlling spermiogenesis and the development of flagellar bending // PLoS Genet. 2013. V. 9. № 12. e1003960.
Schindler R.F., Brand T. The Popeye domain containing protein family – A novel class of cAMP effectors with important functions in multiple tissues // Prog. Biophys. Mol. Biol. 2016. V. 120. № 1–3. P. 28.
Banerjee U., Cheng X. Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: Structure, function and therapeutics // Gene. 2015. V. 570. № 2. P. 157.
de Rooij J., Rehmann H., van Triest M. et al. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs // J. Biol. Chem. 2000. V. 275. № 27. P. 20 829.
Schmidt M., Dekker F.J., Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions // Pharmacol. Rev. 2013. V. 65. № 2. P. 670.
Sugawara K., Shibasaki T., Takahashi H., Seino S. Structure and functional roles of Epac2 (Rapgef4) // Gene. 2016. V. 575. № 2. Pt. 3. P. 577.
Roberts O.L., Dart C. cAMP signalling in the vasculature: the role of Epac (exchange protein directly activated by cAMP) // Biochem. Soc. Trans. 2014. V. 42. № 1. P. 89.
Dao K.K., Teigen K., Kopperud R. et al. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition // J. Biol. Chem. 2006. V. 281. № 30. P. 21 500.
Lezoualc'h F., Fazal L., Laudette M., Conte C. Cyclic AMP Sensor EPAC Proteins and their role in cardiovascular function and disease // Circ. Res. 2016. V. 118. № 5. P. 881.
Borland G., Gupta M., Magiera M.M. et al. Microtubule-associated protein 1B-light chain 1 enhances activation of Rap1 by exchange protein activated by cyclic AMP but not intracellular targeting // Mol. Pharmacol. 2006. V. 69. № 1. P374.
Gloerich M., Ponsioen B., Vliem M.J. et al. Spatial regulation of cyclic AMP-Epac1 signaling in cell adhesion by ERM proteins // Mol. Cell. Biol. 2010. V. 30. № 22. P. 5421.
Rehmann H., Arias-Palomo E., Hadders M.A. et al. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B // Nature. 2008. V. 455. № 7209. P. 124.
Rehmann H., Rueppel A., Bos J.L., Wittinghofer A. Communication between the regulatory and the catalytic region of the cAMP-responsive guanine nucleotide exchange factor Epac // J. Biol. Chem. 2003. V. 278. № 26. P. 23 508.
Fujita T., Umemura M., Yokoyama U. et al. The role of Epac in the heart // Cell. Mol. Life Sci. 2017. V. 74. № 4. P. 591.
Nikolaev V.O., Bunemann M., Hein L. et al. Novel single chain cAMP sensors for receptor-induced signal propagation // J. Biol. Chem. 2004. V. 279. № 36. P. 37 215.
Wong W., Scott J.D. AKAP signalling complexes: focal points in space and time // Nat. Rev. Mol. Cell Biol. 2004. V. 5. № 12. P. 959.
Lee L.C., Maurice D.H., Baillie G.S. Targeting protein-protein interactions within the cyclic AMP signaling system as a therapeutic strategy for cardiovascular disease // Future Med. Chem. 2013. V. 5. № 4. P. 451.
Garg J., Feng Y.X., Jansen S.R. et al. Catecholamines facilitate VEGF-dependent angiogenesis via β2-adrenoceptor-induced Epac1 and PKA activation // Oncotarget. 2017. V. 8. № 27. P. 44732.
Belacel-Ouari M., Zhang L., Hubert F. et al. Influence of cell confluence on the cAMP signalling pathway in vascular smooth muscle cells // Cell Signal. 2017. V. 35. P. 118.
Kato Y., Yokoyama U., Yanai C. et al. Epac1 Deficiency Attenuated Vascular Smooth Muscle Cell Migration and Neointimal Formation // Arterioscler. Thromb Vasc. Biol. 2015. V. 35. № 12. P. 2617.
Cullere X., Shaw S.K., Andersson L. et al. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase // Blood. 2005. V. 105. № 5. P. 1950.
Rodrigues S.F., Granger D.N. Blood cells and endothelial barrier function // Tissue Barriers. 2015. V. 3. № 1–2. e978720.
Sayner S.L., Alexeyev M., Dessauer C.W., Stevens T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier // Circ. Res. 2006. V. 98. № 5. P. 675.
Pannekoek W.J., Post A., Bos J.L. Rap1 signaling in endothelial barrier control // Cell Adh. Migr. 2014. V. 8. № 2. P. 100.
Béraud-Dufour S., Gautier R., Albiges-Rizo C. et al. Interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1 // FEBS J. 2007. V. 274. № 21. P. 5518.
Sehrawat S., Ernandez T., Cullere X. et al. AKAP9 regulation of microtubule dynamics promotes Epac1-induced endothelial barrier properties // Blood. 2011. V. 117. № 2. P. 708.
Post A., Pannekoek W.J., Ponsioen B. et al. Rap1 Spatially Controls ArhGAP29 To Inhibit Rho Signaling during Endothelial Barrier Regulation // Mol. Cell Biol. 2015. V. 35. № 14. P. 2495.
Parnell E., Smith B.O., Palmer T.M. et al. Regulation of the inflammatory response of vascular endothelial cells by EPAC1 // Br. J. Pharmacol. 2012. V. 166. № 2. P. 434.
Sands W.A., Woolson H.D., Milne G.R. et al. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells // Mol. Cell Biol. 2006. V. 26. № 17. P. 6333.
Vestweber D. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation // Arterioscler. Thromb Vasc. Biol. 2008. V. 28. № 2. P. 223.
Kooistra M.R., Corada M., Dejana E., Bos J.L. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin // FEBS Lett. 2005. V. 579. № 22. P. 4966.
Rampersad S.N., Ovens J.D., Huston E. et al. Cyclic AMP phosphodiesterase 4D (PDE4D) Tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability // J. Biol. Chem. 2010. V. 285. № 44. P. 33 614.
Korayem A.H., Mujica P.E., Aramoto H. et al. Endothelial cAMP deactivates ischemia-reperfusion-induced microvascular hyperpermeability via Rap1-mediated mechanisms // Am. J. Physiol. Heart Circ. Physiol. 2017. V. 313. № 1. P. H179.
Kopperud R.K., Rygh C.B., Karlsen T.V. et al. Increased microvascular permeability in mice lacking Epac1 (Rapgef3) // Acta Physiol. (Oxf). 2017. V. 219. № 2. P. 441.
Birukova A.A., Zagranichnaya T., Fu P. et al. Prostaglandins PGE(2) and PGI(2) promote endothelial enhancement via PKA- and Epac1/Rap1-dependent Rac activation // Exp. Cell Res. 2007. V. 313. № 11. P. 2504.
Hong J., Doebele R.C., Lingen M.W. et al. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1 // J. Biol. Chem. 2007. V. 282. № 27. P. 19 781.
Doebele R.C., Schulze-Hoepfner F.T., Hong J. et al. A novel interplay between Epac/Rap1 and -activated protein kinase kinase 5/extracellular signal-regulated kinase 5 (MEK5/ERK5) regulates thrombospondin to control angiogenesis // Blood. 2009. V. 114. № 20. P. 4592.
Amano H., Ando K., Minamida S. et al. Adenylate cyclase/protein kinase A signaling pathway enhances angiogenesis through induction of vascular endothelial growth factor in vivo // Jpn. J. Pharmacol. 2001. V. 87. № 3. P. 181.
Chrzanowska-Wodnicka M., Kraus A.E., Gale D. et al. Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice // Blood. 2008. V. 111. № 5. P. 2647.
Lakshmikanthan S., Sobczak M., Chun C. et al. Rap1 promotes VEGFR2 activation and angiogenesis by a mechanism involving integrin αvβ3 // Blood. 2011. V. 118. № 7. P. 2015.
Fang Y., Olah M.E. Cyclic AMP-dependent, protein kinase A-independent activationextracellular signal-regulated kinase 1/ following adenosine receptor stimulation in human umbilical vein endothelial cells: role of exchange protein activated by cAMP 1 (Epac1) // J. Pharmacol. Exp. Ther. 2007. V. 322. № 3. P. 1189.
Namkoong S., Kim C.K., Cho Y.L. et al. Forskolin increases angiogenesis through the coordinated cross-talkPKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling // Cell Signal. 2009. V. 21. № 6. P. 906.
Wang S., Zhang Z., Qian W. et al. Angiogenesis and vasculogenic mimicry are inhibited by 8-Br-cAMP through activation of the cAMP/PKA pathway in colorectal cancer // Onco Targets Ther. 2018. V. 11. P. 3765.
Bond M., Wu Y.J., Sala-Newby G.B., Newby A.C. Rho GTPase, Rac1, regulates Skp2 levels, vascular smooth muscle cell proliferation, and intima formation in vitro and in vivo // Cardiovasc. Res. 2008. V. 80. № 2. P. 290.
Hewer R.C., Sala-Newby G.B., Wu Y.J. et al. PKA and Epac synergistically inhibit smooth muscle cell proliferation // J. Mol. Cell Cardiol. 2011. V. 50. № 1. P. 87.
Kimura T.E., Duggirala A., Hindmarch C.C. et al. Inhibition of Egr1 expression underlies the anti-mitogenic effects of cAMP in vascular smooth muscle cells // J. Mol. Cell Cardiol. 2014. V. 72. P. 9.
Kawano Y., Yoshimura T., Kaibuchi K. Smooth muscle contraction by small GTPase Rho // Nagoya J. Med. Sci. 2002. V. 65. № 1–2. P. 1.
Lubomirov L.T., Reimann K., Metzler D. et al. Urocortin-induced decrease in Ca2+ sensitivity of contraction in mouse tail arteries is attributable to cAMP-dependent dephosphorylation of MYPT1 and activation of myosin light chain phosphatase // Circ. Res. 2006. V. 98. № 9. P. 1159.
Murthy K.S., Zhou H., Grider J.R., Makhlouf G.M. Inhibition of sustained smooth muscle contraction by PKA and PKG preferentially mediated by phosphorylation of RhoA // Am. J. Physiol. Gastrointest Liver Physiol. 2003. V. 284. № 6. P. G1006.
Zieba B.J., Artamonov M.V., Jin L. et al. cAMP-responsive Rap1 exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity // J. Biol. Chem. 2011. V. 286. № 19. P. 16681.
Cuíñas A., García-Morales V., Viña D. et al. Activation PKA and Epac cyclic AMP depletes intracellular stores and reduces availability for vasoconstriction // Life Sci. 2016. V. 155. P. 102.
Shi Y., Chen X., Wu Z. et al. cAMP-dependent protein kinase phosphorylation produces interdomain movement in SUR2B leading to activation of the vascular KATP channel // J. Biol. Chem. 2008. V. 283. № 12. P. 7523.
Purves G.I., Kamishima T., Davies L.M. et al. Exchange protein activated by cAMP (Epac) mediates cAMP-dependent but protein kinase A-insensitive modulation of vascular ATP-sensitive potassium channels // J. Physiol. 2009. V. 587. Pt. 14. P. 3639.
Roberts O.L., Kamishima T., Barrett-Jolley R. et al. Exchange protein activated cAMP (Epac) induces vascular relaxation by activating Ca2+-sensitive K+ channels in rat mesenteric artery // J. Physiol. 2013. V. 591. № 20. P. 5107.
Humphries E.S., Kamishima T., Quayle J.M., Dart C. Calcium/calmodulin-dependent kinase 2 mediates Epac-induced spontaneous transient outward currents in rat vascular smooth muscle // J. Physiol. 2017. V. 595. № 18. P. 6147.
Stott J.B., Barrese V., Greenwood I. Kv7 Channel Activation Underpins EPAC-Dependent Relaxations of Rat Arteries // Arterioscler. Thromb Vasc. Biol. 2016. V. 36. № 12. P. 2404.
García-Morales V., Cuíñas A., Elíes J., Campos-Toimil M. PKA and Epac activation mediates cAMP-induced vasorelaxation by increasing endothelial NO production // Vascul. Pharmacol. 2014. V. 60. № 3. P. 95.
Chotani M.A., Mitra S., Eid A.H. et al. Distinct signaling pathways differentially regulate alpha2C-adrenoceptor expression: role in serum induction in human arteriolar smooth muscle cells // Am. J. Physiol. Heart Circ. Physiol. 2005. V. 288. № 1. P. H69.
Eid A.H., Chotani M.A., Mitra S. et al. Cyclic AMP acts through Rap1 and JNK signaling to increase expression of cutaneous smooth muscle alpha2C-adrenoceptors // Am. J. Physiol. Heart Circ. Physiol. 2008. V. 295. № 1. P. H266.
Jeyaraj S.C., Unger N.T., Eid A.H. et al. Cyclic signaling activates RhoA to induce α(2c)-adrenoceptor translocation to the cell surface of microvascular smooth muscle cells // Am. J. Physiol. Cell Physiol. 2012. V. 303. № 5. P. C499.
Motawea H.K., Jeyaraj S.C., Eid A.H. et al. Cyclic signaling mediates translocationmicrovascular smooth muscle α2C-adrenoceptors through the actin-binding protein filamin-2 // Am. J. Physiol. Cell Physiol. 2013. V. 305. № 8. P. C829.
Wang H., Robichaux W.G., Wang Z. et al. Inhibition of Epac1 suppresses mitochondrial fission and reduces neointima formation induced by vascular injury // Sci. Rep. 2016. V. 6. P. 36552.
Qiao J., Mei F.C., Popov V.L. et al. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP // J. Biol. Chem. 2002. V. 277. № 29. P. 26 581.
Yokoyama U., Minamisawa S., Quan H. et al. Epac1 is upregulated during neointima formation and promotes vascular smooth muscle cell migration // Am. J. Physiol Heart Circ. Physiol. 2008. V. 295. № 4. P. H1547.
Дополнительные материалы отсутствуют.
Инструменты
Физиология человека