

Физиология человека, 2020, T. 46, № 4, стр. 111-134
Роль регуляторных белков Ерас в физиологии и патологии сердечно-сосудистой системы. Часть II. Роль белков Ерас в физиологии и патологии сердца
С. А. Крыжановский 1, *, Т. Д. Никифорова 1, М. Б. Вититнова 1, А. Д. Дурнев 1
1 ФГБНУ НИИ фармакологии имени В.В. Закусова
Москва, Россия
* E-mail: sak-538@yandex.ru
Поступила в редакцию 03.07.2019
После доработки 10.01.2020
Принята к публикации 20.03.2020
Аннотация
Во второй части обзора приведены современные представления о роли белков Ерас в регуляции инотропной и ритмической функции сердца, межклеточного сопряжения кардиомиоцитов и их апоптоза. Подробно рассмотрен вклад белков Ерас в развитие гипертрофии, фиброза и ремоделирования миокарда, а также их роль в генезе острого ишемического повреждения сердечной мышцы и нарушений сердечного ритма.
Исторически полагали, что в кардиомиоцитах единственным эффектором сАМР является протеинкиназа А (РКА), которая, взаимодействуя с внутриклеточными мишенями, регулирует процессы, обеспечивающие поддержание сократительного статуса и ритмической активности кардиомиоцитов, их электромеханическое сопряжение и т.д. Однако после открытия в 1998 г. белков Ерас стало ясно, что в кардиомиоцитах существует альтернативный, не зависимый от РКА, путь регуляции внутриклеточных сигнальных путей.
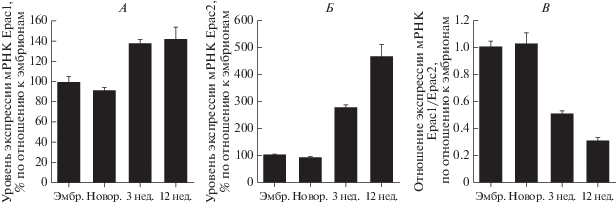
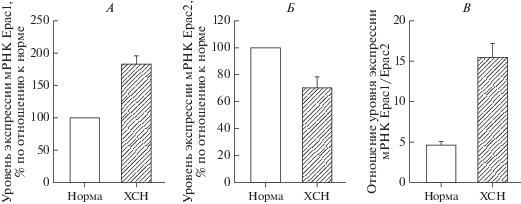
В кардиомиоцитах экспрессируются как белки Ерас1, так и белки Ерас2. В экспериментах in vitro, выполненных на культуре кардиомицитов мышей, показано, что экспрессия белков Ерас1 у взрослых животных по сравнению с новорожденными увеличивается на 42% (р < 0.01), тогда как белков Ерас2 – в 3.7 раза (р < 0.001) [1]. При расчете отношения уровня экспрессии мРНК Epac1/ Epac2 показано, что с возрастом в сердце происходит существенное относительное снижение экспрессии белка Ерас1 (р < 0.001) (рис. 1), на основании чего авторы пришли к заключению, что с возрастом белки Ерас2 в миокарде становятся доминирующей изоформой. В кардиомиоцитах локализация белков Ерас различна, с чем во многом и связаны различия в их функциональной активности. Белки Ерас1 располагаются вблизи внутренней поверхности клеточной мембраны, на мембране митохондрий, на перинуклеарной мембране и, возможно, в ядре клетки, тогда как белки Ерас2 локализуются в области Z-линий рядом с Т-трубочками, вблизи которых сосредоточено скопление цистерн саркоплазматического ретикулума (SR) [2].
Рис. 1.
Уровень экспрессии белков Ерас в сердце в зависимости от возраста (по [1]). Объяснение в тексте.
Роль белков Ерас в регуляции инотропной функции сердца
Известно, что инотропная функция сердца находится под контролем симпатической нервной системы. Выделившийся из пресинаптических окончаний нейромедиатор норэпинефрин активирует встроенные в клеточную мембрану кардиомиоцитов постсинаптические сопряженные с G-белком β1-адренорецепторы, что сопровождается увеличением внутриклеточной концентрации сАМР и активацией ее эффектора РКА. Активированная РКА фосфорилирует трансмембранные медленные потенциалзависимые Са2+-каналы L-типа, в результате чего в цитозоль кардиомиоцитов поступает незначительное количество так называемых триггерных внеклеточных ионов Са2+, которые инициируют конформационные изменения (активацию) Са2+-освобождающих каналов, чувствительных к рианодину (RyR2; син. рианодиновые рецепторы 2 типа), встроенных в мембрану SR и расположенных рядом с Т-трубочками. (Этот феномен в литературе обозначается аббревиатурой CICR (Calcium-induced calcium release) – инициированое ионами Са2+ высвобождение ионов Са2+.) Поскольку концентрация ионов Са2+ в цистернах SR на несколько порядков превышает его концентрацию в цитоплазме, ионы Са2+ диффундируют во внутриклеточное пространство, в результате чего концентрация ионов Са2+ в цитоплазме увеличивается с 10–7 до 10–6–10–5 М, что обеспечивает их взаимодействие с регуляторным глобулярным белком – тропонином С (TnC). Образовавшийся комплекс Са2+/TnC сдвигает фибриллярный белок тропомиозин с активного центра волокон актина, что позволяет последнему присоединиться к миозину и инициировать сокращение кардиомиоцита. Сразу же после завершения сокращения в мембране SR в результате сАМР-опосредованной активации РКА фосфорилируется ключевой регулятор расслабления кардиомиоцитов белок фосфоламбан (PLB или PLN), который активирует Са2+-зависимую АТРазу (Sarco/endoplasmic reticulum Ca2+-ATPase, SERCA2) – кальциевый насос SR. SERCA2 стимулирует активный транспорт ионов Са2+ из цитоплазмы в цистерны SR, где он связывается белком кальсеквестрином. По окончании работы кальциевого насоса SR концентрация ионов Са2+ в цитозле кардиомиоцитов снижается до 10–8 М, а в цистернах SR возрастает до 10–3 М. Помимо этого РКА, фосфорилируя нити актина, снижает их чувствительность к ионам Са2+ и тем самым способствует расслаблению кардиомиоцитов – лузитропный эффект РКА.
В экспериментах in vitro, выполненных на изолированных кардиомиоцитах желудочков взрослых крыс, показано, что активация белков Ерас селективным агонистом 8-CPT не влияла ни на триггерный входящий кальциевый ток (ICa), протекающий через трансмембранные Ca2+-каналы L-типа (т.е. не инициирует CICR), ни на продолжительность потенциала действия кардиомиоцитов [3]. Однако в этих экспериментах было показано, что активированные 8-CPT белки Ерас инициируют выброс ионов Са2+ из SR посредством фосфорилирования RyR2 (Ryanodine receptor 2) по Ser2815. Посредником в передаче сигнала с белков Ерас на RyR2 служит цитозольная Ca2+/кальмодулин-зависимая протеикиназа II (CaMKIIß), поскольку ее блокада селективным ингибитором KN-93 препятствовала фосфорилированию RyR2 и выбросу ионов Са2+ из SR. Также в этом исследовании было показано, что блокада РКА ее эндогенным ингибитором PKI не влияет на Ерас-опосредованное фосфорилирование RyR2. Таким образом, авторы этого исследования показали, что белки Ерас могут, не зависимо от РКА, инициировать сокращение кардиомиоцитов. В экспериментах in vitro, выполненных на изолированных кардиомиоцитах желудочков взрослых мышей, показано, что белки Ерас инициируют выброс ионов Са2+ из SR посредством активации Ерас/Rap1/фосфолипаза Сϵ (PLCϵ) сигнального пути [4]. Авторы продемонстрировали, что PLCϵ необходима для Ерас-опосредованного выброса ионов Са2+ из SR, поскольку ее блокада селективным ингибитором U73122 ингибировала высвобождение ионов Са2+ в цитозоль кардиомиоцитов. Также в этом исследовании было показано, что блокада РКА ее ингибиторами (Rp-cAMPs, 8-Br-Rp-cAMPs и/или H89) не влияет на Ерас-опосредованный выброс ионов Са2+ из SR. Несколько позже эти же авторы показали, что активация Ерас/Rap1/PLCϵ сигнального каскада влечет за собой активацию протеинкиназы С эпсилон (PKCϵ) и последующее фосфорилирование (активацию) CaMKIIß по Thr286 и, как следствие этого, активацию RyR2 [5]. В экспериментах in vitro, выполненных на изолированных кардиомиоцитах желудочков взрослых крыс, было продемонстрировано, что белки Ерас, вне зависимости от влияния на функциональную активность SR, могут стимулировать сократительный ответ кардиомиоцитов [6]. Показано, что активация белков Ерас1 селективным агонистом 8-CPT уже с 1-ой мин сопровождалась уменьшением длины (сокращением) и увеличением силы сокращения миофиламентов (р < 0.05) кардиомицитов, которое достигало плато через 5 мин после внесения в культуру клеток 8-CPT, тогда как активация РКА ее агонистом – Bnz-cAMP приводила к уменьшению силы сокращения. Авторы исследования пришли к заключению о том, что белки Ерас и РКА оказывают противоположное действие на Са2+-опосредованную силу сокращения миофиламентов и их чувствительность к ионам Са2+. В том же исследовании было показано, что повышение чувствительности миофиламентов к ионам Са2+ связано со способностью регуляторных белков Ерас фосфорилировать (активировать) тропонин I (TnI) и сердечный миозин-связывающий белок С (myosin-binding protein C, cardiac-typе, сMyBP-C); последний фосфори-лируется по Ser282. Фосфорилирование TnI и сMyBP-C не связано с активацией белком Ерас малой GТFазы Rap1 и происходит просредством активации сопряженного с Ерас не выявленного эффектора, который активирует сопряженный с белком Ерас PLCϵ/PKCϵ/CaMKIIß сигнальный каскад. Помимо этого показано, что Ерас-опосредованное фосфорилирование сMyBP-C происходит не зависимо от PKA. В экспериментах in vitro, выполненных на изолированных кардиомиоцитах желудочков взрослых крыс, определяли влияние белков Ерас на внутриклеточную ([Ca2+]i) и ядерную ([Ca2+]n) концентрацию ионов Са2+ [7]. Показано, что активация белков Ерас селективным агонистом 8-CPT сопровождается снижением пиковой систолической и увеличением диастолической ([Ca2+]i), тогда как в ядре кардиомиоцитов увеличивается систолическая и диастолическая ([Ca2+]n). Авторы исследования показали, что Ерас-опосредованная модуляция концентрации ионов Са2+ в цитозоле связана с активацией PLC/инозитол 1,3,5-трифосфат (IP3)/CaMKII сигнального каскада, поскольку ингибирование PLC – U73122, или IP3 – 2-APB, или CaMKII – KN-93 блокировало модулирующее действие белков Ерас. Аналогичным образом происходит модуляция концентрации ионов Са2+ в ядре кардиомиоцитов, однако этот сигнальный путь может быть короче, поскольку IP3 может влиять на ядерную концентрацию ионов Са2+ посредством активации встроенных в ядерную мембрану IP3-рецепторов (IP3R). Также в этом исследовании было показано, что Ерас-опосредованная активация ядерных IP3R и CaMKII могут индуцировать транслокацию в ядро клетки гистондеацетилазы 5 (Histone deacetylase 5, HDAC5) – белка, обладающего функцией транскрипционного репрессора, регулирующего в частности рост/гипертрофию кардиомиоцитов, с последующей активацией прогипертрофического транскрипционного фактора усиления миоцитов 2 (myocyte enhancer factor 2, MEF2) [7]. В экспериментах in vitro, выполненных на изолированных сегментах миокарда желудочков мышей, показано, что базальная сила сокращений полоски миокарда, взятой у нокаутных по белкам Ерас1 мышей, была значительно меньше чем у интактных, соответственно 73 ± 7.1 и 112 ± 14.7 мг/мм2 (р < 0.05) [8]. Далее было показано, что если величина инотропного ответа на неселективный β-адреностимулятор изопротеренол была одинакова в обеих группах, то продолжительность последующей за сокращением релаксации полоски миокарда, полученной от нокаутных мышей, была значимо больше – соответственно 54 ± 1.5 и 61 ± 2.0 мс (р < 0.05). Изучение механизмов, лежащих в основе увеличения периода релаксации, продолжили в экспериментах in vitro, выполненных на культуре клеток кардиомиоцитов взрослых мышей, в которых было показано, что как РКА, так и белки Ерас1 регулируют процессы релаксации кардиомиоцитов просредством активации белка фосфоламбана (PLN), фосфорилируя его по Ser16. Однако, если при активации РКА селективным агонистом 6-Bnz-cAMP фофорилировалось 40% от общего PLN, то при активации белков Ерас1 8-CPT – 60% PLN. Ерас1-опосредованное фосфорилирирование PLN происходит путем активации PLC/PKCϵ/CaMKII сигнального каскада (рис. 2). На основании полученных данных авторы сделали заключение о том, что лузитропное (релаксирующее) действие РКА и белков Ерас1 является аддитивным, однако последним принадлежит ведущая роль. Не менее важным результатом этого исследования является то, что в кардиомиоцитах, полученных от нокaутных по белкам Ерас1 и интактных мышей, интенсивность активированного изопротеренолом ICa, протекающего через медленные трансмембранные потенциалзависимые Са2+-каналы L-типа, одинакова, что свидетельствует о том, что инотропное/лузитропное действие белков Ерас, в отличие от РКА, не реализуется по CICR пути. В этом же исследовании продемонстрировано, что в кардиомиоцитах, полученных от нокаутных по белкам Ерас1 животных, в цитозоле базальная и пиковая концентрация значимо (р < 0.01) ниже, чем у интактных – соответственно 99 ± 11.8 против 139 ± 6.8 нM и 559 ± 33.2 против 883 ± 35.3 нM. Эти результаты хорошо согласуются с данными, полученными в экспериментах in vivo и свидетельствующими о том, что у нокаутных по белкам Ерас1 фракция выброса левого желудочка сердца, характеризующая его инотропную функцию, значимо (р < 0.01) ниже [8].
Рис. 2.
Схематическое отображение Epac- и РКА-опосредованной регуляции инотропной функции сердца (по [8]). Объяснение в тексте.
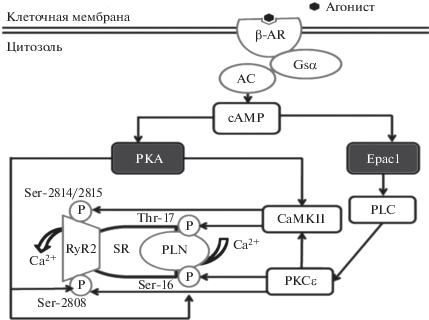
Таким образом, подводя итог выше изложенному, можно говорить о том, что инотропное/лузитропное действие белков Ерас, в отличие от РКА, не связано с CICR и в нормальных физиологических условиях реализуется следующими путями: в результате активации катехоламинами β1-адренорецепторов и сопряженного с ними Gs-белка активируется аденилациклаза, которая инициирует синтез сАМР и последующую активацию белков Ерас. Далее события разворачиваются по следующим направлениям: белки Ерас активируют локализованную на внутренней поверхности клеточной мембраны PLCϵ, которая гидролизует мембранный фосфолипид – фосфатидилинозитол 4,5-дифосфат (рhosphatidylinositol, PIP2), в результате чего образуется диацилглицерол (DAG) и IP3. Затем IP3 активирует IP3-R, что приводит к высвобождению из SR незначительного количества ионов Са2+, которые сенсибилизируют RyR2 и активируют CaMKIIß, что приводит к фосфорилированию RyR2 и последующему выбросу ионов Са2+ из цистерн SR. Параллельно с этим CaMKIIß при участии PKC фосфорилирует тропонин I (TnI) и миозин-связывающий белoк C (MyBP-C), в результате чего повышается чувствительность миофиламентов к ионам Са2+, что на фоне резкого увеличения концентрации ионов Са2+ в цитоплазме инициирует сокращение кардимиоцитов, в реализации которого принимают участие Са2+-связывающий белок кальмодулин (СаМ) и Са2+-зависимая серин-треонин фосфатаза – кальциневрин (Cn) [9]. В момент достижения пика сокращения CaM-KIIß фосфорилирует PLN, который активирует Са2+-зависимую АТРазу (SERCA2), стимулирующую активный обратный транспорт ионов Са2+ из цитоплазмы в цистерны SR и последующее расслабление кардиомиоцитов. Помимо этого, локализованная на ядерной мембране PLCϵ активирует IP3, который, в свою очередь, активирует ядерные IP3-Rn, в результате чего возрастает внутриядерная концентрация ионов Са2+, приводящая к активации и ядерной CaMKIIn, которая, в свою очередь, активирует HDAC5/MEF2 сигнальный каскад. Следует отметить, что в настоящее время физиологическая роль Ерас-опосредованной активации PLCϵ/IP3-Rn/CaMKIIn/ HDAC5/MEF2 сигнального каскада остается не ясной. Также остается дискутабельным и вопрос о вкладе той или иной изоформы белка Ерас в регуляцию инотропной функции сердца, однако большинство исследователей склонны полагать, что в физиологических условиях сократительную функцию кардиомиоцитов регулируют РКА и белки Ерас1 [8, 10].
Роль белков Ерас в регуляции межклеточного сопряжения кардиомиоцитов
Нексусы или щелевые контакты (gap junction, GJ) обеспечивают электрическое и химическое сопряжение кардиомиоциов. Нексус состоит из кластеров каналов, каждый из которых представлен парой коннексонов – политопных интегральных мембранных белков, расположенных напротив друг друга в мембранах соседних клеток. Коннексоны 4 раза пересекают мембрану и состоят из двух внеклеточных петель (EL-1 и EL-2) и цитоплазматической петли (CL). Каждый коннексон образован 6 молекулами белков – коннексинов (Cx), расположенных гексагонально вокруг центрального гидрофильного канала, шесть белковых коннексиновых субъединиц которого сгруппированы вокруг гидрофильной поры, пронизывающей мембрану, и образуют коннексон. Два полуканала соседних клеток, расположенные напротив друг друга, соединяются и образуют непрерывный межклеточный канал. В кардиомиоцитах идентифицированы три изоформы Cx – Сх40, Сх43 и Сх45. Kоннексины Сх43, которые играют основную роль в межклеточной коммуникации, преимущественно представлены в нексусах сократительных кардиомицитов желудочков, а Сх40 и Сх45 – в проводящих кардиомиоцитах. Нарушение функциональной активности GJ влечет за собой сократительную дисфункцию и инициирует нарушение ритма сердечных сокращений.
Еще в конце ХХ века было описано, что сАМР регулирует экспрессию в миокарде коннексинов Сх43 и Сх45 [11]. Позднее в экспериментах in vitro, выполненных на культуре неонатальных кардиомицитов крыс, показали, что экспрессия коннексинов Сх43 регулируется сАМР/Rap1 сигнальным каскадом [12]. Помимо этого, в экспериментах, выполненных на неонатальных кар-диомицитах крыс, показали, что добавление в культуру клеток неселективного β-адреностимулятора изопротеренола инициирует РКА-опосредованную экспрессию коннексинов Сх43 [13]. Однако в экспериментах in vitro, также выполненных на культуре клеток неонатальных кардиомицитов крыс – NRCMs, было продемонстрировано, что блокада РКА селективным ингибитором H89 уменьшала, но не подавляла сАМР-опосредованную экспрессию коннексинов Сх43 [14]. Далее было показано, что 8-CPT-иницированная активация сигнального каскада Ерас/Rap1 сопровождается накоплением коннексинов Сх43 в области GJ. При этом увеличение экспрессии коннексинов Сх43 на фоне активации 8-CPT каскада Ерас/Rap1 было значимо больше (р < 0.05), чем при активации РКА ее агонистом – 6 Bnz-cAMP. Авторы этого исследования делают вывод о том, что белки Ерас преимущественно увеличивают экспрессию коннексинов Сх43, тогда как PKA облегчает/увеличивает проводимость GJ без существенного влияния на продукцию коннексинов Сх43. Аналогичные выводы были сделаны на основании и более поздних экспериметов in vivo [15]. Также отмечалось, что белки Ерас инициируют образование de novo GJ, активируя N-кадгерин-опосредованное формирование якорных межклеточных контактов, ассоциированных с микрофиламентами (adherens junctions, AJ) [14]. В экспериментах in vitro, выполненных на культуре клеток неонатальных кардиомицитов крыс – NNRCM, показано, что инициированное неселективным β-адреностимулятором изопротеренолом фосфорилирование (активация) коннексинов Сх43 осуществляется посредством активации сАМР/Ерас1/PKCϵ сигнального каскада, поскольку блокада активируемого изопротеренолом сАМР/РКА сигнального каскада эндогенным ингибитором РКА – PKI не предотвращала фосфорилирование коннексинов Сх43 [16]. Авторы этого исследования пришли к заключению, что фосфорилирование коннексинов Сх43 кардиомиоцитов связано с сАМР-опосредованной активацией белков Ерас1, тогда как вклад РКА в этот процесс незначителен.
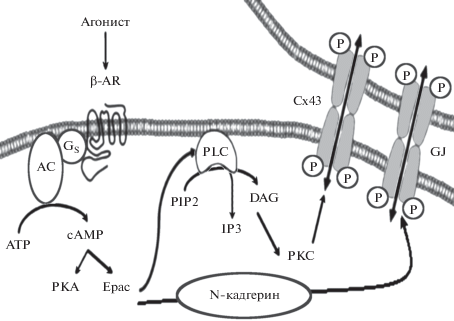
Таким образом, имеющиеся в настоящее временя данные позволяют заключить, что в физиологических условиях в кардиомиоцитах белки Ерас1 не зависимо от РКА инициируют экспрессию играющих ключевую роль в межклеточной коммуникации коннексинов Сх43 и тем самым оптимизируют электрическое и химическое сопряжение клеток сердца. Этот эффект белков Ерас1 реализуется посредством активации катехоламинами сАМР/Ерас1/PLCϵ/PIP2/DAG/PKCϵ сигнального каскада, а также N-кадгерин-опосредованного формирования AJ (рис. 3).
Рис. 3.
Схематическое отображение роли белков Epac в регуляции электрического и химического сопряжения клеток сердца (по [15]). Объяснение в тексте.
Роль белков Ерас в регуляции процессов апоптоза
Еще в конце ХХ века было показано, что хроническая (в течение 24 ч) стимуляция интактных кардиомиоцитов неселективным β-адреностимулятором изопротеренолом вызывает апоптоз кардиомиоцитов просредством активации РКА [17]. Впоследствие результаты этого исследования были подтверждены в эксперитментах in vitro и in vivo, в которых было показано, что апоптоз кардиомицитов, инициируемый РКА, ассоциируется с активацией β1-адренорецепторов, тогда как сопряженные с β2- и β3-адренорецепторами сигнальные каскады имеют антиапоптотическую направленность [18]. В экспериментах in vitro, выполненных на клеточной культуре эмбриональных (H9c2) и неонатальных (NRCMs) кардиомицитов крыс, 8-CPT-опосредованная активация белков Ерас подавляла NO-индуцированный апоптоз просредством активации Ерас/Rap1/RAC-α-серин-треонин-протеинкиназа (RAC-α serine-threonine-protein kinase, Akt; син. протеинкиназа В, РКВ) сигнального каскада [19]. Эти результаты были подтверждены в экспериментах in vitro, выполненных на клеточной культуре эмбриональных кардиомиоцитов – H9c2, в которых 8-CPT-опосредованная активация белков Ерас1 ингибировала апоптоз, инициированный гипергликемией и активными формами кислорода (ROS) в результате активации Ерас1/фосфоинозитид-3-киназа (рhosphoinositide 3-kinases; PI3K)/Akt сигнального пути [20]. Также в этом исследовании было показано, что Ерас1-опосредованная активация Akt происходит за счет ее фосфорилирования по Ser473. В экспериментах in vitro, выполненных на клеточной культуре неонатальных кардиомицитов крыс, у которых апоптоз вызывали 24-часовой инкубацией кардимиоцитов с H2O2, в сравнительном аспекте были изучены механизмы, лежащие в основе антиапоптотического действия РКА и белков Ерас1 [21]. На первом этапе исследования отмечалось, что в условиях оксидативного стресса активация рецепторов глюкагон-подобного пептида-1 (Glucagon-like peptide-1, GLP-1; син. энтероглюкагон) – GLP-1R селективным агонистом exendin-4 подавляет апоптоз и оказывает антиоксидантное действие. Эти эффекты GLP-1R реализуются путем активации аденилатциклаза (АС)/сАМР сигнального пути, поскольку ингибирование АС ее селективным антагонистом дидезоксиаденозином – ddA блокирует антиапоптотическое и антиоксидантное действие exendin-4. Аналогичные результаты были получены при внесении в культуру клеток эндогенного супрессора белков Ерас1 – siRNAs, что позволило авторам сделать заключение о том, что антиапоптотическое и антиоксидантное действие exendin-4 реализуется просредством активации сА-МР/Ерас1 сигнального каскада. Это заключение было подтверждено и в экспериментах, в которых 8-CPT-опосредованная активация белков Ерас1 в подвергнутых оксидативному стрессу кардиомиоцитах сопровождалась подавлением образования ROS и апоптоза. Далее было показано, что антиапоптотическое действие белков Ерас1 связано с активацией Ерас1/Rap/ PLCϵ/PKCϵ сигнального каскада и последующим ингибированием каспазы 3, а также увеличением экспрессии антиапоптотического белка В клеточной лимфомы 2 (B-cell lymphoma 2, Bcl-2), а Ерас1-опосредованное антиоксидантное действие связано с увеличением производства таких эндогенных антиоксидантов как каталаза, глутатион пероксидаза-1 (GPx-1) и марганец-содержащая супероксиддисмутаза (Mn-SOD) (рис. 4). В этих же экспериментах отмечали, что РКА не влияет на экспрессию эндогенных антиоксидантов, однако обладает антиапоптотической активностью, которая реализуется по не зависимому от белков Ерас1 сигнальному каскаду – РКА/CREB (cAMP response element-binding protein – CREB) (рис. 4). В экспериментах in vitro, также выполненных на клеточной культуре неонатальных кардиомицитов крыс, были подтверждены выше приведенные данные о том, что активация сАМР/Ерас1/Rap1 сигнального пути подавляет апоптоз кардиомиоцитов посредством увеличения экспрессии Bcl-2 и подавления активности каспазы 3 [22]. Вместе с этим, в этой работе было продемонстрировано, что активация в кардиомиоцитах сАМР/РКА/CREB сигнального каскада не влияет на интенсивность процессов апоптоза, а скорее всего, регулирует их инотропную функцию. В экспериментах in vitro, выполненных на митохондриях, выделенных из кардиомиоцитов взрослых крыс, был изучен вклад митохондриальных белков Ерас в регуляцию апоптоза [23]. Известно, что в митохондриях экспрессируется растворимая изоформа аденилатциклазы – sAC, которая в отличие от аденилатциклазы, расположенной на внутренней поверхности клеточной мембраны – tmACs, не активируется G-белками и форсколином, но может быть активирована гидрокарбонатом ${\text{HCO}}_{3}^{ - }$ и/или ионами Са2+ [24, 25]. В кардиомиоцитах взрослых крыс экспрессируется так называемая усеченная изоформа sAC – sACt [23]. В условиях патологии гиперактивация sAC инициирует митохондрия-опосредованный апоптоз [26]. В основе этого процесса лежит активация потенциалзависимого Са2+-канала внутренней мембраны митохондрий (mitochondrial calcium uniporter, MCU), который транспортирует ионы Са2+ через внутреннюю митохондриальную мембрану к митохондриальному матриксу. Далее поступившие ионы Са2+ инициируют открытие поры, изменяющей проницаемость мембраны митохондрий (мitochondrial рermeability тransition Pore, mPTP), состоящей из потенциалзависимого анионного канала (VDAC) и ADP/ATP-антипортера. Длительное открытие mPTP сопровождается перегрузкой митохондрий ионами Са2+, диссипацией ее мембранного потенциала (Δd) и, как следствие этого, резким увеличением митохондриальной проницаемости (mitochondrial permeability transition, MPT). На первом этапе исследований было показано, что HCO$_{3}^{--}$-опосредованная активация sACt, но не активация tmACs форсколином, сопровождается увеличением экспрессии сАМР в митохондриях. Затем было показано, что сА-МР-опосредованная активация митохондриальных белков Ерас1 стимулировала процессы окислительного фосфорилирования и увеличивала продукцию АТР (рис. 5) даже в присутствии высоких концентраций ионов Са2+. В этих условиях блокада белков Ерас1 их селективным антагонистом CE3F4, но не блокада белков Ерас2 селективным антагонистом ESI-05, сопровождалась активацией MCU и последующим увеличением MPT (рис. 5). Также в этом исследовании было показно, что РКА, в отличие от белков Ерас1, не принимает участие в регуляции MPT. На основании полученных данных авторы делают заключение о том, что ${\text{HCO}}_{3}^{ - }$/Са2+-опосредованная активация sACt/сАМР/Ерас1 сигнального каскада, оптимизируя митохондриальный метаболизм, препятствует дестабилизации MPT и тем самым оказывает антиапоптотическое действие. В модельных экспериментах in vitro, выполненных на выделенных из сердец взрослых крыс кардиомиоцитах, были изучены механизмы кардиопротективного действия пептидa урокортин-1 (Ucn-1), который реализует свои протективные эффекты, активируя сАМР [27]. Перед началом исследования кардиомиоциты подвергали 30-минутной ишемии, затем 18-часовой реперфузии. Отмечалось, что Ucn-1, в частности, реализует свои кардиопротективные эффекты путем активации сАМР/Ерас2 сигнального каскада, сопровождающейся увеличением экспрессии X-связанного ингибитора белков апоптоза (X-linked inhibitor of apoptosis protein, XIAP; син. IAP3) и подавлением экспрессии проапоптотического белка BAD. В случае блокады белков Ерас2 их селективным антагонистом ESI-05 не было отмечено увеличения экспрессии XIAP и уменьшения экспрессии BAD.
Рис. 4.
Схематическое отображение Epac- и РКА-опосредованной регуляции апоптоза кардиомиоцитов (по [21]). Объяснение в тексте.
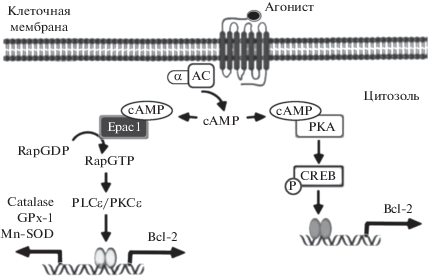
Рис. 5.
Схематическое отображение роли митохондриальных белков Epac в регуляции продукции АТР и препятствии дестабилизации MPT (по [23]). Объяснение в тексте.
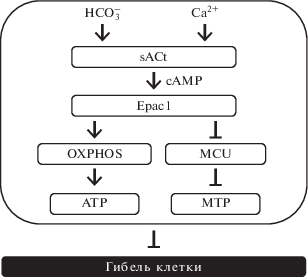
Таким образом, имеющиеся литературные данные свидетельствуют о том, что активация локализованных в цитозоле кардиомиоцитов белков Ерас1 подавляет NO-и ROS-индуцированный апоптоз посредством активации Ерас1/PLCϵ/ PIP2/DAG/PKCϵ сигнального каскада (рис. 4), тогда как митохондриальные белки Ерас1 реализуют свои антиапоптотические эффекты посредством ингибирования MCU, что препятствует дестабилизации MPT (рис. 5). Антиапототическое действие также продемонстрировано и для белков Ерас2, которые в условиях ишемия/реперфузия инициируют увеличение экспрессии антиапоптотического белка XIAP и подавляют экспрессию проапоптотического белка BAD.
Вместе с тем, следует отметить, что в 2017 г. были опубликованы результаты исследования, в котором корейские ученные на основе результата анализа 134 публикаций, используя нормированные обыкновенные дифференциальные уравнения, построили математическую модель сети сигнальных путей в клетке, согласно которой были выделены 5 наиболее важных контекстно-независимых взаимодействий, регулирующих апоптоз и гипертрофию, одним из которых являлся Epac/CaMKII сигнальный каскад [28]. Однако из текста статьи не ясно, что инициирует в кардиомиоцитах Epac/CaMKII сигнальный каскад – апоптоз или гипертрофию.
Роль белков Ерас в развитии гипертрофии и ремоделирования миокарда
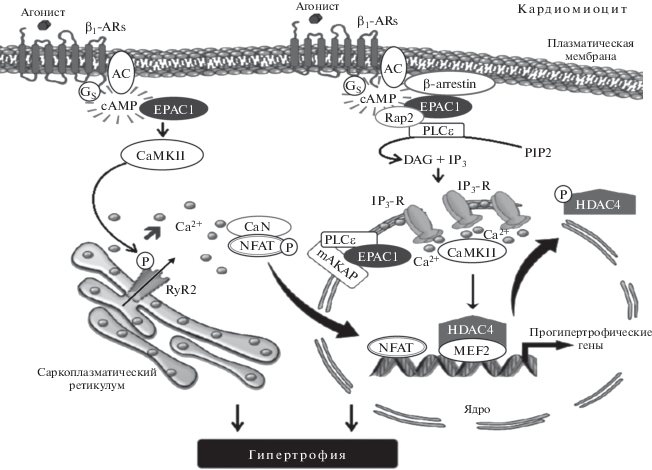
Роль симпатической нервной системы в формировании гипертрофии/ремоделирования миокарда была достаточно подробно изучена еще в 80–90 гг. ХХ века [29, 30]. Однако, уже начиная с 2000 г. стали появляться сообщения о том, что гипертрофия кардиомиоцитов не связана с активацией β-адренорецептор/сАМР/РКА сигнального каскада [31, 32], а реализуется независимо от РКА путем активации CaМKIIδ и/или CaMKIIγ [33]. В 2005 г. впервые было продемонстрировано, что активация белков Ерас способствует гипертрофии кардиомиоцитов [34]. На первом этапе этого исследования было продемонстрировано, что внесение в культуру неонатальных кардиомиоцитов агониста белков Ерас 8-CPT или аденовируса Ad.EpacWT, инициирующего суперэкспрессию белков Epac1, приводило к двукратному увеличению площади поверхности кардиомиоцитов (р < 0.001) и такому же увеличению синтеза белка, что, по мнению авторов, “придает кардиомиоцитам все признаки гипертрофированного фенотипа”. 8-CPT-опосредованная активация белков Ерас или инициация их суперэкспрессии Ad.EpacWT стимулирует продукцию/активность прогипертрофического ядерного транскрипционного фактора NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells). Активация NF-kB белками Ерас опосредуется кальций/кальмодулин-зависимой серин/треонин фосфатазой – кальциневрином (CaN), поскольку фармакологическая блокада CaN его ингибитором циклоспорином A препятствует экспрессии NF-kB. При изучении уровня экспрессии белков Ерас1 и Ерас2 в биоптатах тканей, полученных из сердец мышей, у которых гипертрофию миокарда вызывали коарктацией аорты, было показано, что по сравнению с интактными животными в них значимо (р < 0.05) увеличивается только экспрессия мРНК белков Ерас1, тогда как уровень экспрессии мРНК белков Ерас2 не изменяется [1]. Аналогичные результаты были получены и при изучении экспрессии белков Ерас в миокарде человека. Биоптаты миокарда левого желудочка человека получали из изъятых при трансплантации сердец пациентов, страдающих IV классом хронической сердечной недостаточности (ХСН) по NYHA (средняя величина фракции выброса предтрансплантата левого желудочка 22 ± 4%), не получавших инотропной поддержки, по крайней мере, в течение 7 дней перед трансплантацией. У этих пациентов, по сравнению с не страдающими ХСН, экспрессия мРНК белков Ерас1 в 2 раза выше (р < 0.05), тогда как экспрессия мРНК белков Ерас2 не изменена [35] (рис. 6). Эти данные были подтверждены и при изучении вклада белков Ерас в развитие гипертрофии миокарда, вызванной радиотерапией у онкологических больных [36]. В выше упомянутом исследовании, выполненном на животных с коарктацией аорты, также отмечалось, что параллельно с экспрессией мРНК белка Ерас1 увеличивается и экспрессия мРНК малых GTPаз Rap1A и Rap2A [1]. В экспериментах in vitro, выполненных на культуре кардиомиоцитов мышей, полученных как у интактных, так и у перенесших за 5 дней до взятия клеток коарктацию грудного отдела аорты (гипертрофированные клетки) животных, показано, что у последних экспрессия белков Ерас1 значимо (р < 0.01) выше [35]. Внесение в культуру клеток интактных и оперированных животных агониста белков Ерас – 8-CPT инициировал рост как интактных, так и гипертрофированных кардиомиоцитов, однако рост кардиомиоцитов, полученных от мышей с коарктацией аорты, был значимо больше (р < 0.01). Эти результаты напрямую коррелируют с данными, полученными в ранее цитируемых исследованиях, однако, если в них сообщалось, что гипертрофия кардиомиоцитов реализуется посредством активации Ерас1/Rap сигнального каскада, то в последней работе инициированная белками Ерас1 гипертрофия кардиомиоцитов опосредуется малыми GTPазами Ras, поскольку ингибирование Ras блокировало гипертрофическое действие белков Ерас1, а ингибирование GTPаз Raр на него не влияло. Далее в этом исследовании было показано, что 8-CPT-опосредованная активация Ерас1/Ras сигнального прогипертрофического каскада инициирует последовательную активацию Ca2+-чувствительных белков CaN и CaMKII. Впоследствие эти же авторы подробно изучили вклад малой GTPазы Ras в Ерас1-опосредованную гипертрофию кардиомиоцитов [37]. В экспериментах in vitro, выполненных на культу-ре неонатальных кардиомиоцитов, отмечалось, что активация белков Ерас их агонистом или же инициация их экспрессии аденовирусом Ad.EpacWТ сопровождалась быстрой активацией GTP-азы H-Ras (син. преобразователь белка p21), которая, как известно, является одной из ключевых малых GTPаз, индуцирующих гипертрофию миокарда [38]. Аналогичные результаты были получены и при внесении в клеточную среду конститутивно активированной формы Epac1 – Ad.EpacΔcAMP. Степень активации H-Ras, иницированой селективными агонистами белков Ерас1, не отличалась от таковой, вызванной катехоламинами (норэпинефрин). Также было показано, что доминантная негативная форма GTPазы H‑Ras – RasS17N, внесенная в клеточную культуру, препятствовала реализации прогипертрофического действия Ad.EpacWТ, что также подтверждает данные о ключевой роли GTPазы H-Ras в Ерас-опосредованной гипертрофии кардиомиоцитов. На следующем этапе исследований было показано, что белки Ерас1 активируют GTPазы H-Ras посредством иx взаимодействия с С-концевым каталитическим доменом СDC25, тогда как белки Ерас, лишенные домена СDC25 – EpacΔCdc25, теряли способность активировать H-Ras. Поскольку известно, что нижестоящим эффектором белков Ерас1 является фосфолипаза С (PLC), инициирующая гидролиз расположенного на внутренней поверхности клеточной мембраны фосфолипида PIP2, сопровождающийся образованием инозитол 1,4,5-трисфосфата (IP3) и диацилглицерола (DAG), а последний активирует протеинкиназу С (PKC), которая способствует выходу в цитозоль кардиомиоцитов ионов Ca2+ из цистерн SR, активируя встроенные в его мембрану канальные IP3 рецепторы (IP3-R), был оценен вклад этого сигнального каскада в развитие гипертрофии кардиомиоцитов. Было показано, что прогипертрофическое действие белков Ерас1, опосредованное их активацией 8-CPT или инициацией их суперэкспрессии Ad.EpacWT, не реализуется в случае или блокады PLC ее фармакологическим ингибитором U73122, или ингибирования IP3-R блокатором Xestospongin C, или связывания ионов Ca2+ их внутриклеточным хелатором BAPTA-AM. На следующем этапе был продемонстрирован вклад Ca2+-чувствительных белков CaN и CaMKIIδ в прогипертрофическое действие белков Ерас1. Увеличение площади поверхности кардиомиоцитов, индуцированное аденовирусом, кодирующим конститутивно активированную форму белков Epac1 – Ad.EpacΔcAMP, было блокировано или фармакологическим ингибитором CaN – CsA, или фармакологическим ингибитором CaMKII – KN-93, что свидетельствует о том, что CaN и CaMKIIδ являются следующим звеном Ерас-опосредованного прогипертрофического сигнального каскада. На следующем этапе этого исследования продемонстрировали, что 8-CPT-опосредованная активация белков Ерас1 инициирует фосфорилирование цитозольной CaMKIIδ, которая активирует цитоплазматический фактор транскрипции NFAT (ядерный фактор активации Т-клеток, nuclear factor of activated T-cells), который, перемещаясь в ядро клетки, связывается с элементами ответа NFAT и модулирует экспрессию прогипертрофических генов. Авторы исследования полагают, что белки Ерас1, помимо цитозольной, могут активировать и ядерную CaMKIIδ, поскольку ими показано, что 8-CPT-опосредованная активация белков Ерас1 инициирует экспорт из ядра клетки деацетилазы гистонов 4 (нistone deacetylase, HDAC4), что сопровождается перемещением в цитозоль клетки стимулятора 1-ой стадии миогенеза – миоцит-специфического усиливающего фактора-2 (мyocyte-specific enhancer factor 2, MEF-2). Известно, что HDAC4 является репрессором MEF-2, образуя с ним неактивный комплекс HDAC4/MEF-2, стабильность которого находится под контролем ядерной CaMKIIδ. Позднее это предположение было подтверждено в экспериментах in vitro, выполненных на кардиомиоцитах, полученных от нокаутных по Ерас1 мышей [2]. В этих кардиомиоцитах, в отличие от полученных от интактных мышей, внесение в культуру клеток селективного агониста белков Ерас1 – 8-[Pharos-575]-2'-O-methyladenosine-3',5'-cyclic monophosphate (Φ-O-Me-cAMP) не сопровождалось ни фосфорилированием перинуклеарной CaMKIIδ, ни ядерным экспортом HDAC и, соответственно, активацией MEF-2. Также авторы этого исследования высказали предположение, что активация Ерас1-опосредованного сигнального каскада преимущественно регулирует активность перинуклеальной CaMKIIδ, тогда как активация сопряженного с белком Ерас2 сигнального каскада модулирует активность цитозольной CaMKIIδ, в своем большинстве расположенной вблизи Т-трубочек. В экспериментах in vitro, выполненных на культуре неонатальных кардиомиоцитов, при помощи иммуногистохимического окрашивания с использованием конфокальной микроскопии было показано, что внесение в клеточную культуру неселективного β-адреностимулятора изопротеренола вызывает быструю (1 мин) транслокацию активированной PKCε из цитозоля в перинуклеарное пространство [39]. Аналогичное действие оказывала и 8-CPT-опосредованная активация белков Ерас1. При этом количество PKCε в перинуклеарной зоне, по сравнению с контролем, увеличивалось на 168 ± 23% (р < 0.05). Активация PKCε инициирует экспрессию фосфорилированной внеклеточной сигнал-регулируемой киназы (extracellular signal-regulated kinase, рERK1/2; син. митоген-активированная протеинкиназа, MAPK) и последующую гипертрофию кардиомиоцитов, которая блокируется ингибитором белков Ерас – R279K. Также в этом исследовании были подтверждены ранее полученные данные [40] о том, что изопротеренол-опосредованная гипертрофия кардиомиоцитов не связана с активацией РКА, поскольку ее блокада ингибитором PKI не влияет на прогипертрофическое действие изопротеренола. В марте 2019 г. опубликованы результаты исследования, в котором изучена возможность использования оригинального селективного антагониста белков Ерас1 производного тиено[2,3-b]пиридин соединения AM-001 (3-амино-N-(4-фторфенил)-4-фенил-6-(тиофен-2-ил)тиено[2,3-b]пиридин-2-карбоксамид) для защиты сердца от катехоламинового стресса [41]. На первом этапе исследования в экспериментах in vitro, выполненных на культуре неонатальных кардиомиоцитов мыши, находящихся в условиях гипоксии/реоксигенации, отмечалось, что соединение AM-001, внесенное в клеточную культуру, блокировало вызванную активацией 8-CPT белков Ерас1 гипертрофию и гибель кардиомиоцитов. Далее в острых модельных экспериментах in vivo, воспроизводящих ишемию/реперфузию миокарда, было показано, что соединение AM-001, введенное в/в мышам за 5 мин до реперфузии, эффективно предотвращало реперфузионное повреждение ткани сердца – если у контрольных животных площадь инфаркта составила 50 ± 3% от площади левого желудочка, то у мышей, получавших соединение AM-001 – 36 ± 3% (p < 0.01). На последнем этапе исследования, также выполненного в модельных экспериментах in vivo, была изучена возможность использования соединения AM-001 для защиты сердца от патологического ремоделирования, вызванного длительной (14 дней) инфузией изопротеренола. Соединение AM-001 вводили в/в с 3-го по 14-ый день эксперимента. Систематическая экспериментальная терапия селективным блокатором белков Ерас1 соединением AM-001 эффективно препятствует ремоделированию миокарда: по сравнению с контролем у животных, получавших соединение AM-001, значимо меньше масса миокарда (p < 0.001), конечно-систолический и конечно-диастолический диаметр левого желудочка (p < 0.05), а фракция выброса (сократимость миокарда) значимо (p < 0.01) выше. Авторы этого исследования делают вывод о том, что селективная блокада белков Ерас1 в условиях катехоламинового стресса эффективно препятствует гипертрофии/ремоделированию миокарда.
Рис. 6.
Уровень экспрессии белков Ераc в сердце у здоровых людей и пациентов с хронической сердечной недостаточностью (по [35]). Объяснение в тексте.
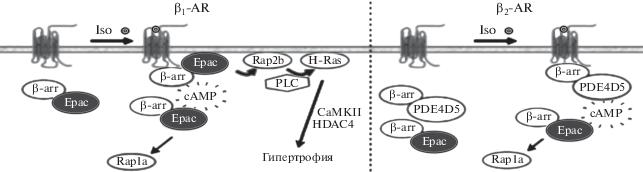
В экспериментах in vitro, выполненных на культуре неонатальных кардиомиоцитов мыши и культуре клеток HEK 293, которые, как известно, стабильно экспрессируют β1- и β2-адренорецепторы (β1-AR и β2-AR), изучали взаимосвязь между этими рецепторами и белками Ерас1 [42]. На первом этапе исследования было продемонстрировано, что в покоящейся клетке белки Ерас1 конститутивно взаимодействовали с β1-AR и β2-AR, а при их стимуляции изопротеренолом взаимодействие белков Ерас1 с β1-AR резко возрастало, тогда как с β2-AR практически не изменялось. Если в базальных условиях белки Ерас1 локализовались в цитоплазме клетки, то после стимуляции β1-AR они мигрировали к внутренней поверхности клеточной мембраны в область расположения β1-AR, стимуляция же β2-AR не вызывала транслокации белков Ерас1 из цитозоля к мембране. Вместе с тем, при совместной стимуляции β1-AR и β2-AR белки Ерас1 транслоцировались к обоим подтипам β-адренорецепторов. В результате β1-AR-опосредованной активации белков Ерас1 активируется Rap2В/H-Ras сигнальный каскад, инициирующий гипертрофию кардиомиоцитов, которая не развивается в случае замены GTPазы Rap2В на ее биологически инертную форму Rap2B–Si (Rap2B by SiRNA). Также было показано, что если в клетку, экспрессирующую β1-AR, трансфицировать белок Ерас1, то в ней активность GTPазы H-Ras возрастает в 2.5 раза, в случае же трансфицирования белка Ерас1 в клетку, экспрессирующую β2-AR, активность H-Ras не изменяется. Затем с использованием метода резонансного переноса энергии в биолюминесцентных системах (BRET) in vitro было продемонстрировано, что для транслокации белков Ерас1 из цитозоля к β1-AR и последующей активации Rap2В/H-Ras прогипертрофического сигнального каскада необходимо взаимодействие белка Ерас1 с регуляторным белком β-аррестином 2 (β-arr2), который контролирует активность сопряженных с G-белками трансмембранных рецепторов посредством регуляции связи с их цитоплазматической петлей (рис. 7). Тем же методом было показано, что в случае активации сопряженного с β2-AR Ерас1/Rap1А сигнального каскада не происходит взаимодействия белков Ерас1 с β-arr2, поскольку оно блокируется специфической для сАМР 3',5-цикло-фосфодиэстеразой 4В (cAMP-specific 3',5'-cyclic phosphodiesterase 4B, PDE4D5) (рис. 7).
Рис. 7.
Схематическое отображение Ерас-опосредованных сигнальных каскадов, инициируемых стимуляцией β1- и β2-адренорецепторами (по [42]). Объяснение в тексте.
Таким образом, имеющиеся в литературе данные свидетельствуют о том, что избыточная активация/экспрессия белков Ерас1, опосредованная β1-адреностимуляцией, что клинически соответствует ситуации, наблюдаемой у пациентов, страдающих хронической сердечной недостаточностью различного генеза, приводит к развитию гипертрофии/ремоделирования миокарда. В основе этого патологического процесса лежит активация сопряженного с белками Ерас1 прогипертрофического сигнального каскада: Ерас1/Rap2/H-Ras/ PLCϵ/PIP2/DAG/PKCϵ/IP3/Ca2+/CaN/CaMKIIδ/ HDAC4/MEF-2 (рис. 8). Свое прогипертрофическое действие белки Ерас1 реализуют не зависимо от РКА.
Рис. 8.
Схематическое отображение сопряженных с белками Ерас прогипертрофических сигнальных каскадов (по [73]). Объяснение в тексте.
Следует отметить, что в литературе представлены результаты исследования, в котором приводятся данные о том, что активация белков Ерас1 в условиях септического шока препятствует развитию сердечной недостаточности [43]. Этот кардиопротективный эффект реализуется за счет способности белков Ерас1 активировать супрессор цитокинов 3 (suppressors of cytokine signaling 3, SOCS3), что в свою очередь блокирует интерлейкин 6 (IL-6)-опосредованный Jak/STAT провоспалительный сигнальный каскад и последующую гиперэкспрессию индуцибильной NO синтазы (iNOS).
Роль белков Ерас в развитии фиброза миокарда
Известно, что сердечные фибробласты (CFs), входящие в состав внеклеточного матрикса, составляют две трети от общей популяции сердечных клеток и формируют “каркас” для кардиомиоцитов [44]. В условиях патологии (инфаркт миокарда, сердечная недостаточность, кардиомопатия и т.д.) фибробласты трансформируются в миофибробласты, которые активируются различными факторами роста, цитокинами, а также механическими стимулами и инициируют выработку коллагенов, преимущественно I и III типов, и последующее замещение сократительных кардиомиоцитов соединительной тканью, т.е. развитие фиброза миокарда. В литературе имеются данные о том, что активация сАМР/РКА сигнального каскада в CFs сопровождается подавлением синтеза коллагена. Так, например, активация сАМР форсколином или активация РКА ее агонистом – 8-хлор-3',5'-циклическим аденозинмонофосфатом (8-ClcAMР, син. CPT-cAMP) в клеточной культуре сердечных фибробластов, выделенных из миокарда взрослых крыс, сопровождаются значительным подавлением (р < 0.05) активированного или ангиотензином II (Ang II), или трансформирующим фактором роста-β (transforming growth factor-β, TGF-β) синтеза коллагена [45]. В настоящее время вклад белков Ерас в развитие фиброза миокарда остается не ясным, поскольку согласно литературным данным они могут проявлять как про-, так и антифиброзную активность. Впервые об экспрессии белков Ерас1, но не белков Ерас2, в CFs было сообщено в исследовании U. Yokoyama et al. [46]. В этом же исследовании в экспериментах in vitro, выполненных на культуре полученных от взрослых интактных крыс CFs, показано, что 8-CPT-опосредованная активация Ерас1/Rap1 сигнального каскада инициирует миграцию фибробластов, тогда как активация РКА ее агонистом N6-phenyl cAMP (Phe-cAMP) блокирует их миграцию. Далее в экспериментах in vitro, выполненных на культуре CFs, полученных из периинфарктной зоны миокарда мышей через одну неделю после воспроизведения инфаркта, отмечалось снижение экспрессии белков Epac1 с параллельным увеличением экспрессии профибротического TGF-β1. Авторы исследования полагают, что функциональное снижение экспрессии белков Ерас1 в условиях инфаркта миокарда стимулирует/облегчает процесс замещения соединительной тканью некротизированного участка сердечной мышцы. Несколько позже те же авторы высказали предположение, что снижение экспрессии белков Ерас1, инициированное профибротическими агентами (TGF-β, Ang II и др.) может способствовать развитию фиброза миокарда [47]. В экспериментах in vitro, выполненных на культуре CFs, полученных от неонатальных крыс, мышей с инфарктом миокарда через 21 день после перевязки коронарной артерии и CFs, полученных у пациентов с инфарктом миокарда, оценивали вклад белков Ерас1 в регуляцию фиброгенеза [48]. На первом этапе исследования было выявлено, что ключевым белком, инициирующим фиброгенез в ишемированных сердцах, является кальций/кальмодулин-зависимая фосфодиэстераза 1A 3',5'-циклических нуклеотидов (сalcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A, PDE1A), поскольку ее экспрессия значительно увеличивалась в биоптатах ткани, взятых из периинфарктной области (по сравнению с уровнем экспрессии PDE1A в интактных фибробластах), а ее блокада селективным ингибитором IC86340 препятствовала индуцированной Ang II экспрессии таких индукторов фиброгенеза как коллаген 1, фибронектин (Fn) и ингибитор активатора плазминогена (PAI-I). Далее было показано, что PDE1A реализует свои профибротические эффекты посредством активации cAMP/ Epac1/Rap1 сигнального каскада. На основании полученных данных авторы пришли к заключению, что они “выявили ранее неописанный сигнальный механизм PDE1A-cAMP-Epac1-Rap1, лежащий в основе активации миофибробластов при патологическом ремоделировании сердца”. В экспериментах in vitro, выполненных на культуре CFs, полученных от неонатальных мышей (NMCFs), изучали роль индуцированной β-адреностимуляцией экспрессии в фибробластах профиброгенного интерлейкина 6 (IL-6) [49]. На первом этапе было продемонстрировано, что изопротеренол-опосредованная стимуляция β-адренорецепторов инициирует активацию δ-изоформы РКС (PKCδ) с последующей инициацией экспрессии IL-6. Далее отмечалось, что вызванная PKCδ экспрессия IL-6 реализуется за счет активации Epac1/Rap1 сигнального каскада, поскольку нокдаун белков Epac1 подавлял изопротеренол-опосредованную экспрессию IL-6.
Активация сАМР/Epac1/Rap1/PKCδ сигнального каскада завершается фосфорилированием P38 МАР-киназы (P38MAPK), играющей ведущую роль в активации IL-6. Также отмечалось, что профиброгенное действие белков Ерас1 реализуется не зависимо от РКА. При изучении уровня экспрессии белков Ерас1 в биоптатах ткани миокарда взрослых крыс, полученных на 14-ый день после воспроизведения ишемии/реперфузии, показано, что систематическая экспериментальная терапия антагонистом белков Ерас1 витексином подавляет интенсивность профибротических процессов в ишемизированных тканях сердца [50]. Аналогичные результаты были получены и при использовании селективного блокатора белков Ерас1 – AM-001 в модельных экспериментах in vivo, воспроизводящих длительный катехоламиновый стресс [41].
Таким образом, в настоящее время имеются достаточно убедительные данные о том, что в условиях ишемического повреждения сердца активация PDE1A/cAMP/Epac1/Rap/PKCδ/P38MAPK сигнального каскада способствует развитию фиброза миокарда. Однако в литературе представлена и другая точка зрения. Так, в экспериментах in vitro, выполненных на культуре кардиомиобластов (h9c2), полученных из сердец взрослых крыс, показано, что инициированная альдостероном экспрессия белка остеопонтина (OPN), который, как известно, необходим для реализации в миокарде профибротических эффектов Ang II и TGFβ1 [51], в свою очередь, подавляет экспрессию белка Ерас1 [52], тогда как внесение в культуру миофибробластов, лишенных OPN (OPN-KN knout (KO cells), β2-адреностимулятора сальбутомола восстанавливала экспрессию белков Ерас1. Авторы этого исследования пришли к заключению “о неблагоприятной роли OPN в сдерживании β2-AR-зависимой, cAMP/Epac1-опосредованной антифиброзной сигнализации”. Вместе с тем, они никак не обсуждают ранее полученные данные о том, что PDE1A/cAMP/Epac1 сигнальный каскад инициирует экспрессию профибротических цитокинов. В экспериментах in vitro, выполненных на культуре фибробластов, полученных из сердец взрослых крыс, показано, что α-адреностилулятор норэпинефрин (NE) значимо (р < 0.05) подавляет экспрессию белков Ерас1 при одновременном увеличении (в 4.5 раза) экспрессии коллагена I типа [53]. Внесение в культуру клеток селективного антагониста α1-адренорецепторов (α1-AR) празозина предотвращало NE-индуцированное снижение экспрессии Epac1. Затем было выявлено, что неселективный агонист β-адренорецепторов (β-АR) изопротеренол значимо (р < 0.01) увеличивал экспрессию белков Epac1 и уменьшал секрецию коллагена I. Этот эффект изопротеренола был блокирован селективным антагонистом β2-AR – ICI-118551, но не селективным антагонистом β1-AR – CGP-20712A. Блокада белков Ерас1 их антагонистом ESI-09 препятствовала подавлению экспрессии коллагена I, инициированной селективным агонистом β2-AR сальбутамолом. Авторы этого исследования полагают, что адренергическая регуляция Ерас1-опосредованного контроля фиброгенеза разнонаправленна и, с одной стороны, посредством активации α1-AR подавляет экспрессию белков Ерас1, и тем самым стимулирует синтез коллагена, а с другой стороны, путем активации β2-AR стимулирует экспрессию белков Ерас1 и последующую активацию антифибротических сигнальных каскадов.
Роль белков Ерас в генезе острого ишемического повреждения миокарда
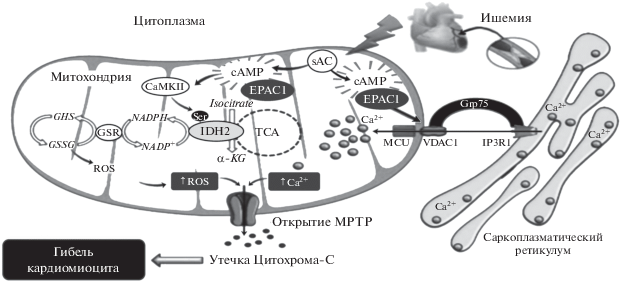
Информация о роли белков Ерас в генезе острого ишемического повреждения миокарда, несмотря на всю важность этой проблемы, крайне незначительна. В модельных экспериментах in vivo было выявлено, что в биоптатах миокарда мышей с острым ишемическим повреждением сердца (45 мин ишемии + 24 ч реперфузии) значимо (р < 0.01), по сравнению с интактными животными, увеличена экспрессия белков Ерас1, что напрямую коррелирует с результатами, полученными при изучении экспрессии белков Ерас1 в биоптатах ишемизированного миокарда человека [54]. В модельных экспериментах in vivo, воспроизводящих острый инфаркт миокарда у нокаутных по белку Ерас1 мышей, показано, что у них, по сравнению с интактными животными, значимо (р < 0.05) меньше площадь ишемического повреждения – соответственно 33 ± 4 и 53 ± 4% [54]. Далее в модельных экспериментах in vitro, выполненных на кардиомиоцитах, полученных от нокаутных по белку Ерас1 мышей, показано, что у них, в отличие от интактных животных, 4-часовая гипоксия и последующая 2-часовая реоксигенация (т.е. воспроизведение условий ишемия/реперфузия) не сопровождались повреждением клеточной мембраны и снижением продукции АТР. Внесение в модельную культуру кардиомиоцитов, полученных от нокаутных по белку Ерас1 мышей, его агониста 8-CPT не инициировало повреждение клеток, также как и предварительное внесение в модельную культуру интактных мышей селективного ингибитора белков Ерас1 – CE3F4 защищало клетки от ишемического повреждения. Далее методом иммуноблоттинга было выявлено, что белки Ерас1 локализуются не только в цитозоле кардиомиоцитов, но и в области внутренней мембраны и матрикса митохондрий. Для того, чтобы оценить вклад митохондриальной изоформы белка Ерас1 (MitEpac1), была сконструирована мутантная форма белка с удаленной митохондриальной последовательностью – Epac1Δ2-37. Поскольку мутант Epac1Δ2-37, также как и нативный белок Ерас1, активировал эффектор Rap1, то можно было говорить о том, что потеря митохондриальной последовательности не влияет на цитозольные эффекты белка Ерас1. Затем было показано, что интактные кардиомиоциты с трансфицированным белком Epac1Δ2-37, в отличие от интактных кардиомиоцитов, содержащих немодифицированный белок Ерас1, не были повреждены в условиях модели ишемия/реперфузия [54]. При изучении механизмов, лежащих в основе кардиотоксического действия MitEpac1, отмечалось, что MitEpac1 инициирует перегрузку митохондрий ионами Ca2+ за счет активации комплекса, состоящего из потенциалчувствительного анионного канала 1 (VDAC1), глюкозо-регулируемого шаперона 75 (GRP 75) и рецептора инозитол-1,3,5-трифосфата (IP3R1). Известно, что комплекс VDAC1/GRP75/IP3R1 сконцентрирован на митохондрии-связанных участках мембраны SR (mitochondria-associated SR membranes, MAMs), где он регулирует обмен ионов Ca2+ между SR и митохондриями. Также было продемонстрировано, что активация комплекса VDAC1/GRP75/IP3R1 и последующее за этим открытие поры, изменяющей проницаемость мембраны митохондрий (mPTP), не сопровождается активацией RyR2, что позволяет говорить о том, что вклад цитозольной изоформы белка Ерас1 в обусловленую ишемией гибель кардиомиоцитов незначителен. Поскольку в ишемия/реперфузия-обусловленном повреждении кардиомиоцитов помимо ионов Ca2+ значительная роль принадлежит ROS, было изучено влияние белков Ерас1 на их продукцию в ишемизированных кардиомиоцитах [54]. В модельных экспериментах in vitro, воспроизводящих условия ишемия/реперфузия, выполненных на кардиомиоцитах, полученных от нокаутных по белку Ерас1 мышей, показано, что у них, в отличие от интактных животных, образование ROS значимо (р < 0.05) снижено. Аналогичные данные были получены при 8-CPT-опосредованнной активации белков Ерас1. Известно, что одним из ключевых белков, защищающих кардиомиоциты от избыточной продукции ROS, является локализованный в матриксе митохондрий NADP-зависимый фермент изоцитратдегидрогеназа 2 (isocitrate dehydrogenase 2, IDH2), катализирующий окислительное декарбоксилирование изоцитрата в α-кетоглутарат (α-KG) и последующую продукцию NADPH, необходимого для регенерации двух митохондриальных антиоксидантов, защищающих мтДНК от окислительного стресса: митохондриального глутатиона (mitochondrial glutathion, GSH) и тиредоксина. При помощи метода ко-иммунопреципитации показано, что в интактных кардиомиоцитах 8-CPT-опосредованнная активация белков Ерас1 увеличивает серин-специфическое фосфорилирование IDH2, вследствие чего снижается продукция NADPH, тогда как в кардиомиоцитах с трансфицированным белком Epac1Δ2-37 (белки Ерас1 с удаленным митохондриальным доменом) 8-CPT не вызывал фосфорилирования IDH2, что дает возможность предполагать, что именно белки MitEpac1 инициируют ROS-обусловленное повреждение клеток. Также с использованием метода ко-иммунопреципитации было выявлено, что в митохондриях белки MitEpac1 находятся в составе макромолекулярного комплекса, включающего в себя помимо белка MitEpac1 CaMKIIδ и IDH2. Далее в экспериментах, выполненных на изолированных интактных митохондриях, отмечено, что блокада CaMKIIδ селективным ингибитором KN93 препятствует MitEpac1-опосредованному фосфорилированию IDH2, что свидетельствует о том, что CaMKIIδ является связующим звеном между MitEpac1 и IDH2.
Таким образом, в настоящее время представлено, что в условиях острой ишемии миокарда активация белка MitEpac1 сопровождается гибелью кардиомиоцитов, которая в условиях целостного организма манифестируется увеличением зоны инфаркта. В основе этого патологического процесса лежит как MitEpac1-опосредованная активация VDAC1/GRP75/IP3R1 сигнального каскада, приводящего к открытию митохондриальной поры – mPTP и последующей фатальной пере-грузке митохондрий ионами Ca2+, так и MitEpac1-опосредованное фосфорилирование IDH2, сопровождающееся гиперпродукцией ROS (рис. 9).
Рис. 9.
Схематическое отображение роли митохондриальных белков Epac в остром ишемическом повреждении миокарда (по [54]). Объяснение в тексте.
Роль белков Ерас в развитии нарушений сердечного ритма
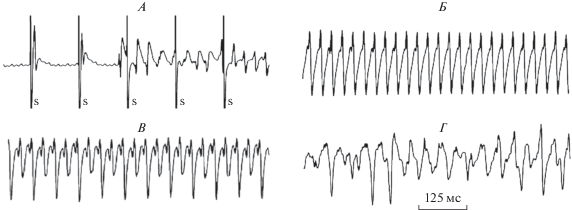
Впервые о наличии у белков Ерас аритмогенного потенциала сообщил в 2008 г. S.S. Hothi et al. [55]. В экспериментах in vitro, выполненных на спонтанно сокращающемся изолированном по методу Лангендорфа сердце мыши, перфузируемом раствором Кребса-Хензелайта, было описано, что агонист белков Ерас 8-CPT не влиял на частоту сердечных сокращений, но в ряде случаев инициировал триггерную активность. Тригерная активность возникала как во время фазы реполяризации (afterdepolarisations, EADs), так и после ее завершения. В аналогичных экспериментах, выполненных с навязыванием сердечного ритма (стационарная стимуляция), показано, что агонист белков Ерас – 8-CPT, внесенный в перфузат, изначально инициировал спонтанный эктопический ритм (рис. 10, А), который сменялся одно- (рис. 10, Б) или двухнаправленной (рис. 10, В) желудочковой тахикардией (ЖТ), переходящей в фибрилляцию желудочков (рис. 10, Г). Далее с использованием метода программированной электрической стимуляции (PES), который используется в клинике для изучения характера и электрофизиологических механизмов нарушений ритма сердца, было показано, что, если в контрольных экспериментах (n = 20) PES ни в одном случае не вызывала пароксизмов ЖТ, то в опытах, выполненных на тех же сердцах, на фоне 8-CPT, предварительно внесенного в перфузат, во всех случаях (n = 20) развивалась ЖТ. В экспериментах с использованием селективного ингибитора РКА – H-89 было продемонстрировано, что аритмогенное действие белков Ерас не зависит от РКА.
Рис. 10.
Нарушения ритма сердца, в условиях его программной стимуляции, инициированные белками Ерас (по [55]). Объяснение в тексте.
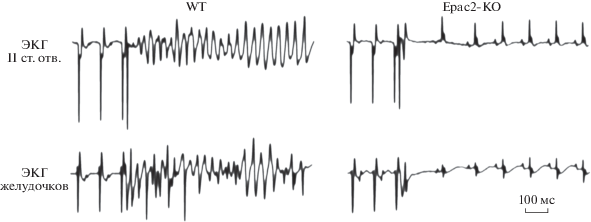
Известно, что аритмогенные синдромы (синдром удлиненного интервала QT [LQT3 и LQT5], синдром Бругада и др.) как в клинике, так и воспроизведенные в эксперименте, манифестируются электрофизиологическими предикторами. Однако в случае белков Ерас не было выявлено каких-либо электрофизиологических предвестников инициируемых ими нарушений сердечного ритма. Так, например, по сравнению с контролем на фоне 8-CPT-опосредованной активации белков Ерас ни в эндокарде, ни в эпикарде не изменялась продолжительность потенциала действия: APD90, соответственно 51.8 ± 2.2 и 51.9 ± ± 2.2 мс (р > 0.05; n = 10) и соответственно 40.4 ± 1.2 и 44.0 ± 1.7 мс (р > 0.05; n = 10). Помимо этого, не было показано отличий ни в трансмуральном, ни в апико-базальном градиенте реполяризации, ни в продолжительности эффективного рефрактерного периода желудочков. На основании полученных результатов авторы этого исследования пришли к заключению, что Ерас-опосредованные нарушения сердечного ритма не манифестируются электрофизиологическими предикторами. Далее было показано, что как в условиях спонтанно сокращающегося изолированного сердца, так и в условиях PES, на фоне блокады CaMKII ее селективным ингибитором KN-93 активация белков Ерас или неселективным β-адреностимулятором изопротеренолом, или их агонистом 8-CPT не инициировала развитие нарушений сердечного ритма, что позволяет говорить о том, что аритмогенное действие белков Ерас реализуется посредством активации CaMKII. В экспериментах in vitro, выполненных на культуре кардиомиоцитов, полученных из левого желудочка сердца мыши, с помощью конфокальной лазерной микроскопии оценивали влияние белков Ерас на динамику перемещения ионов Ca2+ в системе SR/цитозоль клетки [55]. Как в покоящихся, так и сокращающихся стимулированных кардиомиоцитах 8-CPT-опосредованная активация белков Ерас инициировала аномальную аритмогенную диастолическую “утечку” из SR ионов Ca2+ (р < 0.001), которая полностью блокировалась селективным ингибитором CaMKII – KN-93. В экспериментах in vivo и in vitro оценивали вклад изоформ белков Ерас в формирование аритмогенеза [56]. Животные (мыши) были разделены на 4 группы: интактные, нокаутные по белку Ерас1ko (Ерас1ko), нокаутные по белку Ерас2ko (Ерас2ko) и нокаутные по обоим изоформам белков (Ерас Dko). На первом этапе исследования между группами животных не было выявлено различий ни по функциональным, ни по электрофизиологическим характеристикам миокарда. Далее на культуре кардиомиоцитов, полученных из левого желудочка, было показано, что 8-CPT-опосредованная активация белков Ерас в кардиомиоцитах, полученных от интактных и Ерас1ko мышей вызывала значимую (р < 0.01) диастолическую, не зависимую от РКА, “утечку” ионов Ca2+ из цистерн SR (CaSpF), которая отсутствовала в кардиомиоцитах, полученных от Ерас2ko и Ерас Dko мышей. Затем было показано, что, в отличие от интактных животных, программированная электрическая стимуляция левого желудочка сердца у мышей Ерас2ko не сопровождалась развитием желудочковой тахикардии и/или фибрилляции желудочков (р < 0.05) (рис. 11). На основании полученных данных было сделано заключение о том, что аритмогенные эффекты белков Ерас обусловлены активацией сопряженных с белками Ерас2, но не Ерас1, сигнальных каскадов. В экспериментах in vitro, выполненных на интактных кардиомиоцитах, было отмечено, что аритмогенные эффекты белков Ерас2 реализуются посредством активации CaM-KIIδ, которая в свою очередь фосфорилирует RyR2 по Ser2814, поскольку блокада CaMKIIδ селективным ингибитором KN-93 предотвращала 8-CPT опосредованную CaSpF. Также на интактных кардиомиоцитах было показано, что блокада β1-адренорецепторов их селективным антагонистом CGP-20712A, но не блокада β2-адренорецепторов их селективным антагонистом ICI-118.551, ингибировала 8-CPT- или изопротеренол-опосредованную CaSpF, что свидетельствует о том, что Ерас2-опосредованный аритмогенез сопряжен с активацией β1-адренорецепторов. Близкие данные были получены и в более поздних исследованиях [57–59]. В экспериментах in vitro, выполненных на культуре кардиомиоцитов ARVMs, полученных из желудочков взрослых крыс, показано, что изопротеренол-опосредованная CaSpF подавляется селективным ингибитором белков Ерас2 – ESI-05, но не селективным ингибитором белков Ерас1 – CE3F4 [60]. В экспериментах in vitro, выполненных на выделенных из желудочков сердец мышей и кроликов интактных кардиомиоцитах, были подробно изучены компоненты сигнального пути, инициирующие Ерас2-опосредованную CaSpF [61]. На первом этапе исследования было продемонстрировано, что неселективный β-адреностимулятор изопротеренол более чем в 2 раза увеличивал CaSpF (с 0.22 ± 0.048 до 0.46 ± 0.12). Ингибитор нейрональной NO-синтазы (nNOS, NOS1) – L-NPA, внесенный в культуру клеток, не влиял на базальную “утечку” ионов Ca2+ из SR, но полностью блокировал CaSpF, инициированную как изопротеренолом, так и белком Ерас2. Изопротеренол- или Ерас2-опросредованная активация CaMKII/ RyR2 и последующая CaSpF также блокируется L-NPA. Помимо этого было показано, что блокада РКА селективным ингибитором H-89 не оказывала существенного влияния на изопротеренол-стимулированную активность CaMKII. Поскольку в литературе имеются данные о том, что изопротеренол/NOS1-опосредованная активация CaMKII реализуется посредством активации PI3K/Akt сигнального каскада [62], на следующем этапе исследования оценили возможный вклад этого сигнального каскада в Ерас2-опосредованный аритмогенез. Блокада PI3K селективным ингибитором LY924002 препятствовала Ерас2-опосредованой CaSpF.
Рис. 11.
Влияние программной стимуляции на ритмическую активность сердца у интактных и нокаутных по белку Ерас2 мышей (по [56]). Объяснение в тексте.
Таким образом, к настоящему времени накоплены достаточно убедительные данные о том, что Ерас2-опосредованный аритмогенез связан с инициацией аномальной диастолической утечки ионов Ca2+ из цистерн SR, которая реализуется посредством активации β1-AR/cAMP/Epac2/ PI3K/Akt/NOS1/CaMKIIδ/RyR2 сигнального каскада. Вместе с тем, имеются данные о том, что белки Ерас могут провоцировать развитие нарушений сердечного ритма и посредством других механизмов.
Известно, что активация (возбуждение) встроенных в клеточную мембрану кардиомиоцитов быстрых селективных потенциалзависимых Na+-каналов сопровождается “лавинообразным” кратковременным входящим током ионов Na+ (INaТ), генерирующим фазу 0 (фазу деполяризации) потенциала действия. После того как заряд внутренней поверхности клеточной мембраны достигнет уровня +20 – +30 мВ, Na+-каналы закрываются, что блокирует INaТ. Однако в кардиомиоцитах желудочков в следующую за фазой 0 фазу реполяризации незначительная часть Na+-каналов остается открытыми, генерируя не инактивирующий или стойкий входящий ток ионов Na+, который носит название поздний Na+ ток – INaL [63]. Известно, что патологическое увеличение INaL связано с повышенным риском развития злокачественных нарушений сердечного ритма [64]. В экспериментах in vitro, выполненных на культуре кардиомиоцитов, полученных из желудочков сердца мышей, показано, что изопротеренол- или 8-CPT-опосредованная активация белков Ерас сопровождается последующей активацией CaMKIIδ, которая фосфорилирует NaV1.5-каналы, расположенные в области T-трубочек, что влечет за собой увеличение INaL [65]. Также было показано, что CaMKIIδ-опосредованное увеличение INaL не зависит от РКА, поскольку ее ингибрование PKI не влияло на способность CaMKIIδ фосфорилировать NaV1.5-канал. В экспериментах in vitro, выполненных на культуре кардиомиоцитов, полученных из желудочков сердца кролика, было показано, что 8-CPT-опосредованная активация белков Ерас имитирует эффекты β1-адреностимуляции, увеличивая INaL в позднюю фазу плато и 3-ю фазу реполяризации потенциала действия [66]. Этот эффект белков Ерас реализуется посредством активации CaMKIIδ вне зависимости от РКА, которая также может увеличивать INaL по CaMKII-несопряженным сигнальным путям. Авторы обоих исследований полагают, что Ерас-опосредованное увеличение INaL реализуется по β1-AR/сАМР/Ерас2/CaMKII сигнальному пути.
В экспериментах in vitro, выполненных на полученных из предсердий и желудочков сердца мыши изолированных кардиомиоцитах, Ерас-опосредованная активация RyR2 рецепторов и последующая за этим диастолическая утечка ионов Са2+ влечет за собой угнетение функциональной активности быстрых трансмембранных потенциалзависимых Na+-каналов, что приводит к подавлению INaТ [67]. В интактных кардиомиоцитах предсердий и желудочков INaТ составляет соответственно –20.23 ± 1.48 pA/μм и –29.8 ± ± 2.4 pA/μм, тогда как после внесения в среду агониста белков Ерас – 8-CPT он соответственно уменьшался до –11.21 ± 0.91 pA/μм и –19.3 ± ± 1.6 pA/μм (р < 0.006 по отношению к интактным). Этот эффект был полностью блокирован предварительным внесением в среду антагониста RyR2 рецепторов дантролена. В этом же исследовании продемонстрировано, что уменьшение INaТ сопровождается замедлением (как в предсердиях, так и в желудочках) распространения потенциала действия, с чем авторы связывают проаритмические эффекты белков Ерас.
Недавно были опубликованы результаты исследования, из которых следует, что в условиях сердечной недостаточности (СН), индуцированной у мышей изопротеренолом (30 мг/кг/сут, 21 сут), в предсердиях резко увеличивается плотность медленных потенциалзависимых Са2+-каналов L типа, что сопровождается статистически значимым (p < 0.05) увеличением входящего ICaL [68]. Программированная электрическая стимуляция сердца у животных с СН в 80% случаев приводила к развитию фибрилляции предсердий (ФП), тогда как у контрольных животных она развивалась лишь в 12.5% случаев (p < 0.05). Далее методом полимеразной цепной реакции (ПЦР) было показано, что у животных с СН, по сравнению с интактными, в кардиомиоцитах предсердий значимо (p < 0.05), более чем в 2 раза, увеличена экспрессия белков Ерас1. Блокада белков Ерас1 селективным антагонистом CE3F4 у животных СН сопровождалась как значимым снижением ICaL, так и уменьшением количества случаев индуцированной электрической стимуляцией ФП – соответственно 80 и 47% случаев (p < 0.05). Авторы этого исследования приходят к выводу, что в условиях СН вклад белков Ерас1 в развитие ФП связан с их способностью активировать входящий ICaL, протекающий через клеточную мембрану кардиомиоцитов предсердий.
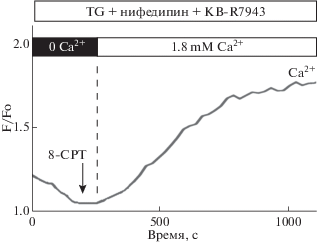
Помимо потенциалзависимых Na+, К+ и Са2+ ионных каналов в клеточную мембрану и мембрану T-трубочек встроены так называемые канонические каналы с транзиторным рецепторным потенциалом (transient receptor potential cation channel subfamily C, TRPC). TRPC относятся к неселективным катионовым ионным каналам, регулирующим движение ионов Na+ и Са2+ (преимущественно Са2+) через плазматические мембраны. В кардиомиоцитах в основном представлены TRPC 3-го и 4-го типов, принимающие участие в регуляции их сократимости и возбудимости. Их избыточная активация и/или гиперэкспресия инициирует развитие гипертрофии миокарда и нарушения сердечного ритма [69, 70]. TRPC-опосредованная гипертрофия кардиомиоцитов связана с активацией PLC/DAG/CaN/NFAT сигнального каскада. Активированный NFAT экспортируется в ядро клетки, где инициирует транскрипцию генов TRPC. Проаритмическое действие активированных TRPC связывают с перегрузкой кардиомиоцитов ионами Са2+. В экспериментах in vitro, выполненных на культуре кардиомиоцитов, полученных из желудочков взрослых крыс, изучили вклад TRPC3 и TRPC4 в аритмогенный потенциал белков Ерас [71]. На первом этапе исследования в цитозоле кардиомиоцитов истощили запасы ионов Са2+ посредством блокады Са2+-зависимой АТРазы (SERCA) селективным ингибитором тапсигаргином (TG) и блокады трансмембранных потенциалзависимых Са2+-каналов L-типа нифедипином, а также блокады Na+/Cа2+ обменника его селективным антагонистом TB–R7943. Далее было продемонстрировано, что инкубация этих кардиомиоцитов в течение 4–6 ч в среде, содержащей ионы Са2+, приводила лишь к незначительному увеличению содержания ионов Са2+ в цитозоле клетки, тогда как добавление в среду агониста белков Ерас 8-CPT вызывала резкое возрастание концентрации ионов Са2+ (рис. 12). Этот эффект 8-CPT был заблокирован неспецифическим антагонистом TRPC3 и TRPC4 – SKF-96365. Такое же действие оказывала и блокада белков Ерас1 их селективным антагонистом CE3F4 или блокада белков Ерас2 ESI-05. Далее было показано, что 8-CPT-опосредованный рост концентрации ионов Са2+ в цитоплазме не сопровождался увеличением экспрессии генов TRPC3 и TRPC4, что свидетельствует о том, что белки Ерас активируют TRPC за счет посттранскрипционных механизмов. На основании полученных данных авторы этого исследования пришли к заключению о том, что обе изоформы белков Ерас принимают участие в аритмогенной активации TRPC3 и TRPC4.
Рис. 12.
Влияние белков Ерас на вход ионов Ca2+ в цитозоль кардиомиоцитов через трансмебранные TRPC3/4 ионные каналы (по [71]). Объяснение в тексте.
Помимо этого, ряд исследователей полагает, что определенный вклад в проаритмическое действие белков Ерас может вносить инициируемая ими в условиях патологии избыточная экспрессия коннексинов Сх43 в области щелевых контактов (GJ) кардиомиоцитов [9, 14, 16], а также Ерас-обусловленная аномальная активность трансмембранных потенциалзависимых К+-каналов задержанного выпрямления IKs, кодируемых геном KCNQ1 [72, 73], в частности посредством подавления медленного компонента калиевого тока замедленного выпрямления (IKs), принимающего участие в формировании 3-ей фазы реполяризации потенциала действия [74].
Таким образом, накопленные к настоящему времени данные свидетельствуют о том, что избыточная активация β1-AR, в свою очередь, инициирует активацию cAMP/Epac2/PI3K/Akt/ NOS1/CaMKIIδ/RyR2 сигнального каскада, приводящую к аритмогенной диастолической “утечке” ионов Са2+ из цистерн саркоплазматического ретикулума в цитозоль кардиомиоцитов и/или сАМР/Ерас2/CaMKII сигнального пути, инициирующего аритмогенное аномальное увеличение интенсивности позднего Na+ тока (INaL), протекающего через клеточную мембрану кардиомиоцитов желудочков. Также аритмогенной перегрузкой кардиомиоцитов ионами Са2+ сопровождается и Ерас-обусловленная активация TRPC3 и TRPC4 каналов (рис. 13). Помимо этого, возможно, что определенный вклад в аритмогенное действие белков Ерас вносит их способность вызывать избыточную экспрессию коннексинов Сх43 в области щелевых контактов (GJ) кардиомиоцитов и инициировать аномальную активность К+-каналов, встроенных в клеточную мембрану кардиомиоцитов (рис. 13).
Рис. 13.
Схематическое отображение сопряженных с белками Ерас проаритмических сигнальных каскадов (по [73]). Объяснение в тексте.
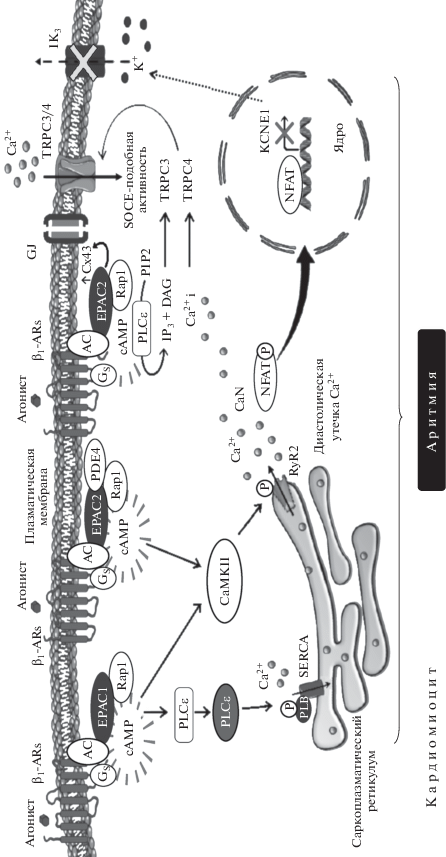
Следует отметить, что в доступной литературе представлено только исследование, в котором приводятся данные о том, что белки Ерас2 проявляют антиаритмическую активность [75]. В опубликованном в 2017 г. исследовании, выполненном на изолированных интактных кардиомиоцитах (ARVMs), выделенных из желудочков взрослых крыс, показано, что ингибирование белков Ерас2 селективным агонистом ESI-05 на 50% понижало содержание GTPазы Rap1 и увеличивало базальную продукцию митохондриями ROS [75]. Авторы этого исследования пришли к заключению, что cопутствующая β1-адренергической стимуляции активация Ерас2/Rap1 сигнального каскада обеспечивает отрицательную обратную связь, которая ослабляет митохондриальную генерацию ROS. Когда этот механизм остро прерван ингибированием Epac2, увеличенная продукция ROS генерирует аритмии. Вместе с тем, авторы никак не комментируют ранее опубликованные результаты, свидетельствующие, с одной стороны, об аритмогенной активности белков Ерас2, а с другой стороны, данные о том, что MitEpac1-опосредованная активация VDAC1/ GRP75/IP3R1 сигнального каскада приводит к фатальной перегрузке митохондрий ионами Ca2+, а MitEpac1-опосредованное фосфорилирование IDH2 сопровождается гиперпродукцией ROS [54]. Также следует отметить, что на современном этапе далеко не ясен вопрос, насколько значителен вклад ROS в генерацию нарушений сердечного ритма.
ЗАКЛЮЧЕНИЕ
Прошло 20 лет с момента открытия нового эффектора сАМР – белков Ерас. За это время в экспериментах in vitro на культуре клеток или на изолированных органах, а затем и в модельных экспериментах in vivo была достаточно подробно изучена их роль в физиологии и патологии сердечно-сосудистой системы. Белки Ерас1 и Ерас2 экспрессируются в эндотелии и гладкомышечных клетках сосудов, кардиомиоцитах и сердечных фибробластах. В настоящее время накоплен достаточно убедительный литературный материал, свидетельствующий о том, что в нормальных физиологических условиях белки Ерас1 регулируют барьерную функцию и, возможно, ангиогенез и пролиферацию эндотелиальных клеток сосудов. Если роль белков Ерас1 в регуляции ангиогенеза и пролиферации эндотелиальных клеток окончательно не ясна, то их место в регуляции пролиферации/ангиогенеза гладкомышечных клеток сосудов представляется достаточно определенным – белки Ерас1, действуя корпоративно с РКА, проявляют антиангиогенную активность. Помимо этого белки Ерас1 участвуют в регуляции тонической активности сосудистого русла – в крупных сосудах активация сопряженных с белками Ерас1 сигнальных путей инициирует вазодилатацию, а в микрососудах кожи, напротив – вазоконстрикцию. В условиях патологии, например, сосудистой травмы, активированные белки Ерас1 стимулируют процессы миграции гладкомышечных клеток сосудов и пролиферацию их в неоинтиме. Несмотря на то, что документально подтверждена экспрессия белков Ерас2 в эндотелиальных и гладкомышечных клетках сосудов, их вклад в регуляцию функциональной активности сосудистого русла не известен. Не менее важен и вклад белков Ерас в регуляцию деятельности сердца. В физиологических условиях сопряженные с белками Ерас1 сигнальные каскады регулируют инотропную и, в определенной мере, лузитропную функцию кардиомиоцитов, а также процессы их межклеточного взаимодействия. Помимо этого, как белки Ерас1, так и белки Ерас2 проявляют выраженную антиапоптотическую активность. В условиях острой ишемии миокарда активация митохондриальной изоформы белков Ерас1 инициирует гибель кардиомиоцитов. В условиях хронической сердечной патологии (хроническая сердечная недостаточность, постинфарктный коронарокардиосклероз и др.) сопутствующая ей избыточная активация β1-адренорецепторов и, соответственно, избыточная активация сАМР/Ерас1-сопряженных сигнальных каскадов играет существенную роль в развитии гипертрофии, ремоделирования и фиброза миокарда, а активация Ерас2-сопряженных сигнальных каскадов инициирует развитие нарушений сердечного ритма.
Вместе с тем, несмотря на достаточно обширный материал, посвященный изучению роли белков Ерас в регуляции деятельности сердечно-сосудистой системы, многие вопросы остаются не ясными. Это связано с тем, что, во-первых, несмотря на то, что для белков Ерас описан ряд эффекторов, их многодоменная структура далеко не исключает наличие и других “партнеров” по связыванию. Во-вторых, остается до конца не ясной взаимосвязь белков Epac с другими сигнальными белками, с которыми они посредством обратной связи регулируют одни и те же биологические процессы. Особенно это касается взаимодействия белков Ерас и РКА, поскольку, например, в кардиомиоцитах, сопряженные с ними сигнальные пути нацелены на одни и те же молекулярные комплексы. И, наконец, получение исчерпывающей информации о биологической роли белков Ерас во многом сдерживается отсутствием в полной мере адекватных фармакологических инструментов, позволяющих селективно активировать, или селективно блокировать как собственно белки Ерас, так и/или их непосредственные эффекторы.
Однако уже сейчас можно говорить о том, что изучение вклада белков Ерас в формирование таких патологических процессов, как стенозирование сосудов, гипертрофия, ремоделирование, фиброз миокарда и нарушение сердечного ритма, создало фундаментальную базу для поиска оригинальных лекарственных средств на основе селективных блокаторов белков Ерас1 и Ерас2.
Благодарности. Авторы выражают глубокую признательность за консультативную помощь в работе над обзором литературы гл. н. с. ФГБНУ “НИИ фармакологии им. В.В. Закусова”, чл.-корр. РАН Ю.В. Вахитовой и за помощь в оформлении работы н. с. ФГБНУ “НИИ фармакологии им. В.В. Закусова” В.В. Барчукову.
Список литературы
Ulucan C., Wang X., Baljinnyam E. et al. Developmental changes in gene expression of Epac and its upregulation in myocardial hypertrophy // Am. J. Physiol. Heart Circ. Physiol. 2007. V. 293. № 3. P. H1662.
Pereira L., Rehmann H., Lao D.H. et al. Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes // Proc. Natl. Acad. Sci. USA. 2015. V. 112. № 13. P. 3991.
Pereira L., Métrich M., Fernández-Velasco M. et al. cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes // J. Physiol. 2007. V. 583. Pt 2. P. 685.
Oestreich E.A., Wang H., Malik S. et al. Epac-mediated activation of phospholipase C(epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes // J. Biol. Chem. 2007. V. 282. № 8. P. 5488.
Oestreich E.A., Malik S., Goonasekera S.A. et al. Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II // J. Biol. Chem. 2009. V. 284. № 3. P. 1514.
Cazorla O., Lucas A., Poirier F. et al. cAMP binding protein Epac regulates cardiac myofilament function // Proc. Natl. Acad. Sci. USA. 2009. V. 106. № 33. P. 14 144.
Pereira L., Ruiz-Hurtado G., Morel E. et al. Epac enhances excitation-transcription coupling in cardiac myocytes // J. Mol. Cell. Cardiol. 2012. V. 52. № 1. P. 283.
Okumura S., Fujita T., Cai W. et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses // J. Clin. Invest. 2014. V. 124. № 6. P. 2785.
Ruiz-Hurtado G., Morel E., Domínguez-Rodríguez A. et al. Epac in cardiac calcium signaling // J. Mol. Cell. Cardiol. 2013. V. 58. P. 162.
Fujita T., Umemura M., Yokoyama U. et al. The role of Epac in the heart // Cell. Mol. Life Sci. 2017. V. 74. № 4. P. 591.
Darrow B.J., Fast V.G., Kléber A.G. et al. Functional and structural assessment of intercellular communication. Increased conduction velocity and enhanced connexin expression in dibutyryl cAMP-treated cultured cardiac myocytes // Circ. Res. 1996. V. 79. № 2. P. 174.
Matsuda T., Fujio Y., Nariai T. et al. N-cadherin signals through Rac1 determine the localization of connexin 43 in cardiac myocytes // J. Mol. Cell. Cardiol. 2006. V. 40. № 4. P. 495.
Salameh A., Frenzel C., Boldt A. et al. Subchronic alpha- and beta-adrenergic regulation of cardiac gap junction protein expression // FASEB J. 2006. V. 20. № 2. P. 365.
Somekawa S., Fukuhara S., Nakaoka Y. et al. Enhanced functional gap junction neoformation by protein kinase A-dependent and Epac-dependent signals downstream of cAMP in cardiac myocytes // Circ. Res. 2005. V. 97. № 7. P. 655.
Lee T.M., Lin S.Z., Chang N.C. Both PKA and Epac pathways mediate N-acetylcysteine-induced Connexin43 preservation in rats with myocardial infarction // PLoS One. 2013. V. 8. № 8. e71878.
Duquesnes N., Derangeon M., Métrich M. et al. Epac stimulation induces rapid increases in connexin43 phosphorylation and function without preconditioning effect // Pflugers. Arch. 2010. V. 460. № 4. P. 731.
Communal C., Singh K., Pimentel D.R., Colucci W.S. Norepinephrine stimulates apoptosis in adult rat ventricular activation of the beta-adrenergic pathway // Circulation. 1998. V. 98. № 13. P. 1329.
Fujita T., Ishikawa Y. Apoptosis in heart failure. The role of the β-adrenergic receptor-mediated signaling pathway and p53-mediated signaling pathway in the apoptosis of cardiomyocytes // Circ. J. 2011. V. 75. № 8. P. 1811.
Kwak H.J., Park K.M., Choi H.E. et al. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways // Cell. Signal. 2008. V. 20. № 5. P. 803.
Wu X.M., Ou Q.Y., Zhao W. et al. The GLP-1 analogue liraglutide protects cardiomyocytes from high glucose-induced apoptosis by activating the Epac-1/Akt pathway // Exp. Clin. Endocrinol. Diabetes. 2014. V. 122. № 10. P. 608.
Mangmool S., Hemplueksa P., Parichatikanond W., Chattipakorn N. Epac is required for GLP-1R-mediated inhibition of oxidative stress and apoptosis in cardiomyocytes // Mol. Endocrinol. 2015. V. 29. № 4. P. 583.
Aoyama M., Kawase H., Bando Y.K. et al. Dipeptidyl peptidase 4 inhibition alleviates shortage of circulating glucagon-like peptide-1 in heart failure and mitigates myocardial remodeling and apoptosis via the exchange protein directly activated by cyclic AMP 1/Ras-related protein 1 axis // Circ. Heart Fail. 2016. V. 9. № 1. e002081.
Wang Z., Liu D., Varin A. et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death // Cell. Death Dis. 2016. V. 7. e2198.
Litvin T.N., Kamenetsky M., Zarifyan A. et al. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate // J. Biol. Chem. 2003. V. 278. № 18. P. 15922.
Acin-Perez R., Salazar E., Kamenetsky M. et al. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation // Cell. Metab. 2009. V. 9. № 3. P. 265.
Kumar S., Kostin S., Flacke J.P. et al. Soluble adenylyl cyclase controls mitochondria-dependent apoptosis in coronary endothelial cells // J. Biol. Chem. 2009. V. 284. № 22. P. 14760.
Calderón-Sánchez E., Díaz I., Ordóñez A., Smani T. Urocortin-1 mediated cardioprotection involves XIAP and CD40-ligand recovery: Role of EPAC2 and ERK1/2 // PLoS One. 2016. V. 11. № 2. e0147375
Kang J.H., Lee H.S., Park D. et al. Context-independent essential regulatory interactions for apoptosis and hypertrophy in the cardiac signaling network // Sci. Rep. 2017. V. 7. № 1. P. 34.
Engelhardt S., Hein L., Wiesmann F., Lohse M.J. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice // Proc. Natl. Acad. Sci. USA. 1999. V. 96. № 12. P. 7059.
Krum H. Sympathetic activation and the role of beta-blockers in chronic heart failure // Aust. N Z J. Med. 1999. V. 29. № 3. P. 418.
Morisco C., Zebrowski D., Condorelli G. et al. The Akt-glycogen synthase kinase 3beta pathway regulates transcription of atrial natriuretic factor induced by beta-adrenergic receptor stimulation in cardiac myocytes // J. Biol. Chem. 2000. V. 275. № 19. P. 14466.
Zheng M., Han Q.D., Xiao R.P. Distinct beta-adrenergic receptor subtype signaling in the heart and their pathophysiological relevance // Acta Phisiol. Sinica. 2004. V. 56. № 1. P. 1.
Colomer J.M., Mao L., Rockman H.A., Means A.R. Pressure overload selectively up-regulates Ca2+/calmodulin-dependent protein kinase II in vivo // Mol. Endocrinol. 2003. V. 17. № 2. P. 183.
Morel E., Marcantoni A., Gastineau M. et al. cAMP-binding protein Epac induces cardiomyocyte hypertrophy // Circ. Res. 2005. V. 97. № 12. P. 1296.
Métrich M., Lucas A., Gastineau M. et al. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy // Circ. Res. 2008. V. 102. № 8. P. 959.
Monceau V., Llach A., Azria D. et al. Epac contributes to cardiac hypertrophy and amyloidosis induced by radiotherapy but not fibrosis // Radiother. Oncol. 2014. V. 111. № 1. P. 63.
Métrich M., Laurent A.C., Breckler M. et al. Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway // Cell Signal. 2010. V. 22. № 10. P. 1459.
Lezoualc’h F., Métrich M., Hmitou I. et al. Small GTP-binding proteins and their regulators in cardiac hypertrophy // J. Mol. Cell. Cardiol. 2008. V. 44. № 4. P. 623.
Li L., Cai H., Liu H., Guo T. β-Adrenergic stimulation activates protein kinase Cε and induces extracellular signal-regulated kinase phosphorylation and cardiomyocyte hypertrophy // Mol. Med. Rep. 2015. V. 11. № 6. P. 4373.
Métrich M., Morel E., Berthouze M. et al. Functional characterization of the cAMP-binding proteins Epac in cardiac myocytes // Pharmacol. Rep. 2009. V. 61. № 1. P. 146.
Laudette M., Coluccia A., Sainte-Marie Y. et al. Identification pharmacological inhibitor Epac1 that protects heart against acute and chronic modelscardiac stress // Cardiovasc. Res. 2019. V. 115. № 12. P. 1766.
Berthouze-Duquesnes M., Lucas A., Saulière A. et al. Specific interactions between Epac1, β-arrestin2 and PDE4D5 regulatereceptor differential on cardiac hypertrophic signaling // Cell. Signal. 2013. V. 25. № 4. P. 970.
Jin H., Fujita T., Jin M. et al. Cardiac overexpression of Epac1 in transgenic mice rescues lipopolysaccharide-induced cardiac dysfunction and inhibits Jak-STAT pathway // J. Mol. Cell. Cardiol. 2017. V. 108. P. 170.
Eghbali M. Cardiac fibroblasts: function, regulation of gene expression, and phenotypic modulation // Basic Res. Cardiol. 1992. V. 87. Suppl. 2. P. 183.
Swaney J.S., Roth D.M., Olson E.R. et al. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase // Proc. Natl. Acad. Sci. USA. 2005. V. 102. № 2. P. 437.
Yokoyama U., Patel H.H., Lai N.C. et al. The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals // Proc. Natl. Acad. Sci. USA. 2008. V. 105. № 17. P. 6386.
Insel P.A., Murray F., Yokoyama U. et al. cAMP and Epac in the regulation of tissue fibrosis // Br. J. Pharmacol. 2012. V. 166. № 2. P. 447.
Miller C.L., Cai Y., Oikawa M. et al. Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart // Basic Res. Cardiol. 2011. V. 106. № 6. P. 1023.
Chen C., Du J., Feng W. et al. β-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCδ/p38 MAPK signalling in neonatal mouse cardiac fibroblasts // Br. J. Pharmacol. 2012. V. 166. № 2. P. 676.
Che X., Wang X., Zhang J. et al. Vitexin exerts cardioprotective effect on chronic myocardial ischemia/reperfusion injury in rats via inhibiting myocardial apoptosis and lipid peroxidation // Am. J. Transl. Res. 2016. V. 8. № 8. P. 3319.
Lorenzen J.M., Schauerte C., Hübner A. et al. Osteopontin is indispensible for AP1-mediated angiotensin II-related miR-21 transcription during cardiac fibrosis // Eur. Heart J. 2015. V. 36. № 32. P. 2184.
Pollard C.M., Desimine V.L., Wertz S.L. et al. Deletion of osteopontin enhances β2-adrenergic receptor-dependent anti-fibrotic signaling in cardiomyocytes // Int. J. Mol. Sci. 2019. V. 20. № 6. P. E1396.
Surinkaew S., Aflaki M., Takawale A. et al. Exchange protein activated by cyclic-adenosine monophosphate (Epac) regulates atrial fibroblast function and controls cardiac remodeling // Cardiovasc. Res. 2019. V. 115. № 1. P. 94.
Fazal L., Laudette M., Paula-Gomes S. et al. Multifunctional mitochondrial Epac1 controls myocardial cell death // Circ. Res. 2017. V. 120. № 4. P. 645.
Hothi S.S., Gurung I.S., Heathcote J.C. et al. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart // Pflugers. Arch. 2008. V. 457. № 2. P. 253.
Pereira L., Cheng H., Lao D.H. et al. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia // Circulation. 2013. V. 127. № 8. P. 913.
Neef S., Heijman J., Otte K. et al. Chronic loss of inhibitor-1 diminishes cardiac RyR2 phosphorylation despite exaggerated CaMKII activity // Naunyn. Schmiedebergs Arch. Pharmacol. 2017. V. 390. № 8. P. 857.
Li M., Hothi S.S., Salvage S.C. et al. Arrhythmic effects of Epac-mediated ryanodine receptor activation in Langendorff-perfused murine hearts are associated with reduced conduction velocity // Clin. Exp. Pharmacol. Physiol. 2017. V. 44. № 6. P. 686.
Lezcano N., Mariángelo J.I.E., Vittone L. et al. Early effects of Epac depend on the fine-tuning of the sarcoplasmic reticulum Ca2+ handling in cardiomyocytes // J. Mol. Cell. Cardiol. 2018. V. 114. P. 1.
Bobin P., Varin A., Lefebvre F. et al. Calmodulin ki-nase II inhibition limits the pro-arrhythmic Ca2+ waves induced by cAMP-phosphodiesterase inhibitors // Cardiovasc. Res. 2016. V. 110. № 1. P. 151.
Pereira L., Bare D.J., Galice S. et al. β-Adrenergic induced SR Ca2+ mediated Epac-NOS // J. Mol. Cell. Cardiol. 2017. V. 108. P. 8.
Curran J., Tang L., Roof S.R. et al. Nitric oxide-dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation // PLoS One. 2014. V. 9. № 2. e87495.
Maltsev V.A., Sabbah H.N., Higgins R.S. et al. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes // Circulation. 1998. V. 98. № 23. P. 2545.
Belardinelli L., Giles W.R., Rajamani S. et al. Cardiac late Na+ current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress // Heart Rhythm. 2015. V. 12. № 2. P. 440.
Dybkova N., Wagner S., Backs J. et al. Tubulin polymerization disrupts cardiac β-adrenergic regulation of late INa // Cardiovasc. Res. 2014. V. 103. № 1. P. 168.
Hegyi B., Bányász T., Izu L.T. et al. β-adrenergic regulation of late Na+ current during action potential is mediated by both PKA and CaMKII // J. Mol. Cell. Cardiol. 2018. V. 123. P. 168.
Valli H., Ahmad S., Sriharan S. et al. Epac-induced ryanodine receptor type 2 inhibits sodium currents in atrial and ventricular cardiomyocytes // Clin. Exp. Pharmacol. Physiol. 2018. V. 45. № 3. P. 278.
Zhang M.X., Zheng J.K., Wang W.W. et al. Exchange-protein activated by cAMP (EPAC) regulates L-type calcium channel in atrial fibrillation of heart failure model // Eur. Rev. Med. Pharmacol. Sci. 2019. V. 23. № 5. P. 2200.
Doleschal B., Primessnig U., Wölkart G. et al. TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1 // Cardiovasc. Res. 2015. V. 106. № 1. P. 163.
Tiapko O., Groschner K. TRPC3 as a Target of Novel Therapeutic Interventions // Cells. 2018. V. 7. № 7. P. E83.
Domínguez-Rodríguez A., Ruiz-Hurtado G., Sabourin J. et al. Proarrhythmic effectsustained activation on TRPC3/4 in rat ventricular cardiomyocytes // J. Mol. Cell. Cardiol. 2015. V. 87. P. 74.
Brette F., Blandin E., Simard C. et al. Epac activator critically regulates action potential duration by decreasing potassium current in rat adult ventricle // J. Mol. Cell. Cardiol. 2013. V. 57. P. 96.
Laudette M., Zuo H., Lezoualc’h F., Schmidt M. Epac function and cAMP scaffolds in the heart and lung // J. Cardiovasc. Dev. Dis. 2018. V. 5. № 1. P. E9.
Aflaki M., Qi X.Y., Xiao L. et al. Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained β-adrenergic activation in guinea pig hearts // Circ. Res. 2014. V. 114. № 6. P. 993.
Yang Z., Kirton H.M., Al-Owais M. et al. Epac2-Rap1 signaling regulates species and susceptibility to cardiac arrhythmias // Antioxid. Redox. Signal. 2017. V. 27. № 3. P. 117.
Дополнительные материалы отсутствуют.
Инструменты
Физиология человека