

Цитология, 2019, T. 61, № 5, стр. 343-356
Методы флуоресцентной микроскопии сверхвысокого разрешения и их использование для визуализации различных клеточных структур
А. Д. Ведяйкин 1, 2, *, М. А. Ходорковский 1, И. Е. Вишняков 2
1 Санкт-Петербургский политехнический университет Петра Великого
195251 Санкт-Петербург, Россия
2 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: misterkotlin@gmail.com
Поступила в редакцию 03.12.2018
После доработки 20.12.2018
Принята к публикации 20.12.2018
Аннотация
Методы флуоресцентной микроскопии сверхвысокого разрешения являются мощным инструментом для визуализации различных структур в клетках как эукариот, так и прокариот, в том числе в клетках, характеризующихся крайне малым размером. В последние годы разработано большое количество таких методов, что позволяет успешно решать различные задачи. В то же время, высокая техническая сложность их реализации по сравнению с традиционной световой микроскопией несколько сужает использование этого инструмента в клеточной биологии. В обзоре рассматриваются и сравниваются между собой основные разновидности методов флуоресцентной микроскопии сверхвысокого разрешения, наиболее широко используемые в биологии. Кроме того, приведены примеры успешного применения описываемых методов с целью визуализации различных структур в клетке, в том числе результаты, полученные авторами данного обзора.
За последние 20 лет было разработано большое количество методов флуоресцентной микроскопии (ФМ) сверхвысокого разрешения (англ. super-resolution microscopy). Такие методы позволяют решить проблему дифракционного предела разрешения традиционной оптической микроскопии, обусловленную волновой природой света. Во второй половине XIX в. было показано, что изображение точечного источника света в оптическом приборе (например, в микроскопе или телескопе) будет иметь конечные размеры d (дифракционное пятно, обычно описываемое функцией Эйри, оптическое разрешение), зависящие от длины волны λ и числовой апертуры NA оптического прибора в соответствии с формулой (1), впервые описанной Эрнстом Аббе (Lipson et al., 1995):
Фундаментальный характер этой формулы, описывающей дифракционный предел, долгое время ограничивал возможности оптической микроскопии и не позволял получать разрешение лучше, чем примерно половина длины волны используемого света. Самые современные микроскопы, построенные по классической схеме, имеют разрешение не лучше 150 нм. Поэтому даже в относительно крупной эукариотической клетке многие структуры и органеллы не могут быть разрешены при помощи традиционной оптической микроскопии. Еще сильнее ограничены возможности традиционной микроскопии в плане визуализации структур в клетках бактерий, особенно таких маленьких, как, например, микоплазмы (класс Mollicutes) (Борхсениус и др., 2016). Неудивительно, что долгое время исследователи пытались обойти дифракционный предел (а следовательно увеличить разрешение) различными способами.
Из приведенной выше формулы очевидно, что увеличения разрешения можно добиться через уменьшение используемой длины волны. Большинство используемых в биологии флуорофоров являются источниками излучения, соответствующего видимой области спектра. Например, для зеленого флуоресцентного белка максимум спектра испускания приходится на длины волн порядка 500 нм (Tsien, 1998). С учетом формулы (1) при использовании объектива с наиболее высокой из широко используемых числовой апертурой 1.49 получаем теоретический предел разрешения около 170 нм. Переход в ультрафиолетовую область затруднен в связи с губительностью такого излучения для клетки, а также с высоким уровнем автофлуоресценции, что делает невозможной визуализацию живых объектов. Кроме того, для соответствующей области спектра практически отсутствуют флуоресцентные метки, что сразу лишает метод известных преимуществ ФМ, таких как избирательность и высокий контраст.
Использование электронной микроскопии (особенно методов криоэлектронной томографии) позволяет получить высокое разрешение, сравнимое с размерами некоторых крупных молекул, однако исследование живых организмов такими методами невозможно. Возможности локализации определенных компонентов или биомолекул в клетке при помощи этого метода ограничены. В таких случаях используют метод иммуноэлектронной микроскопии, который, благодаря специфической метке, позволяет установить, в какой области клетки находится исследуемый антиген. Кроме того, активно развивается метод коррелятивной световой и электронной микроскопии (CLEM, Correlated Light and Electron Microscopy), что позволяет частично совместить преимущества указанных методов (de Boer et al., 2015).
Увеличение числовой апертуры тоже позволяет повысить разрешение, однако эти возможности ограничены максимальным теоретически достижимым телесным углом, равным 4π. Такие методы реализованы и достаточно широко используются, например, метод 4π-микроскопии (Bewersdorf et al., 2006). Однако в связи с высокой технической сложностью этого метода в сочетании с относительно низким разрешением он не будет обсуждаться в данном обзоре. Кроме того, мы не обсуждаем возможность менять показатель преломления, используя различные иммерсионные объективы, однако для полноты картины упомянуть о самой такой возможности необходимо.
Достаточно давно, еще в 1928 г., был предложен метод ближнепольной оптической микроскопии (БОМ), основанный на сканировании объекта при помощи чрезвычайно малой апертуры с размерами порядка нескольких нанометров. На практике БОМ был реализован только в 1986 г. (Betzig et al., 1986). Примерно в это же время появились такие широко известные зондовые методы, как атомно-силовая микроскопия и туннельная микроскопия (Salapaka, Salapaka, 2008), но в этих вариантах отсутствует оптическая система и связанные с ней дифракционные эффекты. Так как размеры апертуры в БОМ, а также расстояние от нее до объекта намного меньше длины световой волны, дифракционный предел практически не сказывается на достигаемом разрешении; метод позволяет достичь разрешения порядка 10 нм. Несмотря на высокое разрешение, метод БОМ не нашел такого широкого применения в биологии, как методы широкопольной оптической микроскопии, так как его использование ограничивается визуализацией поверхности, а визуализация объемных структур практически невозможна.
Однако за последнее время были разработаны несколько методов ФМ, которые позволяют достичь отличного пространственного разрешения, сохраняя при этом основные достоинства флуоресцентной микроскопии. Это привело к настоящему прорыву в области биологии клетки; в 2014 г. новые методы микроскопии были отмечены Нобелевской премией по химии (Mockl et al., 2014). Их можно разбить на две основные группы: детерминистские и стохастические методы визуализации. При этом существует огромное количество разновидностей основных методов, зачастую отличающихся друг от друга лишь названиями. Например, к методу локализационной микроскопии относятся такие аббревиатуры, как SMLM (Single-Molecule Localization Microscopy, наиболее общая аббревиатура, охватывающая все разновидности метода), PALM (PhotoActivated Localization Microscopy) (Betzig et al., 2006), fPALM (fluorescence PALM) (Hess et al., 2006), BALM (Binding Activated Localization Microscopy) (Schoen et al., 2011), STORM (Stochastic Optical Reconstruction Microscopy) (Rust et al., 2006), dSTORM (direct STORM) (Heilemann et al., 2008), SOFI (Super-resolution Optical Fluctuation Imaging) (Dertinger et al., 2012), PAINT (Points Accumulation for Imaging in Nanoscale Topography) (Schnitzbauer et al., 2017) и др. STED (STimulated Emission Depletion) также имеет родственные методы, например, GSD (Ground State Depletion) (Bretschneider et al., 2007), а также метод, имеющий наиболее общее название RESOLFT (REversible Saturable Optical Linear Fluorescence Transitions) (Kwon et al., 2015). Методу SIM (Structured Illumination Microscopy) родственны методики SMI (Spatially Modulated Illumination) (Birk et al., 2009), SSIM (Saturated Structured-Illumination Microscopy), который является комбинацией методов SIM и STED (Gustafsson, 2005) и др.
В настоящем обзоре не стояла задача привести описание всех разновидностей существующих методов микроскопии сверхвысокого разрешения, так как этого не позволяет формат статьи. Здесь на примере SIM, STED и SMLM описаны только основные характеристики методов, которые, тем не менее, в большей или меньшей степени применимы ко всем разновидностям флуоресцентной микроскопии сверхвысокого разрешения. Кроме того, обсуждается практическое использование этих методов для визуализации различных структур в клетке, в том числе на примере работ авторов данного обзора. Дополнительную информацию по методам микроскопии сверхвысокого разрешения можно найти в обзорах других авторов (например, Sahl et al., 2017).
ДЕТЕРМИНИСТСКИЕ МЕТОДЫ ВИЗУАЛИЗАЦИИ
В основе детерминистских методов флуоресцентной микроскопии сверхвысокого разрешения обычно лежит априорная информация о пространственном распределении возбуждающего и (или) испускаемого света. Так, в случае метода SIM – это информация, заложенная в паттерн освещения, а в случае STED – информация о размере области, испускающей свет.
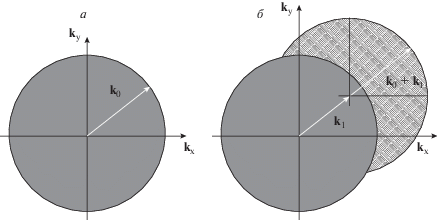
Одним из наиболее удобных и широко используемых в биологии методов микроскопии сверхвысокого разрешения является метод микроскопии структурированного освещения (SIM). Принцип этого метода обстоятельно рассмотрен в работе Густафссона (Gustafsson, 2000). В основе метода лежит последовательное освещение образца заданным паттерном освещения в виде стоячей волны (рис. 1). Несколько ориентаций стоячей волны в паттерне освещения позволяют благодаря так называемому муаровому эффекту получить дополнительную информацию о структуре объекта. Компьютерная обработка полученных изображений (на основании известной информации о форме стоячей волны в паттерне освещения) с использованием обратного преобразования Фурье обеспечивает реконструкцию изображения с улучшенным разрешением. С математической точки зрения, метод SIM использует расширение области наблюдаемых частот в частотном пространстве, что эквивалентно увеличению разрешения (рис. 2). При этом наложение паттерна освещения с частотой k1 на исходное изображение с максимальной частотой k0 увеличивает наивысшую наблюдаемую пространственную частоту (разрешение) с k0 до k0 + k1. Таким образом, чтобы максимизировать разрешение, необходимо, чтобы для освещения были характерны как можно более высокие пространственные частоты k1. Разумеется, значение |k1| ограничено дифракцией на уровне 2NA/λ ≈ k0, поэтому новый предел разрешения k0 + k1 может быть не более ≈2|k0|. Таким образом, SIM позволяет достичь разрешения около 100 нм, что примерно в 2 раза лучше, чем позволяет получить традиционная ФМ, но существенно хуже других методов субдифракционной визуализации, рассматриваемых ниже. С другой стороны, важным достоинством метода SIM является то, что он не требует каких-либо особенностей пробоподготовки по сравнению с методом традиционной ФМ, а также других методов микроскопии сверхвысокого разрешения, которые могут требовать использования, в частности, особых красителей. Кроме того, метод SIM обеспечивает быструю визуализацию, что позволяет исследовать динамические процессы в клетке с частотой до нескольких Гц, и использует освещение достаточно низкой интенсивности, что дает возможность работать с живыми клетками.
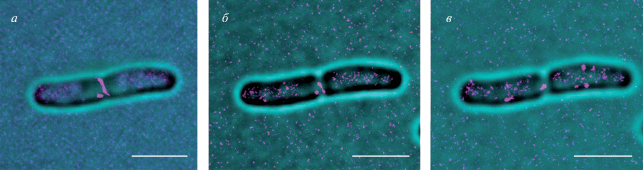
Рис. 1.
Увеличение разрешения метода SIM посредством эффекта муара. Если неизвестная структура образца (а) будет наложена на известный шаблон освещения с регулярным паттерном (б), появится муаровый узор (муаровые полосы, явление, хорошо известное фотографам) (в). Вычислительная обработка (обратное преобразование Фурье) на основе известного распределения на изображении (б) позволяет увеличить разрешение примерно в 2 раза.
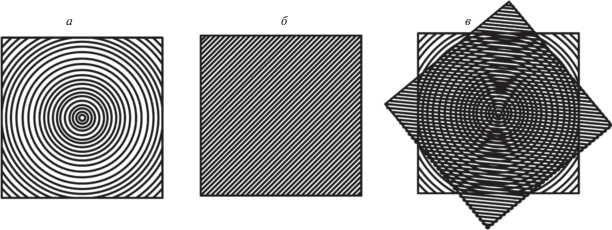
Рис. 2.
Область пространственных частот, которые можно наблюдать при использовании обычного светового микроскопа, ограничена окружностью с радиусом k0 в частотном пространстве (серый круг) (a). Если свет возбуждения содержит пространственную частоту k1 (вектор k1 соответствует структурированной засветке), новый набор информации становится видимым в виде муаровых полос (заштрихованный круг) (б). Эта область имеет ту же форму, что и область, наблюдаемая в обычный микроскоп, но смещена на k1. Максимальная пространственная частота, которая может наблюдаться (в этом направлении), равна k0 + k1.
SIM широко используется для визуализации биологических объектов (Heintzmann, Huser, 2017). Например, при помощи этого метода была успешно осуществлена одновременная визуализация хроматина, ядерных пор и ядерной ламины в миобластах мыши линии C2C12 (Schermelleh et al., 2008), что позволило авторам работы, в частности, визуализировать отдельные ядерные поровые комплексы, которые были колокализованы с каналами в ядерной ламине. Ряд исследователей успешно использовали SIM для изучения структуры бактериального аппарата деления, в частности, Z-кольца, состоящего из филаментов ключевого белка деления бактерий FtsZ. Например, 3D-SIM позволил обнаружить, что Z-кольца в бактериях Bacillus subtilis и Staphylococcus aureus представляют собой неоднородные группы, состоящие из кластеров FtsZ (Strauss et al., 2012), а не непрерывные структуры, как считалось раньше. Динамическая съемка позволила авторам продемонстрировать, что локализация FtsZ в Z-кольце динамична, при этом структура кольца остается неоднородной в течение всего процесса деления клеток. Недавно SIM начали использовать и для визуализации вирусных частиц: например, исследовали созревание и морфогенез вируса, входящего в состав вакцины против оспы (Horsington et al., 2012). Метод 3D-SIM позволил получить изображения оболочки, формируемой белком капсида B5 и представляющей собой сферическую структуру, окружающую ядро вируса, что дало возможность оценивать степень созревания вирусных частиц.
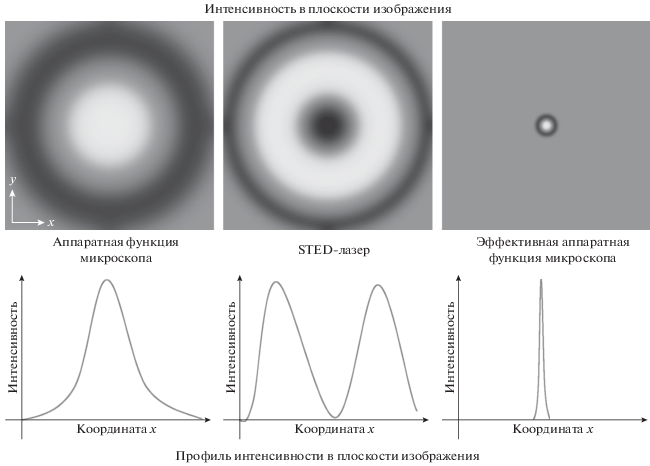
Другой детерминистский метод микроскопии, STED, или микроскопия на основе подавления спонтанного испускания, в отличие от SIM требует существенно большей интенсивности освещения, что несколько ограничивает его применение по отношению к живым клеткам, но зато позволяет получить значительно лучшее разрешение (обычно порядка 10–50 нм). В основе микроскопии STED лежит нелинейный (по интенсивности) отклик флуоресцентных молекул на воздействие света, вызывающего стимулированное испускание. Обычно для реализации метода используют два лазера, формирующих концентрические дифракционно-ограниченные пятна в плоскости образца (рис. 3). Первый лазер – возбуждающий, с меньшей длиной волны. Он имеет профиль интенсивности, близкий к гауссиану, и используется для возбуждения флуоресценции. Второй, с большей длиной волны, индуцирующий (или STED-лазер), имеет профиль интенсивности в виде тора и используется для индукции стимулированного испускания. Индуцирующий лазер позволяет подавить спонтанное испускание на периферии освещаемого участка образца, оставив его лишь в центре. При этом стимулированное излучение имеет большую длину волны, чем спонтанное, поэтому их удается отделить друг от друга спектрально. Подбор интенсивностей возбуждающего и индуцирующего лазеров позволяет добиться условий, при которых испускание происходит лишь из сколь угодно малой (до единиц нанометров) области по центру лазерного пятна. STED является сканирующим методом, по принципу реализации он сходен с конфокальной микроскопией. Кроме того, его можно сравнить с зондовыми методами – совмещенное пятно лазеров представляет собой своего рода “зонд”. Однако в отличие от зондовых методов STED позволяет осуществлять визуализацию по всему объему, а не только на поверхности образца.
Рис. 3.
Обход дифракционного предела разрешения в микроскопии STED. Слева направо: пучок возбуждения флуоресценции (соответствует аппаратной функции микроскопа); STED-лазер; эффективная аппаратная функция микроскопа. Верхний ряд – профиль интенсивности в плоскости XY (цветовая кодировка интенсивности от черного к белому); нижний ряд – профиль интенсивности вдоль координаты x. Нелинейная зависимость вероятности стимулированного излучения от интенсивности приводит к тому, что наложение коаксиальных пучков возбуждения и STED-лазера обеспечивает сколь угодно малую область, из которой происходит спонтанное испускание (эффективную аппаратную функцию).
Технически STED является очень сложным методом. Достаточно отметить, что с момента описания идеи авторами метода в 1994 г. до его практической реализации прошло целых 5 лет (Hell, Wichmann, 1994; Klar, Hell, 1999). Интересно отметить, что изначально идея STED была описана еще в 1986 г. в одном из советских патентов, хотя развития эта идея не получила (Охонин, 1986). STED не так широко как SIM применяется в биологии, прежде всего из-за высокой сложности метода. В одной из работ, например, STED использовали для визуализации мембран живых клеток: анализировали функцию эндосом на примере нейронов гиппокампа, изолированных нервных окончаний и клеток линии PC12 (Hoopmann et al., 2010). STED также был недавно использован для оценки размеров кластеров, формируемых белком FtsZ в клетках Escherichia coli, в том числе в клетках искусственно модифицированной формы (Soderstrom et al., 2018).
Еще одним детерминистским методом микроскопии сверхвысокого разрешения можно назвать микроскопию расширения (ExM, Expansion Microscopy) (Chen et al., 2015). Данный метод основан на физическом увеличении образца за счет расширения матрицы, в которой находится исследуемый фиксированный объект. Несмотря на полную несовместимость с живыми объектами, этот метод представляется очень интересным и практически важным, так как его использование возможно на любых микроскопах и позволяет достичь изотропного увеличения разрешения примерно в 4 раза. Данный метод уже применялся в ряде исследований: например, недавно он был использован для визуализации ядра и белков цитоскелета в клетках Danio rerio (Freifeld et al., 2017).
СТОХАСТИЧЕСКИЕ МЕТОДЫ ВИЗУАЛИЗАЦИИ
В основе стохастических методов микроскопии сверхвысокого разрешения лежит следующий факт: химическая природа множества источников света (флуорофоров) обеспечивает их сложное динамическое поведение, позволяющее подобрать условия, при которых несколько близко расположенных флуорофоров будут излучать свет в разное время, становясь разрешимыми в пространстве в каждый момент времени. Метод локализационной микроскопии SMLM представляет, пожалуй, наибольший интерес среди стохастических методов визуализации. В основе метода локализационной микроскопии лежит возможность локализации одиночной молекулы практически с любой точностью, определяемой в основном числом зарегистрированных фотонов. Эта возможность давно используется в таком методе, как отслеживание одиночных частиц, или SPT (Single Particle Tracking) для исследования движения одиночных флуоресцентно-меченых молекул (например, белков внутри клетки) (Shen et al., 2017). Еще в 1993 г. была показана возможность детектирования одиночной флуоресцентной молекулы при помощи ближнепольной сканирующей микроскопии (Betzig, Chichester, 1993), а уже в 1995 г. эта возможность была использована для того, чтобы в динамике отслеживать движение одиночных флуоресцентно-меченых молекул миозина по актиновому филаменту с субмикроскопической точностью (Funatsu et al., 1995).
Метод SPT не обязательно предполагает использование флуоресцентно-меченых частиц. Например, в одной из первых работ с использованием метода SPT исследовали движение наночастиц золота в цитоплазме клеток PTK-2, при этом использовали визуализацию в проходящем свете (Geerts et al., 1987). Однако именно флуоресцентные методы делают метод SPT по-настоящему удобным. Принципиальная возможность определять положение одиночной молекулы с субмикроскопической точностью используется и в методе локализационной микроскопии, и в других стохастических методах визуализации, но для реализации полноценного метода субмикроскопической визуализации было необходимо научиться разделять флуоресценцию одиночных молекул, что потребовало использования красителей с особыми свойствами.
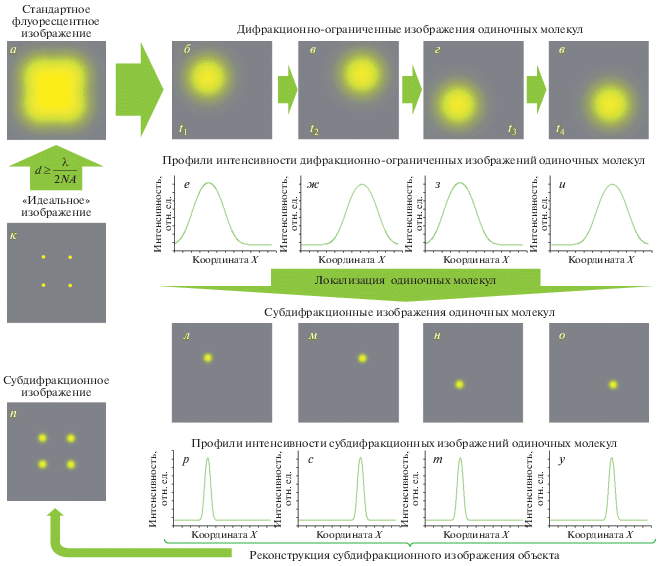
Метод локализационной микроскопии был практически одновременно описан тремя научными группами в 2006 г. (Betzig et al., 2006; Hess et al., 2006; Rust et al., 2006). В указанных работах были использованы различные флуорофоры и способы визуализации, что породило сразу три альтернативных названия метода. В случае PALM использовали фотоактивируемый флуоресцентный белок и метод микроскопии полного внутреннего отражения (TIRF, Total Internal Reflection Fluorescence Microscopy). В случае fPALM применяли фотоактивируемый флуоресцентный белок в сочетании со стандартной микроскопией. В случае STORM использовали фотопереключаемую пару красителей Cy3−Cy5 в сочетании с методом TIRF. Однако суть всех трех методов одинакова: фотофизические и химические свойства флуорофоров позволяют подобрать такие условия эксперимента (свойства среды, интенсивность освещения, плотность флуоресцентного мечения), при которых в каждый момент времени флуоресцирует настолько малая часть молекул, что их изображения в микроскопе не перекрываются между собой (иными словами, на один дифракционный объем приходится не более одной флуоресцирующей молекулы) (рис. 4). Это позволяет определить положение каждой флуоресцентной молекулы (или локализовать ее) с высокой точностью, которая зависит главным образом от числа зарегистрированных фотонов (Ober et al., 2004). Дальнейшая компьютерная обработка позволяет осуществить реконструкцию изображения из локализованных молекул флуорофора с существенно улучшенным разрешением (типично – около 20 нм).
Рис. 4.
Обход дифракционного предела разрешения в локализационной микроскопии (SMLM). Явление дифракции приводит к тому, что “идеальное” изображение (к), фактически отражающее распределение флуорофора, во флуоресцентном микроскопе превращается в дифракционно-ограниченное (а). Метод локализационной микроскопии использует разделение флуоресценции одиночных молекул (б–д) во времени таким образом, что в каждый момент времени (t1–t4) изображения флуоресцентных молекул не накладываются друг на друга. Это позволяет каждую из молекул локализовать с субдифракционной точностью (л–о). В результате компьютерной обработки серий изображений удается реконструировать изображение с улучшенным разрешением (п). Для наглядной демонстрации выигрыша в разрешении на изображениях (е–и) приведены профили интенсивности дифракционно-ограниченных изображений одиночных молекул, а на изображениях (р–у) – соответствующие профили интенсивности субмикроскопических изображений.
С момента изобретения SMLM было предложено множество флуоресцентных меток, обладающих подходящими для метода свойствами (Клементьева и др., 2017; Li, Vaughan, 2018). Использование “ярких” синтетических красителей (фотоактивируемых, либо демонстрирующих обратимый переход в темное состояние) позволяет достичь высочайшего разрешения (вплоть до единиц нанометров), однако использование таких флуоресцентных меток сильно ограничено при изучении живых клеток. Флуоресцентные белки хорошо подходят для in vivo визуализации, хотя их фотофизические свойства несколько ограничивают разрешение, достижимое методом SMLM. Было предложено множество фотоактивируемых, фотопереключаемых белков, которые позволяют осуществлять визуализацию в основном в зеленом и красном спектральных диапазонах (Shcherbakova et al., 2014). Кроме того, активно развивается направление так называемых флуорогенных красителей (такие красители становятся флуоресцентными при соединении с субстратом); описано использование с методом SMLM красителей, специфичных по отношению к ДНК (Szczurek et al., 2017), мембране (Legant et al., 2016), белкам цитоскелета (Sauer, Heilemann, 2017). Перспективным также представляется использование флуорогенных белков (флуоресценция которых появляется при связывании лиганда), в частности, для исследования анаэробных организмов, в которых визуализация при помощи флуоресцентных белков затруднена (Bozhanova et al., 2017).
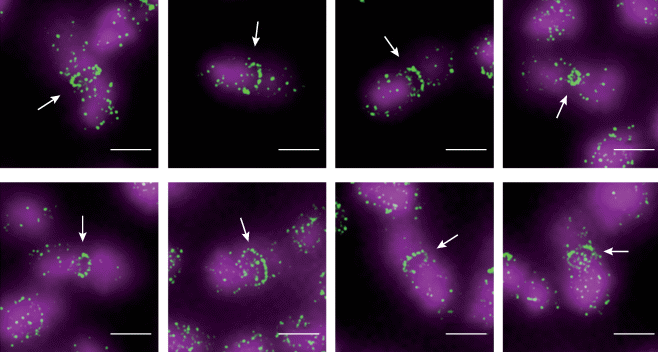
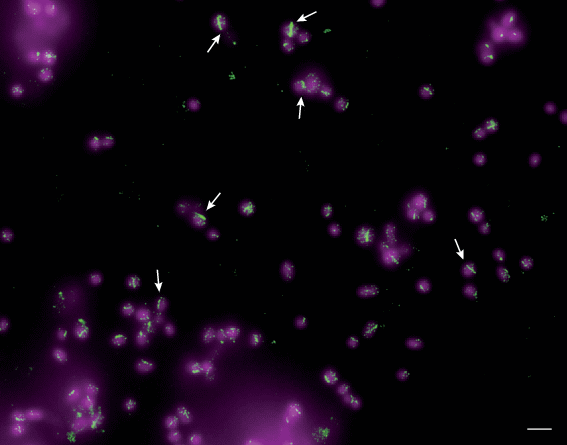
Мы использовали метод локализационной микроскопии для решения нескольких задач, связанных с визуализацией структур в бактериальных клетках. Например, локализационная микроскопия в сочетании с иммунофлуоресцентным мечением позволила визуа-лизировать структуры белка FtsZ в клетках E. co-li с субмикроскопическим разрешением (Vedyaykin et al., 2016b). Было показано, что ключевая структура, формируемая данным белком, Z-кольцо, выглядит как набор из слабо упорядоченных кластеров, напоминая “бусы на нитке” (рис. 5). Высокое разрешение метода позволило также оценить размеры Z-кольца и продемонстрировать его утолщение в ходе цитокинеза.
Рис. 5.
Изображения структур, сформированных белком FtsZ (Z-кольца отмечены стрелками), полученные на фиксированных клетках E. coli при помощи локализационной микроскопии (SMLM, краситель Alexa-647, выделение зеленым цветом) с дополнительной визуализацией ДНК (ФМ, краситель YOYO-1, выделение фиолетовым цветом). Масштабная линейка соответствует 1 мкм.
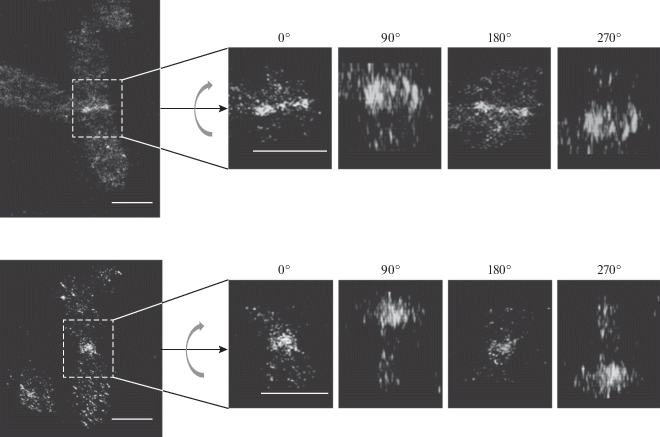
Следует отметить, что в указанной работе нами был использован один из лучших по фотофизическим характеристикам красителей для локализационной микроскопии – Alexа-647, который, однако, обладает одним существенным недостатком: для использования в локализационной микроскопии этот краситель помещается в особую среду, в которой он демонстрирует “мерцание” – явление обратимого перехода в долгоживущее темное состояние. Это приводит к тому, что значительная часть флуоресцентных молекул не участвует в реконструкции, что может приводить к повышению фрагментарности изображения. Для того чтобы убедиться, что фрагментарность структуры Z-кольца вызвана не вносимой стохастичностью метода, нами был использован другой краситель – Cy3b, который в специальных условиях среды демонстрирует явление фотоактивации, что позволяет значительно повысить долю визуализируемых молекул и таким образом снизить вносимую стохастичность визуализации (Vaughan et al., 2012). При визуализации молекул FtsZ с помощью данного метода выяснилось, что Z-кольцо представляет собой неоднородную структуру (рис. 6). Дополнительное подтверждение неоднородности структуры Z-кольца было нами получено при использовании трехмерной локализационной микроскопии (3D-SMLM, рис. 7). 3D-визуализация позволила получить дополнительную информацию об организации кластеров, формируемых белком FtsZ (Vedyaykin et al., 2016а).
Рис. 6.
Субмикроскопические изображения структур, сформированных белком FtsZ, полученные на фиксированных клетках E. coli методом локализационной микроскопии с использованием красителя Cy3b. Масштабная линейка соответствует 2 мкм.
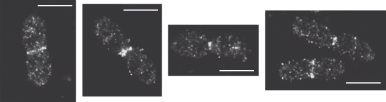
Рис. 7.
3D-реконструкция структур, формируемых белком FtsZ в клетках E. coli (в особенности Z-колец). Представлены 2D‑изображения клеток (слева) и увеличенные фрагменты этих же изображений (справа), соответствующие областям, ограниченным пунктиром. Изображения справа получены с использованием рендеринга с четырьмя различными углами поворота (от 0° до 270°) вокруг осей, обозначенных на рисунке прямыми черными стрелками.
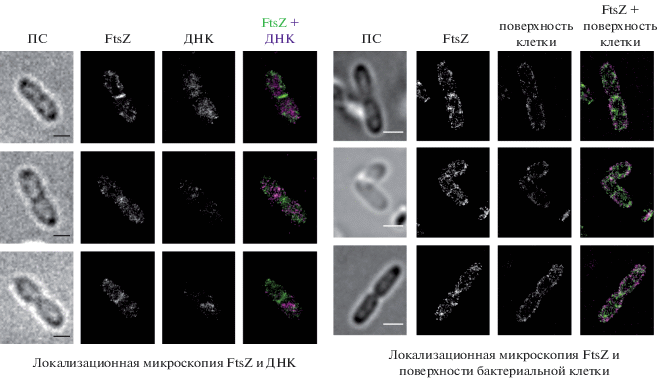
Для более точной интерпретации структур, формируемых белком FtsZ, требуется представление о морфологии клетки, в том числе о положении ДНК и клеточной оболочки, так как с этими элементами связана работа таких систем позиционирования Z-кольца, как нуклеоидная окклюзия и Min-система. Для решения задач, связанных с одновременной визуализацией нескольких элементов, хорошо подходит многоцветная локализационная микроскопия, которая и была нами использована для одновременной визуализации белка FtsZ и ДНК или FtsZ и клеточной поверхности E. coli (рис. 8) (Vedyaykin et al., 2015).
Рис. 8.
Многоцветная локализационная микроскопия с одновременной визуализацией FtsZ и ДНК (слева) или FtsZ и поверхности клетки (справа). ПС – проходящий свет. Масштабная линейка соответствует 1 мкм.
Метод SMLM позволяет работать даже с бактериями очень малых размеров – например, с клетками микоплазм (бактерии класса Mollicutes) (Polinov-skaya et al., 2018). Границы клеток микоплазм сложно определить на совмещенных изображениях, полученных в проходящем свете и при помощи SMLM. Одновременная визуализация ДНК (ФМ) и, например, малого белка теплового шока IbpA (SMLM) позволяет установить его распределение в клетке и указывает на возможность формирования этим белком цитоскелет-подобных структур в клетках фитопатогенной микоплазмы Acholeplasma laidlawii (рис. 9).
Рис. 9.
Изображения структур, сформированных белком IbpA (цитоскелет-подобные структуры отмечены стрелками), полученные на фиксированных клетках A. laidlawii при помощи локализационной микроскопии (SMLM, краситель Alexa-647, выделение зеленым цветом) с дополнительной визуализацией ДНК (ФМ, краситель YOYO-1, выделение фиолетовым цветом). Масштабная линейка соответствует 1 мкм.
Множество техник, основанных на методе SMLM или родственных ему, как отмечалось выше, породило сложную номенклатуру, состоящую из нескольких десятков аббревиатур. В целом, можно разделить все методы на два ключевых направления: методы визуализации одиночных молекул (“настоящий” метод SMLM) и методы анализа множества флуорофоров, изображения которых накладываются друг на друга (MEF, Multi-Emitter Fitting). Последние позволяют достигать улучшенного разрешения (пусть и не такого высокого, как в случае применения “настоящего” метода SMLM) даже при чрезмерно высокой плотности флуорофоров, что существенно расширяет область применения стохастических методов микроскопии сверхвысокого разрешения. Среди методов MEF следует отметить недавно разработанный метод микроскопии радиальных флуктуаций (SRRF, Super-Resolution Radial Fluctuations), основанный на продвинутом анализе серий флуоресцентных изображений, съемка которых ведется аналогично методу SMLM (Gustafsson et al., 2016). Разрешение, получаемое с помощью метода SRRF, зависит от плотности флуорофоров на изображении. В случае визуализации одиночных молекул, изображения которых не перекрываются между собой, метод SRRF позволяет получить разрешение, сравнимое с “классическим” методом SMLM.
Недавно группой исследователей из университета Дьюка в США был сконструирован штамм E. coli, в котором нативный FtsZ замещен белком слияния FtsZ с флуоресцентным белком mNeonGreen, который обеспечивает практически полную функциональность (Moore et al., 2017). К сожалению, данный белок не подходит для визуализации “классическим” методом SMLM, однако использование SRRF позволило нам визуализировать FtsZ с улучшенным разрешением (рис. 10) (Ponomareva et al., 2017). Как и при использовании метода SMLM, в случае применения SRRF нам удалось подтвердить неоднородность структуры Z-кольца E. coli.
Рис. 10.
Визуализация с помощью микроскопии сверхвысокого разрешения (метод SRRF) белка FtsZ в живых клетках E. coli. Слева направо – последовательные изображения с интервалом в 10 мин (а→в), полученные для структур FtsZ (фиолетовый цвет), комбинированные с изображениями клетки в проходящем свете (серо-голубой цвет). Масштабная линейка соответствует 1 мкм.
ЗАКЛЮЧЕНИЕ
Методы флуоресцентной микроскопии сверхвысокого разрешения представляются очень важным и востребованным инструментом для решения различных биологических задач, в особенности связанных с визуализацией внутриклеточных структур. Достаточно большое разнообразие таких методов позволяет подобрать оптимальную методику для определенных нужд с учетом достоинств и недостатков конкретных методов, а также имеющихся методологических ограничений. В заключении нам представляется важным привести положительные и отрицательные стороны каждого из рассмотренных методов в виде небольшой таблицы (табл. 1).
Таблица 1.
Достоинства и недостатки различных методов микроскопии сверхвысокого разрешения
| Метод | Достоинства | Недостатки |
|---|---|---|
| STED и аналогичные методы | – используются любые флуорофоры; – высокое разрешение (до 10 нм) |
– сложность, дороговизна реализации методики; – ограниченное использование при работе с живыми клетками |
| SMLM и аналогичные методы | – высокое разрешение (до 10 нм); – возможность реализации на “стандартном” флуоресцентном микроскопе |
– требуются особые (фотопереключаемые или “мерцающие”) флуорофоры; – ограниченное использование при работе с живыми клетками |
| SIM и аналогичные методы | – полная совместимость с живыми клетками; – используются любые флуорофоры |
– сложность, дороговизна реализации методики; – относительно низкое разрешение (около 100 нм) |
| SRRF | – используются любые флуорофоры | – относительно низкое разрешение (около 100 нм) |
| Микроскопия расширения | – реализуется на стандартном флуоресцентном микроскопе; – используются любые флуорофоры |
– невозможно использование при работе с живыми клетками; – относительно низкое разрешение (около 50–100 нм) |
ФИНАНСИРОВАНИЕ РАБОТЫ
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 17-74-20065).
Список литературы
Борхсениус С.Н., Чернова О.А., Чернов В.М., Вишняков И.Е. 2016. Микоплазмы в биологии и медицине начала XXI в. СПб: Наука. 333 с. (Borchsenius S.N., Chernova O.A., Chernov V.M., Vishnyakov I.E. 2016. Mycoplasmas in biology and medicine at the beginning of the XXI century. SPb: Nauka. 333 p.)
Клементьева Н.В., Божанова Н.Г., Загайнова Е.В., Лукьянов К.А., Мишин А.С. 2017. Флуорофоры для локализационной микроскопии одиночных молекул. Биоорганическая химия. 43(3) : 227–235. (Klementieva N.V., Bozhanova N.G., Zagaynova E.V., Lukyanov K.A., Mishin A.S. 2017. Fluorophores for single-molecule localization microscopy. Russian J. Bioorg. Chem. 43(3) : 227–234.)
Охонин В.А. 1986. Способ исследования микроструктуры образца. Патент SU № 1 374 922. (Okhonin V.A. 1986. Method for studying the microstructure of a sample. SU patent № 1 374 922.)
Betzig E., Chichester R.J. 1993. Single molecules observed by near-field scanning optical microscopy. Science. 262 : 1422–1425.
Betzig E., Lewis A., Harootunian A., Isaacson M., Kratschmer E. 1986. Near Field Scanning Optical Microscopy (NSOM): Development and Biophysical Applications. Biophys J. 49 : 269–279.
Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S., Bonifacino J.S., Davidson M.W., Lippincott-Schwartz J., Hess H.F. 2006. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313 : 1642–1645.
Bewersdorf J., Egner A., Hell S.W. 2006. 4Pi Microscopy. In: Handbook of biological confocal microscopy. Boston, MA, Springer US, 561–570.
Birk U.J., Baddeley D., Cremer C. 2009. Nanosizing by spatially modulated illumination (SMI) microscopy and applications to the nucleus. Methods Mol. Biol. 464 : 389–401.
Bozhanova N.G., Baranov M.S., Klementieva N.V., Sarkisyan K.S., Gavrikov A. S., Yampolsky I.V., Zagaynova E.V., Lukyanov S.A., Lukyanov K.A., Mishin A.S. 2017. Protein labeling for live cell fluorescence microscopy with a highly photostable renewable signal. Chem. Sci. 8 : 7138–7142.
Bretschneider S., Eggeling C., Hell S.W. 2007. Breaking the diffraction barrier in fluorescence microscopy by optical shelving. Phys. Rev. Lett. 98 : 218103.
Chen F., Tillberg P.W., Boyden E.S. 2015. Expansion microscopy. Science. 347 : 543–548.
de Boer P., Hoogenboom J.P., Giepmans B.N.G. 2015. Correlated light and electron microscopy: Ultrastructure lights up! Nature Methods. 12 : 503–513.
Dertinger T., Colyer R., Vogel R., Heilemann M., Sauer M., Enderlein J., Weiss S. 2012. Superresolution optical fluctuation imaging (SOFI). Adv. Exp. Med. Biol. 733 : 17–21.
Freifeld L., Odstrcil I., Förster D., Ramirez A., Gagnon J.A., Randlett O., Costa E.K., Asano S., Celiker O.T., Gao R., Martin-Alarcon D.A., Reginato P., Dick C., Chen L., Schoppik D., Engert F., Baier H., Boyden E.S. 2017. Expansion microscopy of zebrafish for neuroscience and developmental biology studies. Proc. Natl. Acad. Sci. USA. 114 : E10799–E10808.
Funatsu T., Harada Y., Tokunaga M., Saito K., Yanagida T. 1995. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature. 374 : 555–559.
Geerts H., De Brabander M., Nuydens R., Geuens S., Moeremans M., De Mey J., Hollenbeck P. 1987. Nanovid tracking: A new automatic method for the study of mobility in living cells based on colloidal gold and video microscopy. Biophys J. 52 : 775–782.
Gustafsson M.G.L. 2000. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microscopy (Oxford). 198 : 82–87.
Gustafsson M.G.L. 2005. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA. 102 : 13081–13086.
Gustafsson N., Culley S., Ashdown G., Owen D.M., Pereira P.M., Henriques R. 2016. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat. Commun. 7 : 12471.
Heilemann M., van de Linde S., Schuttpelz M., Kasper R., Seefeldt B., Mukherjee A., Tinnefeld P., Sauer M. 2008. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 47 : 6172–6176.
Heintzmann R., Huser T. 2017. Super-resolution structured illumination microscopy. Chem. Rev. 117: 13890–13908.
Hell S.W., Wichmann J. 1994. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Optics Lett. 19 : 780–782.
Hess S.T., Girirajan T.P., Mason M.D. 2006. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91 : 4258–4272.
Hoopmann P., Punge A., Barysch S.V., Westphal V., Bückers J., Opazo F., Bethani I., Lauterbach M.A., Hell S.W., Rizzoli S.O. 2010. Endosomal sorting of readily releasable synaptic vesicles. Proc. Natl. Acad. Sci. USA. 107 : 19 055–19 060.
Horsington J., Turnbull L., Whitchurch C.B., Newsome T.P. 2012. Sub-viral imaging of vaccinia virus using super-resolution microscopy. J. Virol. Methods. 186 : 132–136.
Klar T.A., Hell S.W. 1999. Subdiffraction resolution in far-field fluorescence microscopy. Optics Lett. 24 : 954–956.
Kwon J., Hwang J., Park J., Han G.R., Han K.Y., Kim S.K. 2015. RESOLFT nanoscopy with photoswitchable organic fluorophores. Sci. Rep. 5 : 17 804.
Legant W.R., Shao L., Grimm J.B., Brown T.A., Milkie D.E., Avants B.B., Lavis L.D., Betzig E. 2016. High density three-dimensional localization microscopy across large volumes. Nature Methods. 13 : 359–365.
Li H., Vaughan J.C. 2018. Switchable fluorophores for single-molecule localization microscopy. Chem. Rev. 118 : 9412–9454.
Lipson S.G., Lipson H., Tannhauser D.S. 1995. Optical physics. N.Y., NY, USA, Cambridge University Press. P. XVIII, 497 p.
Mockl L., Lamb D.C., Brauchle C. 2014. Super-resolved fluorescence microscopy: Nobel Prize in Chemistry 2014 for Eric Betzig, Stefan Hell, and William E. Moerner. Angew Chem. Int. Ed. Engl. 53 : 13972–13977.
Moore D.A., Whatley Z.N., Joshi C.P., Osawa M., Erickson H.P. 2017. Probing for binding regions of the FtsZ protein surface through site-directed insertions: Discovery of fully functional FtsZ-fluorescent proteins. J. Bacteriol. 199 : e00553-16.
Ober R.J., Ram S., Ward E.S. 2004. Localization accuracy in single-molecule microscopy. Biophys. J. 86 : 1185–1200.
Polinovskaya V.S., Vedyaykin A.D., Vishnyakov I.E., Ivanov V.A., Khodorkovskii M.A. 2018. Super-resolution microscopy of Mollicutes cells. J. Phys. Conf. Ser. 1038 : 012029.
Ponomareva E.V., Vishnyakov I.E., Morozova N.E., Polinovskaya V.S., Khodorkovskii M.A., Vedyaykin A.D. 2017. Super-resolution microscopy of living bacterial cells. J. Phys. Conf. Ser. 917 : 042014.
Rust M.J., Bates M., Zhuang X. 2006. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods. 3 : 793–795.
Sahl S.J., Hell S.W., Jakobs S. 2017. Fluorescence nanoscopy in cell biology. Nature Rev. Mol. Cell Biol. 18 : 685–701.
Salapaka S.M., Salapaka M.V. 2008. Scanning probe microscopy. IEEE Cont. Sys. Mag. 28 : 65–83.
Sauer M., Heilemann M. 2017. Single-molecule localization microscopy in Eukaryotes. Chem. Rev. 117 : 7478–7509.
Schermelleh L., Carlton P.M., Haase S., Shao L., Winoto L., Kner P., Burke B., Cardoso M.C., Agard D.A., Gustafsson M.G.L., Leonhardt H., Sedat J.W. 2008. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 320 : 1332–1336.
Schnitzbauer J., Strauss M.T., Schlichthaerle T., Schueder F., Jungmann R. 2017. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 12 : 1198–1228.
Schoen I., Ries J., Klotzsch E., Ewers H., Vogel V. 2011. Binding-activated localization microscopy of DNA structures. Nano Lett. 11 : 4008–4011.
Shcherbakova D.M., Sengupta P., Lippincott-Schwartz J., Verkhusha V.V. 2014. Photocontrollable fluorescent proteins for superresolution imaging. Annual Rev. Biophys. 43 : 303–329.
Shen H., Tauzin L.J., Baiyasi R., Wang W., Moringo N., Shuang B., Landes C.F. 2017. Single particle tracking: From theory to biophysical applications. Chem. Rev. 117 : 7331–7376.
Soderstrom B., Badrutdinov A., Chan H., Skoglund U. 2018. Cell shape-independent FtsZ dynamics in synthetically remodeled bacterial cells. Nat. Commun. 9 : 4323 (2018).
Strauss M.P., Liew A.T., Turnbull L., Whitchurch C.B., Monahan L.G., Harry E.J. 2012. 3D-SIM super resolution microscopy reveals a bead-like arrangement for FtsZ and the division machinery: Implications for triggering cytokinesis. PLoS Biol. 10 : e1001389.
Szczurek A., Klewes L., Xing J., Gourram A., Birk U., Knecht H., Dobrucki J.W., Mai S., Cremer C. 2017. Imaging chromatin nanostructure with binding-activated localization microscopy based on DNA structure fluctuations. Nucl. Acids Res. 45 : e56.
Tsien R.Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67 : 509–544.
Vaughan J.C., Jia S., Zhuang X. 2012. Ultrabright photoactivatable fluorophores created by reductive caging. Nat. Methods. 9 : 1181–1184.
Vedyaykin A.D., Gorbunov V.V., Sabantsev A.V., Polinovskaya V.S., Vishnyakov I.E., Melnikov A.S., Serdobintsev P.Y., Khodorkovskii M.A. 2015. Multi-color localization microscopy of fixed cells as a promising tool to study organization of bacterial cytoskeleton. J. Phys. Conf. Ser. 643 : 012020.
Vedyaykin A.D., Sabantsev A.V., Vishnyakov I.E., Morozova N.E., Polinovskaya V.S., Khodorkovskii M.A. 2016a. 3D super-resolution microscopy of bacterial division machinery. J. Phys. Conf. Ser. 741 : 012066.
Vedyaykin A.D., Vishnyakov I.E., Polinovskaya V.S., Khodorkovskii M.A., Sabantsev A.V. 2016b. New insights into FtsZ rearrangements during the cell division of Escherichia coli from single-molecule localization microscopy of fixed cells. Microbiologyopen. 5 : 378–386.
Дополнительные материалы отсутствуют.
Инструменты
Цитология