

Цитология, 2019, T. 61, № 5, стр. 357-369
Провоспалительные механизмы гибели нейтрофильных гранулоцитов
С. Н. Плескова 1, 2, *, Р. Н. Крюков 1
1 Научно-образовательный центр “Физика твердотельных наноструктур”
Нижегородского государственного университета им. Н.И. Лобачевского
603950 Нижний Новгород, Россия
2 Кафедра Нанотехнологии и биотехнологии Нижегородского государственного технического университета
им. Р.Е. Алексеева
603950 Нижний Новгород, Россия
* E-mail: pleskova@mail.ru
Поступила в редакцию 08.11.2018
После доработки 13.02.2019
Принята к публикации 15.02.2019
Аннотация
Нейтрофильные гранулоциты являются центральным фактором неспецифической резистентности организма. Они вооружены широким набором эффекторов, которые могут быстро мобилизоваться, и обладают выраженными защитными свойствами (способностью продуцировать активные формы кислорода и азота, мобилизовать широкий набор гидролитических ферментов, уничтожать чужеродные агенты с помощью катионных белков, продуцировать цитокины и т.д.). Для них зафиксировано более 10 механизмов клеточной гибели, в том числе уникальный механизм нетоз, т.е. способность образовывать внеклеточные ловушки. Поскольку нейтрофил является центральным звеном эксудативно-деструктивного воспаления, механизм клеточной гибели может напрямую влиять на диалектику воспаления, либо способствуя его разрешению и восстановлению повреждения, либо, напротив, усиливая эффекторный каскад и вызывая развитие флогогенных осложнений. В представленном обзоре обсуждаются механизмы гибели нейтрофильных гранулоцитов, способствующие развитию провоспалительных реакций.
Неспецифическая резистентность играет важную роль в защите организма человека от микроорганизмов (Ярилин, 1999). Это обусловлено тем, что в исходе борьбы между макро- и микроорганизмом важен фактор времени, и выигрывает тот из участников процесса, который действует быстрее – микроорганизм, использующий широкий набор факторов патогенности, или макроорганизм, реализующий физиологические защитные реакции. В этом случае флогогенный потенциал является важной составляющей, которая сдерживает агрессию патогенов, давая возможность сформироваться полному комплексу более медленных, но более сильных и прицельных механизмов адаптивного иммунитета. Главным “дирижером” флогогенных реакций является нейтрофильный гранулоцит (НГ), который принимает активное участие в экссудативно-деструктивном воспалении (Маянский, 2006). Воспаление – это защитно-приспособительная реакция организма, направленная на ликвидацию повреждающего (флогогенного) агента и восстановление поврежденной ткани (Белоцкий, Авталион, 2008). Это сосудисто-мезенхимная циклическая реакция со сменой клеточных коопераций и трансформацией клеток и сосудов, которая в норме должна заканчиваться восстановлением повреждения (Маянский, 2006). В ходе воспалительного ответа происходит активация НГ (праймирование), но у людей с нейтропенией или генетическими нарушениями функций НГ наблюдается неконтролируемое воспаление или развитие жизненно-опасных инфекций (Kannengiesser et al., 2008; Moutsopoulos et al., 2014).
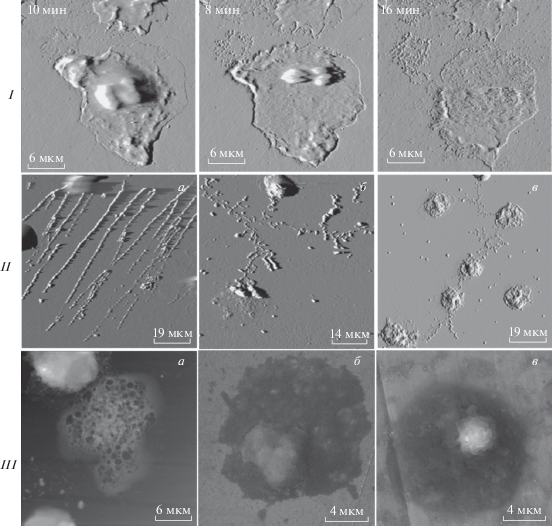
Однако результатом активации НГ не всегда является восстановление гомеостаза и разрешение инфекционного процесса. В случае выраженного праймирования клеток происходит высвобождение медиаторов воспаления, а ткани и органы повреждаются вследствие развития воспалительных процессов (Odobasic et al., 2016). Интересной особенностью является то, что НГ способен регулировать степень развития воспаления даже после своей гибели. Разные механизмы смерти НГ способны вносить разный вклад в развитие, регуляцию и разрешение воспаления (Pleskova, Mikheeva, 2018). В настоящее время Номенклатурный комитет по клеточной гибели (Nomenclature Committee on Cell Death) официально выделяет 12 типов смерти клеток на основании морфологических, биохимических, функциональных, генетических и фармакологических особенностей (Galluzzi et al., 2018). Рассмотрим вклад разных механизмов гибели НГ в реализацию и усиление воспалительных реакций. Морфологические особенности некроза, нетоза (NET – neutrophil extracellular traps) и аутофагии представлены на рис. 1, полученном авторами методами атомно-силовой и сканирующей электронной микроскопии.
Рис. 1.
Основные морфологические типы гибели нейтрофильного гранулоцита (НГ). I – Динамика некротической гибели НГ в режиме реального времени; атомно-силовая микроскопия (АСМ). II – Нетоз НГ (АСМ): а – живой образец; б, в – фиксация метанолом. III – Аутофагия НГ: а – АСМ, фиксация метанолом; б, в – сканирующая электронная микроскопия (2 препарата), фиксация 2.5%-ным глутаровым альдегидом.
НЕКРОЗ – ПРОВОСПАЛИТЕЛЬНЫЙ МЕХАНИЗМ КЛЕТОЧНОЙ ГИБЕЛИ
Некрозом называется гибель НГ, возникающая в результате повреждения плазмолеммы с выходом клеточного содержимого в окружающее пространство (Benarafa, Simon, 2017). Он возникает в результате неадекватных внешних механических, физических, химических, осмотических воздействий и сопровождается набуханием органелл, грубым разрушением плазмолеммы и лизисом клеток (Nikoletopoulou et al., 2013). Клетки, погибающие по механизму некроза, активно вырабатывают такие факторы, как амфотерин и фактор роста гератомы (HDGF), вызывая воспалительный ответ. Оба белка способны связываться с криопирином – NLRP3 (NOD-like receptor family pyrin domain 3) – коровым белком инфламмасомы, результатом чего является ее активация. Активированная инфламмасома, в свою очередь, расщепляет про-IL1β, способствуя массовому высвобождению провоспалительного цитокина IL1β нейтрофилом. HDGF способен также запускать процесс тканевой репарации (Zong, Thompson, 2006). Белком NLRP3 инфламмасома активируется главным образом с помощью АТФ, продуцируемой митохондриями (Iyer et al., 2009).
Помимо внутриклеточной регуляции стратегия внеклеточной атаки микроорганизмов у НГ заложена и в механизме экстрацеллюлярной секреции: выбрасывая многочисленные протеазы, клетка “разрыхляет” базальную мембрану эндотелия, обеспечивает расщепление комплемента, что способствует образованию ряда медиаторов, регулирующих воспаление (Серов, Пауков, 1995). Когда механизма внеклеточной секреции оказывается недостаточно для экстренной мобилизации всех эффекторов в зону воспаления, НГ погибают по механизму некроза. При этом экстрацецеллюлярный выброс медиаторов воспаления возрастает кратно. Некротическая гибель НГ не исчерпывается массивным выделением протеолитических ферментов. При расщеплении фосфолипидов образуются такие медиаторы воспаления, как тромбоксаны и простагландины. При окислительном метаболизме арахидоновой кислоты продуцируются лейкотриены. Наращивание эффекторного каскада, связанного с некротической гибелью НГ, и расширение спектра мишеней может продолжаться до удаления первичных (микробных) и вторичных (эффекторов организма человека) флогогенов, либо до возникновения состояния гомеостатического равновесия (Маянский, 2006).
Массовая некротическая гибель НГ в кровяном русле не совместима с жизнью макроорганизма. Однако в случае воспаления, ограниченного определенной областью, она играет положительную роль, поскольку здоровые фагоциты не успевают удалять все инфицированные и апоптозные клетки, и такая несбалансированная ситуация способствует персистенции инфекции (Segawa, Nagata, 2015). Некротическая гибель НГ приводит к массовому выбросу дистресс-ассоциированных молекулярных паттернов (DAMP) в окружающие ткани и обострению процесса воспаления, что оказывает выраженное позитивное действие, препятствующее хронизации воспаления. Некроз может являться и следствием эндогенных процессов, реализующихся после запуска сигнальных путей и приводящих в конечном итоге к мембранолизу. Такой вариант клеточной гибели называется некроптозом (Wallach et al., 2016).
Некроптоз (онкоз) – это форма клеточной гибели, характеризующаяся набуханием клеток и их органелл с последующим увеличением проницаемости клеточных мембран. Инициатором некроптоза могут выступать некоторые цитокины (например, фактор некроза опухоли (TNF)), компоненты клеточной стенки бактерий, например, липополисахарид (ЛПС). Программа некроптоза обычно запускается при ингибировании или инактивации каспаз (в частности, каспазы-8). Процесс инициируется специфическими протеинкиназами (в основном, протеинкиназой 3 (Benarafa, Simon, 2017). Кроме них некроптоз зависит от доменов киназ смешанного происхождения, т.е. псевдокиназы MLKL (mixed lineage kinase domaine like protein), которая обладает порообразующей активностью (Sun et al., 2012): гиперэкспрессия активированной MLKL или N-концевого протеолитического фрагмента газдермина D может вызывать некротическую гибель клетки (Wallach et al., 2016). Активация специфических ферментов может происходить и в результате реализации клеткой нормальных физиологических защитных функций, в частности, некроптоз наблюдается после фагоцитоза Staphylococcus aureus (Wang et al., 2016). В некоторых случаях отмечена диалектическая преемственность развития воспалительной реакции НГ. В частности, изначально возникающий некроз нефронов сопровождается высвобождением DAMP, которые, в свою очередь, запускают выброс цитокинов и хемокинов, которые рекрутируют НГ в зону первоначального воспаления. НГ, в свою очередь, погибают по механизмам некроптоза и нетоза, что сопровождается прямым цитотоксическим воздействием на нефроны и развитием некровоспаления почек (Mulay et al., 2016).
Ферроптоз – это форма регулируемой клеточной гибели, которая сопровождается мощным железо-зависимым перекисным окислением липидов (ПОЛ) (Yang, Stockwell, 2016; Galluzzi et al., 2018). При ферроптозе преимущественно наблюдаются изменения митохондрий, сопровождающиеся их сжатием, увеличением электронной плотности, уменьшением или полным исчезновением крист и разрывом внешней мембраны (Vanden Berghe et al., 2014). Ферроптоз инициируется эрастином (ингибитором цистеин-глутаматового антипортера), RSL3 (ингибитором глутатионпероксидазы 4) (Dolma et al., 2003; Yang, Stockwell, 2008), и FIN56 (промотором разрушения глутатионпероксидазы 4) (Shimada et al., 2016) и подавляется ферростатинами (Dixon et al., 2012), липрокстатинами (Friedmann Angeli et al., 2014), витамином Е, коэнзимом Q10 и их аналогами, т.е. всеми антиоксидантами, инактивирующими активные формы кислорода (АФК) (Abeysinghe et al., 1996; Seiler et al., 2008; Kagan et al., 2017; Zilka et al., 2017). Ферроптоз сопровождается последовательным высвобождением DAMP, что способствует расширению зоны воспаления (Linkermann et al., 2014). Кроме того, обнаружено, что ингибирование глутатионпероксидазы 4, которое происходит при ферроптозе, приводит к патологическим состояниям, включая острую почечную или печеночную недостаточность (Carlson et al., 2016; Doll et al., 2017), нейродегенеративные заболевания (Hambright et al., 2017) и повышенную чувствительность к инфекциям (Matsushita et al., 2015). Несмотря на то, что НГ имеют ограниченное количество митохондрий, при некоторых условиях (внешний стресс, или разрушение ДНК) реализуется этот механизм клеточной гибели.
Пироптоз – вид регулируемой клеточной гибели, запускаемый внутри- или внеклеточным изменением гомеостаза и связанный с механизмами врожденной неспецифической резистентности. Морфологически сопровождается особой формой конденсации хроматина, набуханием клетки с последующей пермеабилизацией цитоплазматической мембраны (Jorgensen, Miao, 2015). Это каспазозависимый (у человека – каспазы 1, 3; у мышей – каспазы 11, 4, 5) (Aachoui et al., 2013; Wang et al., 2017) механизм клеточной гибели, возникающий при внутриклеточной инвазии и сопровождающийся выделением клеткой интерлейкинов ИЛ-1β и ИЛ-18 (не всегда) и развитием выраженного воспаления (Brough, Rothwell, 2007; Franchi et al., 2009). Пироптоз участвует в патогенезе летального септического шока, инициированного ЛПС (Aziz et al., 2014; Vanden Berghe et al., 2014). Активация провоспалительных каспаз выше определенного специфического порога приводит к каталитическому протеолизу газдермина D и гибели клеток по механизму пироптоза (Kayagaki et al., 2015; Shi et al., 2015). Протеолизированный газдермин D приобретает способность переноситься в наружный слой цитоплазматической мембраны, где селективно связывается с кардиолипином и олигомеризуется с образованием пор (с внутренним диаметром 10–14 нм). Эти поры отвечают за проницаемость мембраны (Ding et al., 2016). Одновременно с протеолизом газдермина D атакуются каналы паннексин 1 (PANX1), что приводит к высвобождению АТФ во внеклеточное пространство и активации пуринергического рецептора P2X 7, что еще больше усиливает диспропорцию ионных градиентов и воспалительную сигнализацию (Yang et al., 2015). Еще одним свидетельством провоспалительной активности пироптоза является то, что каспаза 11 может регулироваться каскадом комплемента, зависящим от карбоксипептидазы В1 при активации TLR4 (toll-like receptor 4) и рецептора 1 интерферона α (IFα) (Napier et al., 2016).
Несмотря на разные молекулярные маркеры инициации и развития некроптоза, ферроптоза или пирроптоза, морфологически невозможно различить клетки, погибшие в результате некроза или одного из этих программируемых вариантов клеточной гибели. Вторичный некроз НГ может наблюдаться in vitro, когда изолированные НГ подвергаются спонтанному апоптозу, а через некоторое время наблюдается пермеабилизация их цитоплазматической мембраны и высвобождение цитоплазматического содержимого.
НГ чувствительны к регулируемой клеточной гибели, возникающей под действием лизоосмотических агентов, которые разрушают гранулярные, особенно лизосомальные мембраны, вызывая апоптоз или другие виды программируемой клеточной гибели (Aits, Jäättelä, 2013; Repnik et al., 2014). В некоторых случаях программа некроптоза важна для развития адекватного воспаления, когда речь идет о внутриклеточных патогенах (например, Mycobacterium tuberculosis, Shigella flexneri). В этих случаях выход патогенов, научившихся выживать, размножаться и питаться за счет резервов клеток, во внеклеточное пространство равносилен встрече микроорганизмов с гуморальными и другими защитными механизмами, т.е. тактика внутриклеточного паразитирования оказывается сломанной (Wallach et al., 2016).
Выявление маркеров регулируемого некроза важно для создания терапевтических препаратов способных контролировать и снижать силу воспалительного ответа.
ДУАЛИЗМ НЕТОЗА В РАЗВИТИИ ВОСПАЛИТЕЛЬНЫХ РЕАКЦИЙ
Нетоз – способность нейтрофилов образовывать сетеподобные структуры, сформированные нитями ДНК и внутриклеточными ферментами. Ранее его обнаруживали только у НГ и считали исключительно одним из вариантов программируемой клеточной гибели. Сейчас известно, что внеклеточные ловушки способны выбрасывать эозинофилы и макрофаги, а сам процесс не всегда заканчивается гибелью клеток. Как правило, в русскоязычной литературе для обозначения данного варианта клеточной гибели используют термин нетоз. Это относительно недавно открытый механизм гибели НГ (Brinkmann et al., 2004). Он может вызываться бактериями (Fuchs et al., 2007; Grinberg et al., 2008), грибами (Urban et al., 2006; Bruns et al., 2010), вирусами (Toussaint et al., 2017), простейшими (Ventura-Juarez et al., 2016), АФК (Fuchs et al., 2007) или многочисленными провоспалительными стимулами, например, ИЛ-8 (IL) (Kaplan, Radic, 2012). Нетоз представляет собой сетеподобные структуры, сформированные ядерным хроматином, которые выбрасываются активированными НГ (Araz´na et al., 2014). Каркасом сетеподобной структуры являются нити ДНК различной толщины. Более крупные глобулярные домены на сетях представляют собой ассоциированые с ДНК цитрулированные гистоны и ферменты, прежде всего, нейтрофильную эластазу (НЭ) и миелопероксидазу (МПО). Полностью гидратированные сети имеют вид облака и могут занимать объем в 10–15 раз больше того пространства, из которого они происходят, т.е. они могут занимать, например, всю легочную альвеолу, однако в суспензии НГ образуют внеклеточные ловушки слабо, видимо, чтобы предотвратить формирование тромбов в сосудистом русле (Brinkmann, Zychlinsky, 2012).
Формирование NET требует адгезии НГ на субстрате за счет интегриновых рецепторов MAC-1 (Neeli et al., 2008). Для возникновения внеклеточных сетей требуется целая цепь молекулярных событий: формирование АФК, миграция НЭ и МПО из гранул в ядро, ферментативное преобразование гистонов и разрыв клетки (Brinkmann, Zychlinsky, 2012). НЭ высвобождается из гранул по неизвестному механизму и проникает в ядро, где разрушает линкер гистона Н1 и преобразует коровые гистоны, делая их чувствительными к воздействию других групп ферментов (Papayannopoulos et al., 2010). Под влиянием НЭ и МПО разделение на эухроматин и гетерохроматин теряется, и нуклеоплазма становится гомогенной. Гистоны претерпевают дальнейшие модификации, хроматин деконденсируется. Фермент PAD4 (пептидиларгининдеимидаза 4) катализирует дезаминирование аргенина в цитруллин в трех коровых гистонах, что ведет к ослаблению их связывания с ДНК. Таким образом, в NET гистоны цитруллированы. Затем ядерная мембрана разрушается, хроматин распределяется внутри клетки и смешивается с антимикробными факторами гранул. В конечном итоге, цитоплазматическая мембрана разрушается, и высвобождаются внеклеточные сети НГ. Все компоненты, образующие NET – и ДНК, и специфически модифицированные гистоны, и образующиеся при формировании сетей АФК, и ассоциированные с внеклеточными ловушками ферменты НГ (около 30) – обладают выраженной антимикробной активностью (Brinkmann, Zychlinsky, 2012). Возможно, есть тесная связь между нетозом и аутофагией, поскольку замечено, что при стимуляции НГ форболмеристатацетатом клетка перед образованием NET формирует крупные вакуоли, сходные с аутофагосомами. Такая цепь развития событий достаточно продолжительна во времени и занимает 3–4 ч с максимумом через 2 ч (Pleskova et al., 2018). Однако был описан и другой, быстрый путь развития NET при стимуляции НГ S. aureus. Его отличительными чертами, в сравнении с классическим нетозом, были следующие: 1) быстрое развитие в течение 5–60 мин; 2) кислородная независимость; 3) отсутствие на ранних стадиях лизиса НГ и разрушения их плазматических мембран; 4) ограниченная протеолитическая активность, при сохраненной способности уничтожать S. aureus; 5) везикулярное высвобождение ДНК; 6) особая морфология в виде “бусин на нити” (Pilsczek et al., 2010).
Основная функция образования внеклеточных сетей – сдерживание инфекции и регуляция воспалительного процесса. Они способны улавливать любые типы патогенов, даже те, которые не могут быть фагоцитированы из-за слишком больших размеров (Lu et al., 2012). Попадание микроорганизмов в сети предотвращает распространение инфекции и создает локальную высокую концентрацию бактерицидных факторов (Zawrotniak, Rapala-Kozik, 2013). В реализации бактерицидного эффекта принимают участие белки и антимикробные пептиды гранул и цитоплазмы. Помимо НЭ, МПО и гистонов в уничтожении микроорганизмов участвуют катепсин G, протеиназа 3, лактоферрин, кальпротектин, дефензины, пептид LL37 (Urban et al., 2009). LL37 и гистоны дезинтегрируют мембрану патогенных клеток, снижая их жизнеспособность (Cho et al., 2009; Méndez-Samperio, 2010). Активность МПО важна для уничтожения S. aureus (Parker, Winterbourn, 2013), а для подавления фунгального роста используются такие хелатирующие протеины НГ, как лактоферрин и кальгранулин (Farnaud, Evans, 2003). Выяснилось, что НГ, образующие сети, могут достаточно быстро двигаться in vivo за счет псевдоподий, увеличивая площадь распространения сетей, при этом начало программы NET не отменяет возможности фагоцитоза (Yipp et al., 2012). NET удаляются после разрешения воспаления, а основным ферментом, который их расщепляет является ДНКаза I. Ферментативно расщепленные фрагменты сетей впоследствии удаляются пришедшими в зону воспаления НГ и макрофагами. ДНКаза являться также одним из факторов патогенности бактерий, которые благодаря ей расщепляют ловушки и уходят от этого бактерицидного механизма защиты (Welty et al., 2005; Buchanan et al., 2006). Другими вариантами защиты от нетоза со стороны бактерий являются смена заряда или формирование капсулы (Wartha et al., 2007).
Образование нейтрофильных ловушек в аномально больших количествах или их медленный клиренс приводит к повреждению тканей и нарушению функций органов. В частности, показана патогенетическая роль нетоза в развитии аллергической астмы (Dworski et al., 2011), кистозного фиброза (Manzenreiter et al., 2011), хронической обструктивной болезни легких (Obermayer et al., 2014), острого повреждения легких. Исследование цитотоксичности нетоза по отношению к эндотелиальным и эпителиальным клеткам человека выявило, что основными факторами, разрушающими ткани, являются гистоны, МПО, НЭ и катепсин G, а легкие являются основной мишенью, поскольку НГ находятся в них дольше, чем в других органах (Kolaczkowska, Kubes, 2013). С другой стороны, невозможность образовывать нетозные сети, например, при мутации хотя бы в одной из субъединиц НАДФН-оксидазного комплекса, приводит к хронической гранулематозной болезни. Такие пациенты страдают от угрожающих жизни рецидивирующих инфекций (Bianchi et al., 2009). Стимуляция НГ пациентов с хронической гранулематозной болезнью пероксидом водорода восстанавливала способность клеток производить NET (Fuchs et al., 2007). У пациентов с врожденной делецией или мутацией в гене, кодирующем МПО, также отмечается невозможность к возникновению NET. Иммунодефициты такого рода сопровождаются рецидивирующими кандидозами, а при полном дефиците МПО возникают серьезные инфекционные и воспалительные осложнения (Metzler et al., 2011).
Таким образом, любая дисфункция в работе нетоза является лишь одним из звеньев в общей патогенетической картине развивающихся воспалительных реакций. С этой точки зрения нетоз нужно рассматривать шире, чем просто механизм клеточной гибели, препятствующий распространению инфекции в организме. Выявленные факты миграции НГ при образовании NET, их активное участие в деструкции эпителиальных и эндотелиальных тканей расширяет представления о регуляции воспалительных процессов. В частности, известно, что собственная (эндогенная) ДНК и РНК воспринимаются организмом как DAMP (Brinkmann, Zychlinsky, 2012).
Экстрацеллюлярная ДНК активирует рецептор TLR-9, который представлен на моноцитах и дендритных клетках. В частности, было показано, что ДНК в комплексе с антимикробными факторами LL37 или амфотерином формирует стабильную структуру, стимулирующую дендритные клетки (Lande et al., 2007). NET способны и напрямую воздействовать на рецепторы TLR-9 дендритных клеток (Lande et al., 2011; Garcia-Romo et al., 2011), а гистоны, содержащиеся во внеклеточных сетях активируют рецепторы TLR-2 и TLR-4 (Semeraro et al., 2011). Кроме того, NET могут праймировать Т-клетки, хотя в настоящее время не установлены рецепторы, через которые происходит эта активация (Tillack et al., 2012).
Здесь уместно провести аналогию с сетью цитокинов. Возможно, формирование NET для клеток является такой же попыткой создать сеть, способствующую системной реакции организма, как для гуморальной системы – создание сети цитокинов, только в случае клеток понятие “сеть” имеет вполне прямой, а не переносный смысл. В настоящее время неоспоримым является факт, что образующиеся внеклеточные сети могут оказывать разнонаправленное модулирующее действие на реализацию воспаления.
Такую же разнонаправленную регуляцию воспаления вызывает коагуляция: умеренная коагуляция предотвращает кровопотерю и способствует локализации микробной инвазии. Вместе с тем, избыточное тромбообразование сопровождается блокадой перфузии и ишемией тканей и органов. Активированный эндотелий помимо высвобождения фактора Виллебранда, необходимого для рекрутирования и адгезии тромбоцитов, продуцирует компоненты, которые при контакте с НГ стимулируют нетоз, что, в свою очередь, усиливает повреждение эндотелия (Gupta et al., 2010). Было показано, что использование ДНКазы I, расщепляющей сети, предотвращает каскад событий, приводящих к тромбозу (Brill et al., 2012). Тромбоз может быть инициирован высвобождением тканевого фактора и цитокинов, выделяемых клетками в процессе воспаления. Было выявлено, что НГ во время формирования NET могут продуцировать тканевой фактор и выделять его в вены (Von Brühl et al., 2012), таким образом, была установлена связь между тромбозом и воспалением (Zawrotniak, Rapala-Kozik, 2013). ДНК и гистоны связывают фактор XII, стимулируя образование фибрина (Von Brühl et al., 2012). Кроме того, НЭ, входящая в состав NET, регулирует коагуляцию благодаря расщеплению ингибиторов тромбообразования и повышению активности фактора Xa (Steppich et al., 2008). NET могут напрямую захватывать и активировать тромбоциты (Ma, Kubes, 2008). Волокна сети связывают тромбоциты и поддерживают их агрегацию (Fuchs et al., 2011).
Первый этап взаимодействия тромбоцитов с ловушками основан на электростатических взаимодействиях между гистонами сети и фосфолипидами или карбогидратами тромбоцитов, а также благодаря связыванию тромбоцитов с TLR (Semeraro et al., 2011). По всей вероятности в процессе также задействованы молекулы адгезии для фактора Виллебранда, фибронектина и фибриногена (Fuchs et al., 2010). Активированные тромбоциты вызывают дальнейшее высвобождение NET, что увеличивает проницаемость эндотелия (Brill et al., 2012). Таким образом, НГ оказываются вовлечены в усиливающуюся петлю обратной связи при тромбообразовании. Синергетический эффект различных функций сетей, т.е. противомикробный и протромбический крайне важен для сохранения гомеостаза при инфекционных заболеваниях, особенно при сепсисе (Zawrotniak, Rapala-Kozik, 2013). В частности, у мышей, инфицированных E. coli, бактерии эффективно изолируются в микрососудах печени в случае нормального функционирования NET, и гораздо хуже в случае блокады образования сетей антителами к ДНК (Geddings, Mackman, 2014).
Таким образом, нетоз играет двойственную роль в процессе воспаления. С одной стороны, он существенно ограничивает степень распространения инфекционных агентов, препятствуя реализации ими патогенного потенциала, и, таким образом, способствуют разрешению воспаления. С другой стороны, NET вовлекаются в несколько цепей самоусиливающихся реакций, и при их гиперактивации возможно развитие клинически значимого воспаления, выходящего за рамки приспособительных гомеостатических реакций.
АУТОФАГИЯ КАК ПРОЦЕСС ПОДДЕРЖАНИЯ ГОМЕОСТАЗА НЕЙТРОФИЛА И МЕХАНИЗМ ГИБЕЛИ, РЕГУЛИРУЮЩИЙ ВОСПАЛЕНИЕ
Аутофагия известна как процесс рециркуляции содержимого цитоплазмы и деградации собственных компонентов клетки (Iba et al., 2013). Она необходима для доставки анаболитов к нуждающимся в них клеточным компартментам, для удаления разрушенных органелл и выживания клеток в условиях стресса (например, истощения питательных веществ), а также для защиты от внутриклеточных патогенов (Klionsky, Emr, 2000; Mizushima, Levine, 2010; Rabinowitz, White, 2010; Tanida, 2011).
В норме аутофагия характеризуется образованием крупных везикул – аутофагосом. Они содержат цитозоль и органеллы, сливаются с лизосомами и деградируют без повреждения клеток (Klionsky, 2004). Захват цитоплазмы начинается с образования уникальной “преаутофагосомной структуры”, из которой вначале формируется мембранный “фагофор” размером 0.3–1.0 мкм, а затем полноценная аутофагосома (Suzuki et al., 2001). Белки цитоплазмы, измененные в результате стресса или недостатка энергетического обеспечения, поврежденные митохондрии, избыточный эндоплазматический ретикулум перемещаются к мембранам благодаря образованию комплексов с белками ULK1/2, Atg13, Atg101, FIP-200. На мембранах органелл эти белки формируют комплекс I, включающий дополнительно белки Vps34, Beclin I, Vps15, Atg14L. Вокруг комплекса I образуется внутренняя мембрана фагофора. Фагофоры формируются случайным образом, а аутофагосомы двигаются целенаправленно вдоль микротрубочек в направлении центра организации микротрубочек, где сконцентрированы лизосомы. Затем происходит образование аутолизосомы, либо напрямую через слияние аутофагосомы с лизосомой, либо последовательно через стадии слияния сначала с эндосомой с образованием амфисомы, а затем с лизосомой (Eskelinen, 2005). Формирование аутофагосомы требует участия LC3 II, образующегося в результате липолизации фосфатидилэтаноламином цитозольного белка LC3 и комплекса белков Atg5–Atg12/Atg16L1. Последующее созревание аутофагосомы в аутофаголизосому осуществляется через слияние с лизосомами за счет комплекса белков II, включающего Vps34, Beclin 1, UVRAG (Levine et al., 2011; Liu et al., 2013).
Фагофор может образовываться de novo или альтернативно, с использованием существующих цитоплазматических мембран. Во втором случае для его образования может быть использована любая из клеточных мембран; возможна также ситуация, при которой несколько типов клеточных мембран участвует в образовании фагофора. Окрашивание фагофоров и аутофагосом осмием показало, что они богаты липидами с высоким содержанием ненасыщенных жирных кислот и практически не содержат белков (Reunanen et al., 1985). Источником мембран для аутофагосом могут служить эндоплазматический ретикулум, в особенности омегасома (Axe et al., 2008), аппарат Гольджи (Mari et al., 2010; Ohashi, Munro, 2010), внешняя мембрана митохондрий (Hailey et al., 2010) и плазматическая мембрана (Ravikumar et al., 2010).
Образование аутофагосом требует участия белков SNARE (Nair et al., 2011). Содержимое аутофагосом соответствует содержимому цитоплазмы, в частности ферменты присутствуют в них в тех же концентрациях. Выделяют три варианта аутофагии: макроаутофагия (основной тип), микроаутофагия (когда захват содержимого цитоплазмы осуществляется путем инвагинации мембраны лизосом) и шаперон-зависимая аутофагия (когда доставка цитоплазматического материала в лизосомы осуществляется с помощью белков-шаперонов) (Mostowy, Cossart, 2012; Salminen et al., 2012; Tang et al., 2012). Особенностью шаперон-зависимой аутофагии является отсутствие серьезной реорганизации лизосомальной мембраны и необязательность формирования везикул. При этом варианте аутофагии происходит направленный транспорт частично денатурированных белков из цитоплазмы сквозь мембрану лизосомы в ее полость, где они окончательно расщепляются. Процесс происходит при участии цитоплазматических белков-шаперонов семейства белков теплового шока Hsp-70, вспомогательных белков и LAMP-2 (лизосом-ассоциированного мембранного белка типа 2A) (Levine et al., 2011; Kuballa et al., 2012; Randow et al., 2013).
Регуляция аутофагии НГ осуществляется под действием АФК, TLR и цитокинов (в частности, TNF-α и IFNAR1) (Tallóczy et al., 2002; Gutierrez et al., 2004; Djavaheri-Mergny et al., 2006). Активация рецепторов TLR не является результатом усиленного респираторного взрыва, однако при ингибировании апоптоза возможна стимуляция аутофагии (Hayashi et al., 2003; François et al., 2005). Учитывая регуляторную роль апоптоза в процессе воспаления, можно полагать, что индукция аутофагии у выживших НГ усиливает воспалительные реакции за счет задержки клеточной гибели и является существенной частью патогенеза сепсиса из-за задержки апоптоза, приводящей к повреждению тканей (Iba et al., 2013). И наоборот, было показано, что для реализации нетоза обязательно присутствие обоих процессов: образование АФК и аутофагия, и блокада хотя бы одного из них вызывает гибель НГ по механизму апоптоза (Remijsen et al., 2011).
Вызвать аутофагию способны вирусы кори, аденовирусы B и D, вирус 6 герпеса человека, нейссерии (Neisseria) и стрептококки группы А через рецепторы CD46 и TLR (Meiffren et al., 2010; Mitroulis et al., 2010). Инфицированные НГ, в свою очередь, выделяют набор цитокинов, стимулирующих аутофагию (Harris, 2011). Индукция аутофагии у НГ при инфекционном процессе является одним из решающих процессов врожденного иммунитета (Choi, Ryter, 2011), поскольку эффективность поражения патогена НГ и при дегрануляции, и при фагоцитозе зависит от своевременной активации у НГ процесса аутофагии (Chargui et al., 2012). Некоторые бактерии в ходе реализации классического фагоцитоза способны выходить из фагосомы в цитоплазму и, тем самым, инициировать процесс аутофагии (Yano, Kurata, 2008). В этом случае свободные бактерии в цитоплазме убиквитинируются, затем эти патоген-ассоциированные молекулярные паттерны (PAMP) распознаются системой внутриклеточного аутофагосомного контроля, которая, привлекая факторы SQSTM1 (р62) и LC3 (легкая цепь 3), и запускает процесс аутофагии (Yoshikawa et al., 2009). Такая система инициации аутофагии вследствие проникновения в цитоплазму микроорганизмов и их продуктов называется клеточной автономной защитной системой (cell-autonomous defense system). Однако некоторые PAMP (особенно у бактерий) эволюционировали так, чтобы уметь блокировать аутофагию (Gutierrez et al., 2004; Deretic, Levine, 2014; Lapaquette et al., 2011; Chargui et al., 2012), а дефекты этого механизма клеточной гибели могут вызвать воспалительные заболевания (Chargui, El May, 2014).
Аутофагия является бинарным процессом и, в зависимости от условий и характера воздействия, она может содействовать как гибели, так и выживанию клеток. Поэтому при инфекционных процессах аутофагия может способствовать как прогрессированию заболевания, так и его предотвращению (Mizushima et al., 2008; Gundara et al., 2012). Разделение цитоплазмы клетки на отдельные, ограниченные мембранами органеллы (компартментализация) предполагает наличие в каждом из них своего набора рецепторов, распознающих чужеродные PAMP и измененные собственные DAMP. Это создает многоступенчатую систему защиты от патогенов, проникших внутрь клетки (Potapnev, 2014).
Аутофагия является механизмом выживания в условиях стресса, однако при воздействии стимула, превышающего компенсаторные возможности клетки, НГ гибнет (Walsh, Edinger, 2010; Green, 2011; Rubinsztein et al., 2011; Galluzzi et al., 2012; Zelenay, Reise Sousa, 2013). Морфологические особенности аутофагического варианта клеточной гибели включают вакуолизацию, деградацию цитоплазматического содержимого, а конденсации хроматина в этом случае не происходит. НГ, демонстрирующие наиболее выраженную вакуолизацию, как правило, быстро погибают по механизму аутофагии. В этом случае их гибель зависит от активности протеинкиназы (и ее рецептора) и протеиназ семейства папаина, а каспазы в процессе не задействованы (Mihalache et al., 2011). Как и в случае апоптоза, клетка, подвергающаяся аутофагии, может быть интернализирована резидентными макрофагами. Только с этой точки зрения аутофагия может рассматриваться как невоспалительный вариант клеточной гибели (Labbé, Saleh, 2008).
ЗАКЛЮЧЕНИЕ
Таким образом, механизм клеточной гибели НГ может существенным образом влиять на развитие воспалительных реакций: только некроз и его программируемые варианты (некроптоз, ферроптоз, пироптоз) однозначно усиливают флогогенные реакции, тогда как нетоз и аутофагия амбивалентны. Подавляющее большинство механизмов клеточной гибели, обладающих провоспалительным потенциалом и вызывающих выраженные флогогенные реакции, относится к регулируемым. Применяя терапевтические средства, способные инактивировать или перенаправить каскадные реакции воспаления, можно существенно уменьшить масштаб зоны поражения, либо наоборот, в случае недостаточной состоятельности воспалительного процесса и высокой вероятности хронизации, регулируемым образом усилить воспаление.
Список литературы
Белоцкий С.М., Авталион Р.Р. 2008. Воспаление. Мобилизация клеток и клинические эффекты. М.: БИНОМ. 240 с. (Belotsky S.M., Avtalion R.R. 2008. Inflammation. Cell mobilization and clinical effects. M.: BINOM. 240 р.)
Маянский А.Н. 2006. Патогенетическая микробиология. Нижний Новгород: Нижегородская гос. мед. академия. 520 с. (Mayansky A.N. 2006. Pathogenic microbiology. Nizhny Novgorod: Medical Academy 520 p.)
Серов В.В., Пауков В.С. 1995. Воспаление. Руководство для врачей. М.: Медицина. 650 с. (Serov V.V., Paukov V.S. 1995. Inflammation. A guide for doctors. M.: Medicine. 650 р.)
Ярилин А.А. 1999. Основы иммунологии. М.: Медицина. 608 с. (Yarilin A.A. 1999. Fundamentals of Immunology. M.: Medicine. 608 р.)
Aachoui Y., Sagulenko V., Miao E.A., Stacey K.J. 2013. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 16 : 319–326. https://doi.org/10.1016/j.mib.2013.04.004
Abeysinghe R.D., Roberts P.J., Cooper C.E., MacLean K.H., Hider R.C., Porter J.B. 1996. The environment of the lipoxygenase iron binding site explored with novel hydroxypyridinone iron chelators. J. Biol. Chem. 271 : 7965–7972. https://doi.org/10.1074/jbc.271.14.7965
Aits S., Jäättelä M. 2013. Lysosomal cell death at a glance. J. Cell Sci. 126 : 1905–1912. https://doi.org/10.1242/jcs.091181
Araz’na M., Pruchniak M.P., Demkow U. 2014. Reactive oxygen species, granulocytes, and NETosis. Advs. Exp. Med. Biol. Neurosci. Respiration. https://doi.org/10.1007/5584_2014_12
Axe L., Walker S.A., Manifava M., Chandra P., Roderick H.L., Habermann A., Griffiths G., Ktistakis N.T. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182 : 685–701. https://doi.org/10.1083/jcb.200803137
Aziz M., Jacob A., Wang P. 2014. Revisiting caspases in sepsis. Cell Death Dis. 5 : e1526. https://doi.org/10.1038/cddis.2014.488
Benarafa C., Simon H.-U. 2017. Role of granule proteases in the life and death of neutrophils. Biochem. Biophys. Res. Commun. 482 : 473–481. https://doi.org/10.1016/j.bbrc.2016.11.086
Bianchi M., Hakkim A., Brinkmann V., Siler U., Seger R.A., Zychlinsky A., Reichenbach J. 2009. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 114 : 2619–2622. https://doi.org/10.1182/blood-2009-05-221606
Brill A., Fuchs T.A., Savchenko A.S., Thomas G.M., Martinod K., De Meyer S.F., Bhandari A.A., Wagner D.D. 2012. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thrombosis Haemostasis. 10 : 136–144. doi https://doi.org/10.1111/j.1538-7836.2011.04544.x
Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D.S., Weinrauch Y., Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science. 303 : 1532–1535. https://doi.org/10.1126/science.1092385
Brinkmann V., Zychlinsky A. 2012. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 198 : 773–783. https://doi.org/10.1083/jcb.201203170
Brough D., Rothwell N.J. 2007. Caspase-1-dependent processing of prointerleukin-1beta is cytosolic and precedes cell death. J. Cell Sci. 120 : 772–781. https://doi.org/10.1242/jcs.03377
Bruns S., Kniemeyer O., Hasenberg M., Aimanianda V., Nietzsche S., Thywissen A., Jeron A., Latge J.-P., Brakhage A.A., Gunzer M. 2010. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 6 : e1000873. https://doi.org/10.1371/journal.ppat.1000873
Buchanan J.T., Simpson A.J., Aziz R.K., Liu G.Y., Kristian S.A., Kotb M., Feramisco J., Nizet V. 2006. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 16 : 396–400. https://doi.org/10.1016/j.cub.2005.12.039
Carlson B.A., Tobe R., Yefremova E., Tsuji P.A., Hoffmann V.J., Schweizer U., Gladyshev V.N., Hatfield D.L., Conrad M. 2016. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox. Biol. 9 : 22–31. https://doi.org/10.1016/j.redox.2016.05.003
Chargui A., Cesaro A., Mimouna S., Fareh M., Brest P., Naquet P., Darfeuille-Michaud A., Hébuterne X., Mograbi B., Vouret-Craviari V., Hofman P. 2012. Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS ONE. 7 : e51727. https://doi.org/10.1371/journal.pone.0051727
Chargui A., El May M.V. 2014. Autophagy mediates neutrophil responses to bacterial infection. APMIS. 122 : 1047–1058. https://doi.org/10.1111/apm.12271
Cho J.H., Sung B.H., Kim S.C. 2009. Buforins: histone H2A-derived antimicrobial peptides from toad stomach. Biochim. Biophys. Acta. 1788 : 1564–1569. https://doi.org/10.1016/j.bbamem.2008.10.025
Choi A.J., Ryter S.W. 2011. Autophagy in inflammatory diseases. Int. J. Cell Biol. 2011 : 732798. https://doi.org/10.1155/2011/732798
Deretic V., Levine B. 2014. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 5 : 527–549. doi https://doi.org/10.1016/j.chom.2009.05.016
Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., Sun H., Wang D.C., Shao F. 2016. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 535 : 111–116. https://doi.org/10.1038/nature20106
Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., Stockwell B.R. 2012. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 149 : 1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Djavaheri-Mergny M., Amelotti M., Mathieu J., Besançon F., Bauvy C., Souquère S., Pierron G., Codogno P. 2006. NF-‑κB activation represses tumor necrosis factor-α-induced autophagy. J. Biol. Chem. 281 : 30373–30382. doi https://doi.org/10.1074/jbc.M602097200
Doll S., Proneth B., Tyurina Y.Y., Panzilius E., Kobayashi S., Ingold I., Irmler M., Beckers J., Aichler M., Walch A., Prokisch H., Trümbach D., Mao G., Qu F., Bayir H., Füllekrug J., Scheel C.H., Wurst W., Schick J.A., Kagan V.E., Angeli J.P., Conrad M. 2017. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13 : 91–98. https://doi.org/10.1038/nchembio.2239
Dolma S., Lessnick S.L., Hahn W.C., Stockwell B.R. 2003. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 3 : 285–296. https://doi.org/10.1016/S1535-6108(03)00050-3
Dworski R., Simon H.-U., Hoskins A., Yousefi S. 2011. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J. Allergy Clin. Immunol. 127 : 1260–1266. https://doi.org/10.1016/j.jaci.2010.12.1103
Eskelinen E.L. 2005. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 1 : 1–10. https://doi.org/10.4161/auto.1.1.1270
Farnaud S., Evans R.W. 2003. Lactoferrin – a multifunctional protein with antimicrobial properties. Mol. Immunol. 40 : 395–405. https://doi.org/10.1016/S0161-5890(03)00152-4
Franchi L., Eigenbrod T., Muñoz-Planillo R., Nuñez G. 2009. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10 : 241–247. https://doi.org/10.1038/ni.1703
François S., El Benna J., Dang P. M., Pedruzzi E., Gougerot-Pocidalo M.A., Elbim C. 2005. Inhibition of neutrophil apoptosis by TLR agonists in whole blood: Involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J. Immun. 174 : 3633–3642. doi https://doi.org/10.4049/jimmunol.174.6.3633
Friedmann Angeli J.P., Schneider M., Proneth B. Et al. 2014. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16 : 1180–1191. https://doi.org/10.1038/ncb3064
Fuchs T.A., Abed U., Goosmann C., Hurwitz R., Schulze I., Wahn V., Weinrauch Y., Brinkmann V., Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176 : 231–241. https://doi.org/10.1083/jcb.200606027
Fuchs T., Bhandari A., Wagner D.D. 2011. Histones induce rapid and profound thrombocytopenia in mice. Blood. 118 : 3708–3714. https://doi.org/10.1182/blood-2011-01-332676
Fuchs T.A., Brill A., Duerschmied D, Schatzberg D., Monestier M., Myers D.D., Wrobleski S.K., Wakefield T.W., Hartwig J.H., Wagner D.D. 2010. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad Sci. USA. 107 : 15 880–15 885. https://doi.org/10.1073/pnas.1005743107
Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W. et al. 2018. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25 : 486–541. https://doi.org/10.1038/s41418-017-0012-4
Garcia-Romo G.S., Caielli S., Vega B., Connolly J., Allantaz F., Xu Z., Punaro M., Baisch J., Guiducci C., Coffman R.L., Barrat F.J., Banchereau J., Pascual V. 2011. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3 : 73ra20. https://doi.org/10.1126/scitranslmed.3001201
Geddings J.E., Mackman N. 2014. New players in haemostasis and thrombosis. Thromb. Haemost. 111 : 570–574. doi https://doi.org/10.1160/TH13-10-0812
Green D.R. 2011. The end and after: How dying cells impact the living organism. Immunity. 35 : 441–445. https://doi.org/10.1016/j.immuni.2011.10.003
Grinberg N., Elazar S., Rosenshine I., Shpigel N.Y. 2008. Beta hydroxybutyrate abrogates formation of bovine neutrophil extracellular traps and bactericidal activity against mammary pathogenic Escherichia coli. Infect. Immun. 76 : 2802–2807. https://doi.org/10.1128/IAI.00051-08
Gundara J.S, Zhao J., Robinson B.G., Sidhu S.B. 2012. Oncophagy: Harnessing regulation of autophagy in cancer therapy. Endocr. Relat. Cancer. 19 : R281–R295. https://doi.org/10.1530/ERC-12-0325
Gupta A.K., Joshi M.B., Philippova M., Erne P., Hasler P., Hahn S., Resink T.J. 2010. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 584 : 3193–3197. https://doi.org/10.1016/j.febslet.2010.06.006
Gutierrez M.G., Master S.S., Singh S.B., Taylor G.A., Colombo M.I., Deretic V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 119: 753–766. doi https://doi.org/10.1016/j.cell.2004.11.038
Hailey D.W., Rambold A.S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P.K., Lippincott-Schwartz J. 2010. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 141 : 656–667. https://doi.org/10.1016/j.cell.2010.04.009
Hambright W.S., Fonseca R.S., Chen L., Na R., Ran Q. 2017. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12 : 8–17. https://doi.org/10.1016/j.redox.2017.01.021
Harris J. 2011. Autophagy and cytokines. Cytokine. 56 : 140–144. https://doi.org/10.1016/j.cyto.2011.08.022
Hayashi F., Means T.K., Luster A.D. 2003. Toll-like receptors stimulate human neutrophil function. Blood. 102 : 2660–2669. https://doi.org/10.1182/blood-2003-04-1078
Iba T., Hashiguchi N., Nagaoka I., Tabe Y., Murai M. 2013. Neutrophil cell death in response to infection and its relation to coagulation. J. Intensive Care. 1 : 1–13. https://doi.org/10.1186/2052-0492-1-13
Iyer S.S., Pulskens W.P., Sadler J.J., Butter L.M., Teske G.J., Ulland T.K., Eisenbarth S.C., Florquin S., Flavell R.A., Leemans J.C., Sutterwala F.S. 2009. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA. 106 : 20 388–20 393. https://doi.org/10.1073/pnas.0908698106
Jorgensen I., Miao E.A. 2015. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265 : 130–142. https://doi.org/10.1111/imr.12287
Kagan V.E., Mao G., Qu F., Angeli J.P., Doll S., Croix C.S., Dar H.H., Liu B., Tyurin V.A., Ritov V.B., Kapralov A.A., Amoscato A.A., Jiang J. Anthonymuthu T., Mohammadyani D., Yang Q., Proneth B., Klein-Seetharaman J., Watkins S., Bahar I., Greenberger J., Mallampalli R.K., Stockwell B.R., Tyurina Y.Y., Conrad M., Bayır H. 2017. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13 : 81–90. https://doi.org/10.1038/nchembio.2238
Kannengiesser C., Gérard B., El Benna J., Henri D., Kroviarski Y., Chollet-Martin S., Gougerot-Pocidalo M.A., Elbim C., Grandchamp B. 2008. Molecular epidemiology of chronic granulomatous disease in a series of 80 kindreds: Identification of 31 novel mutations. Hum. Mutat. 29 : E132–E149. https://doi.org/10.1002/humu.20820
Kaplan M.J., Radic M. 2012. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 189 : 2689–2695. https://doi.org/10.4049/jimmunol.1201719
Kayagaki N., Stowe I.B., Lee B.L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T., Liu P.S., Lill J.R., Li H., Wu J., Kummerfeld S., Zhang J., Lee W.P., Snipas S.J., Salvesen G.S., Morris L.X., Fitzgerald L., Zhang Y., Bertram E.M., Goodnow C.C., Dixit V.M. 2015. Caspase-11 cleaves gasdermin D for noncanonical inflammasome signalling. Nature. 526 : 666–71. https://doi.org/10.1038/nature15541
Klionsky D.J. 2004. Cell biology: Regulated self-cannibalism. Nature. 431 : 31–32.
Klionsky D.J, Emr S.D. 2000. Autophagy as a regulated pathway of cellular degradation. Science. 290 : 1717–1721. https://doi.org/10.1126/science.290.5497.1717
Kolaczkowska E., Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nature Rev. Immunol. 13 : 159–175. https://doi.org/10.1038/nri3399
Kuballa P., Nolte W.M., Castoreno A.B., Xavier R.J. 2012. Autophagy and the immune system. Ann. Rev. Immunol. 30 : 611–646. https://doi.org/10.1146/annurev-immunol-020711-074948
Labbé K, Saleh M. 2008. Cell death in the host response to infection. Cell Death Differ. 15 : 1339–1349. doi https://doi.org/10.1038/cdd.2008.91
Lande R., Ganguly D., Facchinetti V., Frasca L., Conrad C., Gregorio J., Meller S., Chamilos G., Sebasigari R., Riccieri V., Bassett R., Amuro H., Fukuhara S., Ito T., Liu Y.J., Gilliet M. 2011. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3 : 73ra19. https://doi.org/10.1126/scitranslmed.3001180
Lande R., Gregorio J., Facchinetti V., Chatterjee B., Wang Y.H., Homey B., Cao W., Wang Y.H., Su B., Nestle F.O., Zal T., Mellman I., Schröder J.M., Liu Y.J., Gilliet M. 2007. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 449 : 564–569. https://doi.org/10.1038/nature06116
Lapaquette P., Glasser A.L., Huett A., Xavier R.J., Darfeuille-Michaud A. 2011. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 12 : 99–113. https://doi.org/10.1111/j.1462-5822.2009.01381.x
Levine B., Mizushima N., Virgin H.W. 2011. Autophagy in immunity and inflammation. Nature. 469 : 323–335. https://doi.org/10.1038/nature09782
Linkermann A., Skouta R., Himmerkus N., Mulay S.R., Dewitz C.,De Zen F., Prokai A., Zuchtriegel G., Krombach F., Welz P.S., Weinlich R., Vanden Berghe T., Vandenabeele P., Pasparakis M., Bleich M., Weinberg J.M., Reichel C.A., Bräsen J.H., Kunzendorf U., Anders H.J., Stockwell B.R., Green D.R., Krautwald S. 2014. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad Sci. USA. 111 : 16836–16841. https://doi.org/10.1073/pnas.1415518111
Liu G., Bi Y., Wang R., Wang X. 2013. Self-eating and self-defense: autophagy controls innate immunity and adaptive immunity. J. Leukoc. Biol. 93 : 511–519. https://doi.org/10.1189/jlb.0812389
Lu T., Kobayashi S.D., Quinn M.T., Deleo F.R. 2012. A NET Outcome. Frontiers Immunol. 3 : 365. https://doi.org/10.3389/fimmu.2012.00365
Ma A.C., Kubes P. 2008. Platelets neutrophils and neutrophil extracellular traps (NETs) in sepsis. J. Thrombosis Haemostasis. 6 : 415–420. https://doi.org/10.1111/j.1538-7836.2007.02865.x
Manzenreiter R., Kienberger F., Marcos V., Schilcher K., Krautgartner W.D., Obermayer A., Huml M., Stoiber W., Hector A., Griese M., Hannig M., Studnicka M., Vitkov L., Hartl D. 2011. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros. 11 : 84–92. https://doi.org/10.1016/j.jcf.2011.09.008
Mari M., Griffith J., Rieter E., Krishnappa L., Klionsky D.J., Reggiori F. 2010. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 190 : 1005–1022. https://doi.org/10.1083/jcb.200912089
Matsushita M., Freigang S., Schneider C., Conrad M., Bornkamm G.W., Kopf M. 2015. T-cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212 : 555–568. https://doi.org/10.1084/jem.20140857
Meiffren G., Joubert P. E., Gregoire I. P., Codogno P., Rabourdin-Combe C., Faure M. 2010. Pathogen recognition by the cell surface receptor CD46 induces autophagy. Autophagy. 6 : 299–300. https://doi.org/10.4161/auto.6.2.11132
Méndez-Samperio P. 2010. The human cathelicidin hCAP18/LL-37: A multifunctional peptide involved in mycobacterial infections. Peptides. 31 : 1791–1798. https://doi.org/10.1016/j.peptides.2010.06.016
Metzler K.D., Fuchs T.A., Nauseef W.M., Reumaux D., Roesler J., Schulze I., Wahn V., Papayannopoulos V., Zychlinsky A. 2011. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood. 117 : 953–959. https://doi.org/10.1182/blood-2010-06-290171
Mihalache C.C., Yousefi S., Conus S., Villiger P.M., Schneider E.M., Simon H.U. 2011. Inflammation-associated autophagy-related programmed necrotic death of human neutrophils characterized by organelle fusion events. J. Immunol. 186 : 6532–6542. https://doi.org/10.4049/jimmunol.1004055
Mitroulis I., Kourtzelis I., Kambas K., Rafail S., Chrysanthopoulou A., Speletas M., Ritis K. 2010. Regulation of the autophagic machinery in human neutrophils. Eur. J. Immunol. 40 : 1461–1472. https://doi.org/10.1002/eji.200940025
Mizushima N., Levine B. 2010. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 12 : 823–830. https://doi.org/10.1038/ncb0910-823
Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. 2008. Autophagy fights disease through cellular self-digestion. Nature. 451 : 1069–1075. https://doi.org/10.1038/nature06639
Mostowy S., Cossart P. 2012. Bacterial autophagy: Restriction or promotion of bacterial replication? Trends Cell Biol. 22 : 283–291. https://doi.org/10.1016/j.tcb.2012.03.006
Moutsopoulos N.M., Konkel J., Sarmadi M., Eskan M.A., Wild T., Dutzan N., Abusleme L., Zenobia C., Hosur K.B., Abe T., Uzel G., Chen W., Chavakis T., Holland S.M., Hajishengallis G. 2014. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci. Transl. Med. 6 : 229–240. doi https://doi.org/10.1126/scitranslmed.3007696
Mulay S.R., Kumar S.V., Lech M., Desai J., Anders H.J. 2016. How kidney cell death induces renal necroinflammation. Semin. Nephrol. 36 : 162–173. https://doi.org/10.1016/j.semnephrol.2016.03.004
Nair U., Jotwani A., Geng J., Gammoh N., Richerson D., Yen W.L., Griffith J., Nag S., Wang K., Moss T., Baba M., McNew J.A., Jiang X., Reggiori F., Melia T.J., Klionsky D.J. 2011. SNARE proteins are required for macroautophagy. Cell. 146 : 290–302. https://doi.org/10.1016/j.cell.2011.06.022
Napier B.A., Brubaker S.W., Sweeney T.E., Monette P., Rothmeier G.H., Gertsvolf N.A., Puschnik A., Carette J.E., Khatri P., Monack D.M. 2016. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J. Exp. Med. 213 : 2365–2382. https://doi.org/10.1084/jem.20160027
Neeli I., Khan S.N., Radic M. 2008. Histone deimination as a response to inflammatory stimuli in neutrophils. J. Immunol. 180 : 1895–1902. https://doi.org/10.4049/jimmunol.180.3.1895
Nikoletopoulou V., Markaki M., Palikaras K., Tavernarakis N. 2013. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta. 1833 : 3448–3459. https://doi.org/10.1016/j.bbamcr.2013.06.001
Obermayer A., Stoiber W., Krautgartner W.-D., Klappacher M., Kofler B., Steinbacher P., Vitkov L., Grabcanovic-Musija F., Studnicka M. 2014. New aspects on the structure of neutrophil extracellular traps from chronic obstructive pulmonary disease and in vitro generation. PLoS ONE. 9 : e97784. https://doi.org/10.1371/journal.pone.0097784
Odobasic D., Kitching R., Holdsworth S.R. 2016. Neutrophil-mediated regulation of innate and adaptive immunity: The role of myeloperoxidase. J. Immunol. Res. 2016 : 2349817. https://doi.org/10.1155/2016/2349817
Ohashi Y., Munro S. 2010. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol. Biol. Cell. 21 : 3998–4008. https://doi.org/10.1091/mbc.E10-05-0457
Papayannopoulos V., Metzler K.D., Hakkim A., Zychlinsky A. 2010. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 191 : 677–691. https://doi.org/10.1083/jcb.201006052
Parker H., Winterbourn C.C. 2013. Reactive oxidants and myeloperoxidase and their involvement in neutrophil extracellular traps. Front. Immunol. 3 : 424, 1–6. https://doi.org/10.3389/fimmu.2012.00424
Pilsczek F.H., Salina D., Poon K.K.H., Fahey C., Yipp B.G., Sibley C.D., Robbins S.M., Green F.H.Y., Surette M.G., Sugai M., Gabriela Bowden M., Hussain M., Zhang K., Kubes P. 2010. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 185 : 7413–7425. https://doi.org/10.4049/jimmunol.1000675
Pleskova S.N., Gorshkova E.N., Kriukov R.N. 2018. Dynamics of formation and morphological features of neutrophil extracellular traps formed under the influence of opsonized Staphylococcus aureus. J. Mol. Recog. e2707. https://doi.org/10.1002/jmr.2707
Pleskova S.N., Mikheeva E.R. 2018. The dependence of an inflammatory response to neutrophil granulocyte death. In: New research on cell aging and death. N.Y.: Nova Science Publishers. 43–77. https://doi.org/10.1016/j.micron.2017.11.011
Potapnev M.P. 2014. Autophagy, apoptosis, necrosis and immune recognition of self and nonself. Immunology. 2 : 95–102. https://doi.org/10.1007/s10517-014-2396-1
Rabinowitz J.D., White E. 2010. Autophagy and metabolism. Science. 330: 1344–1348. https://doi.org/10.1126/science.1193497
Randow F., MacMicking J.D., James L.C. 2013. Cellular self-defense: How cell-autonomous immunity protects against pathogens. Science. 340 : 701–706. https://doi.org/10.1126/science.1233028
Ravikumar B., Moreau K., Jahreiss L., Puri C., Rubinsztein D.C. 2010. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 12 : 747–757. https://doi.org/10.1038/ncb2078
Remijsen Q., Vanden Berghe T., Wirawan E., Asselbergh B., Parthoens E., De Rycke R., Noppen S., Delforge M., Willems J., Vandenabeele P. 2011. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 21 : 290–304. https://doi.org/10.1038/cr.2010.150
Repnik U., Hafner Cesen M., Turk B. 2014. Lysosomal membrane permeabilization in cell death: Concepts and challenges. Mitochondrion. 19 : 49–57. https://doi.org/10.1016/j.mito.2014.06.006
Reunanen H., Punnonen E.L., Hirsimäki P. 1985. Studies on vinblastine-induced autophagocytosis in mouse liver. V. A cytochemical study on the origin of membranes. Histochem. 83 : 513–517.
Rubinsztein D.C., Marino G., Kroemer G. 2011. Autophagy and aging. Cell. 146 : 682–695. https://doi.org/10.1016/j.cell.2011.07.030
Salminen A., Kaarniranta K., Kauppinen A. 2012. Beclin I interactome controls the crosstalk apoptosis, autophagy and inflammasome activation: Impact on the aging process. Ageing Res. Rev. 12 : 520–534. https://doi.org/10.1016/j.arr.2012.11.004
Segawa K., Nagata S. 2015. An apoptotic ‘eat me’ signal: Phosphatidylserine exposure. Trends Cell Biol. 25 : 639–650. https://doi.org/10.1016/j.tcb.2015.08.003
Seiler A., Schneider M., Förster H., Roth S., Wirth E.K., Culmsee C., Plesnila N., Kremmer E., Rådmark O., Wurst W., Bornkamm G.W., Schweizer U., Conrad M. 2008. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIFmediated cell death. Cell Metab. 8 : 237–248. https://doi.org/10.1016/j.cmet.2008.07.005
Semeraro F., Ammollo C.T., Morrissey J.H., Dale G.L., Friese P., Esmon N.L., Esmon C.T. 2011. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood. 118 : 1952–1961. https://doi.org/10.1182/blood-2011-03-343061
Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 526 : 660–665. https://doi.org/10.1038/nature15514
Shimada K., Skouta R., Kaplan A., Yang W.S., Hayano M., Dixon S.J., Brown L.M., Valenzuela C.A., Wolpaw A.J., Stockwell B. 2016. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12 : 497–503. https://doi.org/10.1038/nchembio.2079
Steppich B., Seitz I., Busch G., Stein A., Ott I. 2008. Modulation of tissue factor and tissue factor pathway inhibitor-1 by neutrophil proteases. Thrombosis Haemostasis. 100 : 1068–1075. https://doi.org/10.1160/TH08-05-0293
Sun L., Wang H., Wang Z., He S., Chen S., Liao D. Wang L., Yan J., Liu W., Lei X., Xang X. 2012. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 148 : 213–227. https://doi.org/10.1016/j.cell.2011.11.031
Suzuki K., Kirisako T., Kamada Y., Mizushima N., Noda T., Ohsumi Y. 2001. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20 : 5971–5981. doi https://doi.org/10.1093/emboj/20.21.5971
Tallóczy Z., Jiang W., Virgin H.W., Leib D.A., Scheuner D., Kaufman R.J., Eskelinen E.L., Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc. Natl. Acad Sci. USA. 99 : 190–195. https://doi.org/10.1073/pnas.012485299
Tang D., Kang R., Coyne C.B., Zeh H.J., Lotze M.T. 2012. PAMPs and DAMPS: Signal 0s that spur autophagy and immunity. Immunol. Rev. 249 : 158–175. https://doi.org/10.1111/j.1600-065X.2012.01146.x
Tanida I. 2011. Autophagy basics. Microbiol. Immunol. 55 : 1–11. https://doi.org/10.1111/j.1348-0421.2010.00271.x
Tillack K., Breiden P., Martin R., Sospedra M. 2012. T-lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 188 : 3150–3159. https://doi.org/10.4049/jimmunol.1103414
Toussaint M., Jackson D.J., Swieboda D., Guedán A., Tsourouktsoglou T.D., Ching Y.M., Radermecker C., Makrinioti H., Aniscenko J., Edwards M.R., Solari R., Farnir F., Papayannopoulos V., Bureau F., Marichal T., Johnston S.L. 2017. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat. Med. 23 : 681–691. https://doi.org/10.1038/nm.4332
Urban C.F., Ermert D., Schmid M., Abu-Abed U., Goosmann C., Nacken W., Brinkmann V., Jungblut P.R., Zychlinsky A. 2009. Neutrophil extracellular traps contain calprotectin a cytosolic protein complex involved in host defence against Candida albicans. PLoS Pathogens. 5 : e1000639. https://doi.org/10.1371/journal.ppat.1000639
Urban C.F., Reichard U., Brinkmann V., Zychlinsky A. 2006. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 8 : 668–676. https://doi.org/10.1111/j.1462-5822.2005.00659.x
Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. 2014. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 15 : 135–147. https://doi.org/10.1038/ nrm3737
Vanden Berghe T., Demon D., Bogaert P., Vandendriessche B., Goethals A., Depuydt B., Vuylsteke M., Roelandt R., Van Wonterghem E., Vandenbroecke J., Choi S.M., Meyer E., Krautwald S., Declercq W., Takahashi N., Cauwels A., Vandenabeele P. 2014. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am. J. Respir. Crit. Care Med. 189 : 282–291. https://doi.org/10.1164/rccm.201308-1535OC
Ventura-Juarez J., Campos-Esparza M., Pacheco-Yepez J., López-Blanco J.A., Adabache-Ortíz A., Silva-Briano M., Campos-Rodríguez R. 2016. Entamoeba histolytica induces human neutrophils to form NETs. Parasite Immunol. 38 : 503–509. https://doi.org/10.1111/pim.12332
Von Brühl M.-L., Stark K., Steinhart A. et al. 2012. Monocytes neutrophils and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209 : 819–835. https://doi.org/10.1084/jem.20112322
Wallach D., Kang T.-B., Dillon C.P., Green D.R. 2016. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 352 : aaf2154. https://doi.org/10.1126/science.aaf2154
Walsh C.M., Edinger A.L. 2010. The complex interplay between autophagy, apoptosis and necrotic signals promotes T-cell homeostasis. Immunol. Rev. 236 : 95–109. https://doi.org/10.1111/j.1600-065X.2010.00919.x
Wang X., He Z., Liu H., Yousefi S., Simon H.-U. 2016. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J. Immunol. 197 : 4090–4100. https://doi.org/10.4049/jimmunol.1600051
Wang Y., Gao W., Shi X., Ding J., Liu W., He H., Wang K., Shao F. 2017. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 547 : 99–103. https://doi.org/10.1038/nature22393
Wartha F., Beiter K., Normark S., Henriques-Normark B. 2007. Neutrophil extracellular traps: Casting the NET over pathogenesis. Curr. Opin. Microbiol. 10 : 52–56. doi https://doi.org/10.1016/j.mib.2006.12.005
Welty D.M., Long R.D., Bailey J.R., Parnell M.J., Hoe N.P., Adams G.G., Deleo F.R., Musser J.M. 2005. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc. Natl. Acad. Sci. USA. 102 : 1679–1684. https://doi.org/10.1073/pnas.0406641102
Yang D., He Y., Muñoz-Planillo R., Liu Q., Núñez G. 2015. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 43 : 923–932. https://doi.org/10.1016/j.immuni.2015.10.009
Yang W.S., Stockwell B.R. 2008. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15 : 234–245. https://doi.org/10.1016/j.chembiol.2008.02.010
Yang W.S., Stockwell B.R. 2016. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 26 : 165–176. doi https://doi.org/10.1016/j.tcb.2015.10.014
Yano T., Kurata S. 2008. Induction of autophagy via innate bacterial recognition. Autophagy. 4 : 958–960. https://doi.org/10.4161/auto.6802
Yipp B.G., Petri B., Salina D., Jenne C.N., Scott B.N., Zbytnuik L.D., Pittman K., Asaduzzaman M., Wu K., Meijndert H.C., Malawista S.E., de Boisfleury Chevance A., Zhang K., Conly J., Kubes P. 2012. Infection induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 18 : 1386–1393. https://doi.org/10.1038/nm.2847
Yoshikawa Y., Ogawa M., Hain T., Yoshida M., Fukumatsu M., Kim M., Mimuro H., Nakagawa I., Yanagawa T., Ishii T., Kakizuka A., Sztul E., Chakraborty T., Sasakawa C. 2009. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11 : 1233–1240. https://doi.org/10.1038/ncb1967
Zawrotniak M., Rapala-Kozik M. 2013. Neutrophil extracellular traps (NETs) – formation and implications. Acta Biochem. Polonica. 60 : 277–284.
Zelenay S., Reise Sousa C. 2013. Adaptive immunity after cell death. Trends Immunol. 34 : 329–335. https://doi.org/10.1016/j.it.2013.03.005
Zilka O., Shah R., Li B., Friedmann Angeli J.P., Griesser M., Conrad M., Pratt D.A. 2017. On the mechanism of cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 3 : 232–243. https://doi.org/10.1021/acscentsci.7b00028
Zong W.X., Thompson C.B. 2006. Necrotic death as a cell fate. Genes Dev. 20 : 1–15. https://doi.org/10.1101/gad.1376506
Дополнительные материалы отсутствуют.