

Цитология, 2020, T. 62, № 11, стр. 829-836
Производное пиразола подавляет депозависимый вход Са2+ в перитонеальные макрофаги крысы
Л. С. Миленина 1, *, З. И. Крутецкая 1, **, В. Г. Антонов 2, Н. И. Крутецкая 1
1 Кафедра биофизики Санкт-Петербургского государственного университета
199034 Санкт-Петербург, Россия
2 Кафедра клинической биохимии и лабораторной диагностики Военно-Медицинской академии им. C.М. Кирова
194044 Санкт-Петербург, Россия
* E-mail: l.milenina@spbu.ru
** E-mail: z.krutetskaya@spbu.ru
Поступила в редакцию 02.08.2020
После доработки 10.08.2020
Принята к публикации 13.08.2020
Аннотация
Депозависимый вход Са2+ является повсеместным механизмом регулируемого входа Са2+ в клетки эукариот, активируемым при опустошении внутриклеточных Са2+-депо и участвует в регуляции широкого спектра клеточных процессов. Для выяснения фармакологических характеристик депозависимого входа Са2+ в клетки исследовали влияние производного пиразола иммунодепрессанта YM-58483 на депозависимый вход Са2+ в перитонеальные макрофаги крысы, вызываемый ингибиторами эндоплазматических Са2+-АТФаз тапсигаргином и циклопьязониковой кислотой, а также дисульфидсодержащими иммуномодуляторами глутоксимом и моликсаном. С использованием флуоресцентного Са2+-зависимого зонда Fura-2AM впервые показано, что в перитонеальных макрофагах крысы, как и в других клетках, производное пиразола YM-58483 эффективно ингибирует депозависимый вход Са2+ и является удобным фармакологическим инструментом для изучения депозависимого входа Са2+ в макрофаги. Кроме того, полученные данные дополнительно подтверждают, что вход Са2+, индуцируемый глутоксимом или моликсаном, происходит по депозависимому механизму.
Ион Са2+ является универсальным вторичным мессенджером, действующим в клетках микроорганизмов, растений и животных (Berridge et al., 1998). Изменения в транспорте и внутриклеточной концентрации ионов Са2+ играют ключевую роль в запуске и регуляции общих и специализированных клеточных функций, таких как пролиферация, рост, секреция, сокращение, передача нервного импульса, иммунный ответ и т.д. (Berridge et al., 2000, 2003). В клетках иммунной системы (лимфоцитах, тучных клетках, макрофагах) ионы Са2+ работают на всех стадиях жизни клетки, включая развитие, активацию, дифференцировку, продукцию цитокинов и, наконец, смерть клетки (Vig, Kinet, 2009; Trebak, Kinet, 2019).
Са2+-сигналы в клетке жестко регулируются и являются результатом скоординированного взаимодействия и взаимовлияния (crosstalk) между Са2+-каналами в плазмалемме и внутриклеточными Са2+-депо (Berridge et al., 2000, 2003). Примером таких взаимодействий является депозависимый (или “емкостной”) вход Са2+, активируемый стимулами, уменьшающими концентрацию Са2+ во внутриклеточных Са2+-депо (Putney, 1990, 2017; Albarran et al., 2016).
Депозависимый вход Са2+ впервые был описан более 30 лет назад Джеймсом Патни (Putney, 1990, 2017) в невозбудимых клетках, включая клетки иммунной системы. В настоящее время установлено, что депозависимый вход Са2+ является повсеместным (ubiquitous) механизмом регулируемого входа Са2+ в клетки эукариот и регулирует широкий спектр клеточных процессов (экзоцитоз, экспрессию генов, активацию иммунных клеток, рост и пролиферацию клеток и др.) в норме и патологии (Putney, 2011; Prakriya, Lewis, 2015).
Функциональной единицей депозависимого входа Са2+ является мультимолекулярный белковый комплекс SOCIC (store-operated calcium influx complex), компоненты которого обладают высокой мобильностью, а взаимодействия между ними жестко регулируются (Vaca, 2010; Moreno, Vaca, 2012). Основными компонентами комплекса, необходимыми и достаточными для активации депозависимого входа Са2+, являются Са2+-каналы Orai1 в плазмалемме и Са2+-сенсор STIM1 в мембране Са2+-депо (Prak-riya, Lewis, 2015). При опустошении Са2+-депо, STIM1 олигомеризуется, транслоцируется в участки эндоплазматического ретикулума, расположенные у плазмалеммы, и прямо взаимодействует с белками Orai1, вызывая депозависимый вход Са2+ (Nwokonko et al., 2017; Nguyen et al., 2018; Lunz et al., 2019; Lewis, 2020).
К числу фармакологических модуляторов депозависимого входа Са2+ относятся производные бис(трифторметил)пиразолов (BTPs). Обнаружено, что они являются иммунодепрессантами и ингибируют продукцию цитокинов (интерлейкин-2, -4, -5 и др.) в Т-лимфоцитах (Djuric et al., 2000; Trevillyan et al., 2001; Ishikawa et al., 2003). Показана эффективность этих соединений при исследовании иммунных заболеваний на модельных объектах, таких как грызуны и приматы (Djuric et al., 2000; Ishikawa et al., 2003).
Наиболее изученным из производных пиразола является соединение YM-58483 (3,5-бис(трифторметил)пиразол, известное также как BTP2 или Pyr2). Показано, что в низкой концентрации (мкМ или нМ) YM-58483 эффективно ингибирует депозависимый вход Са2+ в разных клетках (Putney, 2001, 2010; Sweeney et al., 2009; Bogeski et al., 2010; Jairaman, Prakriya, 2013; Tian et al., 2016; Bird, Putney, 2018).
Учитывая важную роль депозависимых Са2+-каналов в функционировании клеток иммунной системы, в настоящей работе выясняли фармакологические характеристики депозависимого входа Са2+ в макрофагах под влиянием соединения YM-58483 в перитонеальных макрофагах крысы.
Для активации депозависимого входа Са2+ в экспериментах использовали два структурно различных ингибитора эндоплазматических Са2+-АТФаз – тапсигаргин (Thastrup et al., 1989) и циклопьязониковую кислоту (ЦПК) (Goeger et al., 1988), вызывающие пассивное опустошение Са2+-депо, а также дисульфидсодержащие иммуномодуляторы – препараты глутоксим® (динатриевую соль окисленного глутатиона с d-металлом в наноконцентрации) и моликсан® (комплекс глутоксима и нуклеозида инозина).
МАТЕРИАЛ И МЕТОДИКА
Выделение и культивирование перитонеальных макрофагов крыс. Эксперименты проводили на культивируемых резидентных перитонеальных макрофагах крыс линии Wistar. Содержание животных и все манипуляции выполняли в соответствии с нормативными документами и требованиями Приказа Минздрава России № 267 от 19.06.03 “Об утверждении правил лабораторной практики в Российской Федерации”.
Резидентные макрофаги выделяли из перитонеальной полости крыс массой 180–250 г по традиционному методу, сразу после выделения клетки имели сферическую форму (диаметр 10–20 мкм) (Conrad, 1981; Randriamampita, Trautmann, 1987). Суспензию клеток помещали в бакпечатки с кварцевыми стeклами (10 × 10 мм) и культивировали в течение 1–3 сут при 37°С в среде 199 (рН 7.2), содержащей 20% сыворотки крови быка, глутамин (3%), пенициллин (100 Ед./мл) и стрептомицин (100 мг/мл). С помощью теста на α-нафтилэстеразу подтверждали, что по меньшей мере 96% клеток в монослоях являлись макрофагами (Monahan et al., 1981).
Эксперименты проводили при температуре (22–24°С) через 1–2 сут после начала культивирования клеток. Кварцевые стeкла с клетками помещали в экспериментальную камеру, заполненную физиологическим раствором следующего ионного состава (мМ): 140 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2 и 5 HEPES-NaOH, рН 7.3–7.4. Бескальциевая среда отличалась тем, что содержала 0 мМ CaCl2 и 1 мМ ЭГТА.
Измерение [Ca2+]i. Использовали флуоресцентный зонд Fura-2AM (Sigma-Aldrich, США). Макрофаги инкубировали в течение 45 мин в физиологическом растворе, содержащем 2 мкМ Fura-2AM, при 22–24°С. Стекла с окрашенными клетками отмывали физиологическим раствором и переносили в экспериментальную камеру флуоресцентного микроскопа Leica DM 4000B (Leica Microsystems, Германия). Возбуждение флуоресценции объекта производили при длинах волн 340 и 380 нм через объектив микроскопа. Для выделения соответствующих участков спектра использовали узкополосные оптические фильтры. Эмиссию регистрировали при длине волны 510 нм при помощи специализированной видеокамеры Leica DFC340FX. Для управления экспериментом использовали систему обработки изображения ImageJ (плагин Micro-Manager 1.4).
Результатом измерений являлось отношение интенсивностей флуоресценции Fura-2AM при облучении светом с длиной волны 340 нм к интенсивности флуоресценции при облучении светом с длиной волны 380 нм (F340/F380), где F340 – интенсивность флуоресценции Fura-2AM, связанного с Са2+, а F380 – интенсивность флуоресценции Fura-2AM, не связанного с Са2+, отражающее изменения [Ca2+]i в клетках во время измерений (Bruce, Elliott, 2000; Xie et al., 2002). Для избежания фотовыгорания, измерения проводили через каждые 20 с, облучая объект в течение 2 с. В экспериментах применяли объектив 10× с апертурой 8 мм. Значения [Ca2+]i рассчитывали по уравнению Гринкевича (Grynkiewicz et al., 1985). Статистический анализ проводили с применением t-критерия Стьюдента. Данные представлены в виде среднего и стандартного отклонения. Каждую регистрацию получали для группы из 40–50 клеток. На рисунках представлены результаты однотипных экспериментов из 6–7 независимых. Достоверными считали различия при P ≤ 0.05.
Использованные реактивы. Все реактивы приобретали в фирме Sigma-Aldrich, США. Маточные растворы Fura-2AM (1 мМ), ЦПК (10 мМ), тапсигаргина (0.5 мМ) и YM-58483 (5 мМ) готовили в диметилсульфоксиде. Препараты глутоксим и моликсан предоставлялись фирмой ФАРМА-ВАМ (Санкт-Петербург). Маточные растворы глутоксима (50 мг/мл) и моликсана (50 мг/мл) готовили в воде.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Влияние YM-58483 на депозависимый вход Са2+, индуцируемый ингибиторами эндоплазматических Са2+-АТФаз. В контрольных экспериментах мы обнаружили, что добавление 0.5 мкМ тапсигаргина к макрофагам, находящимся в бескальциевой среде, вызывает увеличение [Ca2+]i, отражающее мобилизацию Са2+ из внутриклеточных Са2+-депо (рис. 1а). В среднем увеличение [Ca2+]i во время фазы мобилизации составило 60 ± 13 нМ (n = 7; P < 0.05). При последующем введении в наружную среду 2 мМ Са2+ наблюдали депозависимый вход Са2+ в цитозоль (рис. 1а). В среднем увеличение [Ca2+]i во время входа Са2+ составило 159 ± 22 нМ (n = 7; P < 0.05). Аналогичные результаты мы получили при использовании 10 мкМ ЦПК (рис. 1б): в среднем увеличение [Ca2+]i во время фазы мобилизации Са2+ из депо, вызываемой ЦПК, составило 61 ± 14 нМ ( n= 7; P < 0.05), а во время входа Са2+ – 170 ± 20 нМ (n = 7; P < 0.05) (рис. 1б).
Рис. 1.
Влияние соединения YM-58483 на депозависимый вход Са2+, индуцируемый тапсигаргином и циклопьязониковой кислотой в перитонеальных макрофагах крысы. а, б – макрофаги стимулировали 0.5 мкМ тапсигаргина (а) или 10 мкМ циклопьязониковой кислоты (ЦПК, б) в номинально бескальциевой среде, затем вход Са2+ инициировали введением в наружную среду 2 мМ Са2+; на фоне развившегося входа Са2+ добавляли 5 мкМ YM-58483; в – клетки стимулировали 10 мкМ ЦПК в бескальциевой среде, после окончания фазы мобилизации Са2+ из депо добавляли 5 мкМ YM-58483; через 5 мин вход Са2+ инициировали введением в наружную среду 2 мМ Са2+. Здесь и на рис. 2 каждая регистрация получена для группы из 40–50 клеток и представляет собой типичный вариант из 6–7 независимых экспериментов.
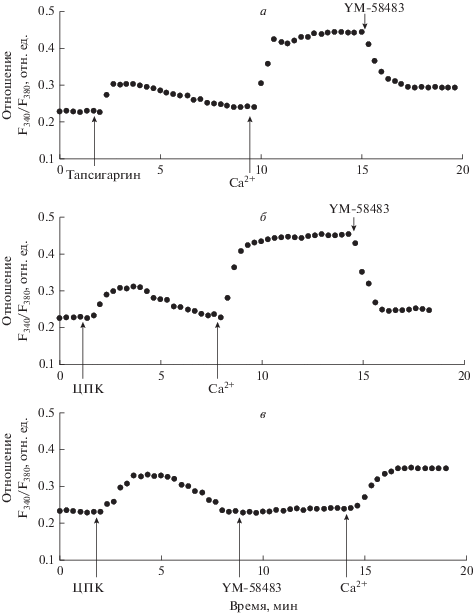
Мы впервые обнаружили, что при добавлении 5 мкМ YM-58483 на фоне развившегося депозависимого входа Са2+, вызываемого тапсигаргином или ЦПК, наблюдается практически полное подавление депозависимого входа Са2+. Так, при добавлении 5 мкМ YM-58483 на фоне входа Са2+, индуцированного тапсигаргином, подавление депозависимого входа Са2+ составило 80.2 ± 15.3% (n = 7; P < 0.05) (рис. 1а). При добавлении 5 мкМ YM-58483 на фоне входа Са2+, индуцированного ЦПК – 90.1 ± 14.9% (n = 7; P < 0.05) (рис. 1б). Это свидетельствует о том, что YM-58483 эффективно подавляет уже развившийся депозависимый вход Са2+ в макрофаги.
Во втором варианте экспериментов мы исследовали влияние YM-58483 на уже активированные опустошением депо механизмы входа Сa2+ в макрофаги. Макрофаги стимулировали 10 мкМ ЦПК в номинально бескальциевой среде. После окончания фазы мобилизации Ca2+ из депо, вызванной ЦПК, клетки инкубировали в течение 5 мин в присутствии 5 мкМ YM-58483, после чего вводили 2 мМ Ca2+ в наружную среду (рис. 1в). Обнаружено, что и в этих условиях 5 мкМ YM-58483 вызывает значительное подавление входа Са2+ (на 64.5 ± 15.3%, n = 7; P < 0.05). Аналогичные результаты были получены с применением 0.5 мкМ тапсигаргина (не представлено).
Полученные данные свидетельствуют о том, что YM-58483 оказывает влияние как на активацию, так и на поддержание депозависимого входа Са2+ в макрофагах.
Влияние YM-58483 на депозависимый вход Са2+, индуцируемый дисульфидсодержащими иммуномодуляторами. Фармакологические аналоги окисленного глутатиона (глутоксим и моликсан) используются как иммуномодуляторы и цитопротекторы в комплексной терапии бактериальных, вирусных и онкологических заболеваний (Борисов и др., 2001; Соколова и др., 2002; Антушевич и др., 2013; Толстой и др., 2019). Эти препараты оказывают комплексное влияние на процессы редокс-регуляции в клетках, однако тонкие биофизические механизмы их действия далеки от полного понимания. Ранее (Курилова и др., 2008, 2012) нами впервые было показано, что глутоксим и моликсан увеличивают [Ca2+]i, вызывая мобилизацию Са2+ из тапсигаргинчувствительных Са2+-депо и последующий депозависимый вход Са2+ в перитонеальные макрофаги крыс.
В настоящей работе контрольные эксперименты показали, что инкубация макрофагов в течение 20 мин с глутоксимом или моликсаном (по 100 мкг/мл; рис. 2а, б) в бескальциевой среде вызывает медленно нарастающее увеличение [Ca2+]i, отражающее мобилизацию Са2+ из внутриклеточных Са2+-депо. Через 20 мин после добавления агентов, [Ca2+]i в среднем увеличивалась от базального уровня, равного 92 ± 16, до 136 ± 21 нМ (n = 7; P < 0.05) для глутоксима и 124 ± 20 нМ (n = 6; P < 0.05) для моликсана. При введении в наружную среду 2 мМ Са2+ наблюдали дальнейшее повышение [Ca2+]i , отражающее депозависимый вход Са2+ в цитозоль (рис. 2). В среднем увеличение [Ca2+]i во время входа Са2+ составило 270 ± 32 (n = 7; P < 0.05) и 295 ± 33 нМ (n = 6; P < 0.05) для глутоксима и моликсана соответственно.
Рис. 2.
Влияние YM-58483 на депозависимый вход Са2+, вызываемый глутоксимом или моликсаном в перитонеальных макрофагах крысы. К макрофагам, находящимся в номинально бескальциевой среде, добавляли 100 мкг/мл глутоксима (а) или моликсана (б), через 20 мин вход Са2+ инициировали введением в наружную среду 2 мМ Са2+; на фоне развившегося входа Са2+ добавляли 5 мкМ YM-58483.
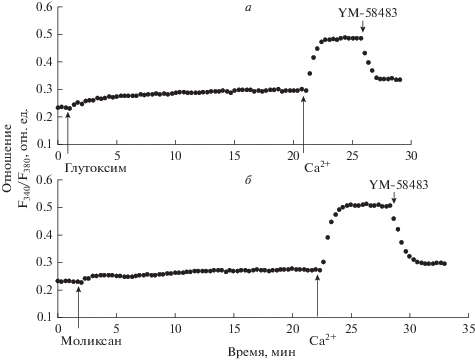
Мы впервые обнаружили, что при добавлении 5 мкМ YM-58483 на фоне развившегося депозависимого входа Са2+, вызываемого глутоксимом или моликсаном, наблюдается практически полное подавление депозависимого входа Са2+ (рис. 2). Так, при добавлении 5 мкМ YM-58483 на фоне входа Са2+, индуцированного 100 мкг/мл глутоксима, подавление депозависимого входа Са2+ составило 81.2 ± 16.1% (n = 7; P < 0.05) (рис. 2а). При добавлении 5 мкМ YM-58483 на фоне входа Са2+, индуцированного 100 мкг/мл моликсана, подавление входа Са2+ составило 90.0 ± ± 15.3% (n = 7; P < 0.05) (рис. 2б).
Это свидетельствует о том, что YM-58483 оказывает влияние на депозависимый вход Са2+, индуцируемый не только тапсигаргином или ЦПК, но и дисульфидсодержащими иммуномодуляторами глутоксимом и моликсаном, а также дополнительно подтверждает, что вход Са2+, вызываемый глутоксимом или моликсаном, происходит по депозависимому механизму.
Полученные нами результаты о подавлении YM-58483 депозависимого входа Са2+ в макрофагах согласуются с данными, представленными в литературе. Так, обнаружено, что YM-58483 подавляет уже развившийся депозависимый вход Са2+, индуцируемый тапсигаргином в Т-лимфоцитах человека (Ishikawa et al., 2003) и эндотелиальных клетках проксимальных тубул линии НК-2 почки человека (Zeng et al., 2017), а также ингибирует развившийся вход Са2+, вызываемый тапсигаргином и ЦПК в нейронах дорзальных корешков спинного мозга мыши (Xia et al., 2014; Wei et al., 2017).
Обнаружено также, что добавление YM-58483 после опустошения Са2+-депо тапсигаргином и до введения в наружную среду 2 мМ Са2+ приводит к подавлению депозависимого входа Са2+ в Т-клетках линии Jurkat человека (Ishikawa et al., 2003), одонтобластах крысы (Kimura et al., 2018) и лейкемических базофилах крысы (линия RBL-1) (Rahman, Rahman, 2017).
Кроме того, показано, что YM-58483 не влияет на мобилизацию Са2+ из депо, но практически полностью подавляет депозависимый вход Са2+, вызываемый тапсигаргином в мышиных амелобластах LS8 (Nurbaeva et al., 2015) и нейронах дорзальных корешков спинного мозга мыши (Xia et al., 2014), а также вход Са2+, вызываемый ЦПК в клетках MDA-MB-231 рака молочной железы (Azimi et al., 2017). YM-58483 практически полностью подавляет депозависимый вход Са2+, вызываемый тапсигаргином в клетках INS-1E инсулиномы крысы (Sabourin et al., 2015), клетках HEК293 эмбриональной почки человека и B-лимфоцитах DT40 цыпленка (He et al., 2005), а также депозависимый вход Са2+, вызываемый фитогемаглютинином, и продукцию интерлейкина (IL) 2 в Т-клетках линии Jurkat человека (Takezawa et al., 2006). В Т-лимфоцитах периферической крови и Т-клетках Jurkat YM-58483 ингибирует депозависимый вход Са2+ и продукцию IL-2 и IL-5 (Zitt et al., 2004).
Механизм влияния YM-58483 на депозависимый вход Са2+ до конца не изучен. Учитывая, что мультибелковый комплекс депозависимого входа Са2+ включает по меньшей мере два компонента – белки STIM1 и Orai1, фармакологические агенты могут регулировать депозависимый вход Са2+, воздействуя на любой из этих белков. Так, YM-58483 может модулировать депозависимый вход Са2+, воздействуя на Са2+-сенсор STIM1, может прямо блокировать пору Orai1-каналов, или влиять на взаимодействие белков STIM1 и Orai1. Методом конфокальной микроскопии на клетках эмбриональной почки HEK293 обнаружено, что YM-58483 не влияет на кластеризацию белков STIM1 или кластеризацию белков Orai1 (Sadaghiani et al., 2014; Rahman, Rahman, 2017). С использованием метода пэтч-клэмп показано, что YM-58483 ингибирует депозависимые Са2+-каналы при приложении только к наружной поверхности Т-клеток линии Jurkat человека (Zitt et al., 2004). В связи с этим полагают, что YM-58483 может ингибировать депозависимый вход Са2+, прямо взаимодействуя с белками Orai1 (Zitt et al., 2004; Derler et al., 2013; Jairaman, Prakriya, 2013; Rahman, Rahman, 2017). Обнаружено также, что ингибирующее действие YM-58483 связано с наличием в его структуре трифторметильной группы в положении С3 и С5 пиразольного кольца (Law et al., 2011).
Таким образом, в настоящей работе мы впервые на перитонеальных макрофагах крысы показали, что производное пиразола YM-58483 эффективно ингибирует депозависимый вход Са2+, вызываемый ингибиторами эндоплазматических Са2+-АТФаз (тапсигаргин, ЦПК) и иммуномодуляторами глутоксимом и моликсаном.
Полученные нами данные о модуляции YM-58483 депозависимого входа Са2+ в перитонеальных макрофагах могут иметь значение для терапии заболеваний, опосредованных нарушением функционирования комплекса депозависимого входа Са2+. Так, существует ряд заболеваний, связанных с уменьшением или увеличением депозависимого входа Са2+. Показано, что мутации генов, кодирующих белки STIM1 и Orai1, вызывающие уменьшение депозависимого входа Са2+ (loss-of-function), приводят к тяжелому комбинированному иммунодефициту (SCID), аутоиммунным заболеваниям, миопатиям и эктодермальной дисплазии, в то время как мутации, вызывающие увеличение депозависимого входа Са2+ (gain-of-function), вызывают синдром Сторморкена и синдром тромбоцитов Йорка, которые характеризуются тромбоцитопенией, легким геморрагическим диатезом и миопатией с трубчатыми агрегатами (Shaw, Feske, 2013; Lacruz, Feske, 2015; Vaeth, Feske, 2018; Feske, 2019). В связи с этим повысился интерес исследователей к разработке низкомолекулярных блокаторов депозависимых Са2+-каналов (Sweeney et al., 2009; Tian et al., 2016).
Фармакологические агенты, влияющие на комплекс депозависимого входа Са2+, имеют практическую значимость для регуляции функционирования клеток иммунной системы и терапии воспалительных и аутоиммунных заболеваний. Так, фармакологические модуляторы депозависимого входа Са2+ уже используются для терапии широкого спектра заболеваний, таких как воспалительное заболевание кишечника, астма, псориаз, острый панкреатит, ревматоидный артрит и злокачественные опухоли (Tian et al., 2016; Liu et al., 2017; Stauderman, 2018; Waldron et al., 2019). Проводится скрининг новых низкомолекулярных блокаторов депозависимого входа Са2+, которые могли бы использоваться в терапии заболеваний, связанных с нарушением депозависимого входа Са2+ (Rahman, Rahman, 2017). YM-58483 может стать одним из них. Так, обнаружено, что YM-58483 обладает анальгетическим действием при хронической и острой боли (Gao et al., 2013), а также при ревматоидном артрите у мышей (Gao et al., 2015; Miyoshi et al., 2018). Полагают, что YM-58483 может стать представителем нового класса анальгетических и противовоспалительных средств (Gao et al., 2013).
Таким образом, полученные нами данные свидетельствуют о том, что, как и для клеток других типов, YM-58483 может быть удобным фармакологическим инструментом для изучения депозависимого входа Са2+ в макрофагах и перспективным агентом для фармакологических исследований не только как иммунодепрессант, но и как препарат для лечения заболеваний, связанных с нарушением механизмов депозависимого входа Са2+. Кроме того, результаты дополнительно подтверждают, что вход Са2+, индуцируемый глутоксимом или моликсаном, происходит по депозависимому механизму.
Список литературы
Антушевич А.А., Антонов В.Г., Гребенюк А.Н., Антушевич А.Е., Ладанова Т.В., Бурова Е.Б. 2013. Патофизиологические основы эффективности глутоксима как средства сопровождения лучевой терапии рака ротоглотки. Вестник Рос. Военно-мед. акад. Т. 3. № 43. С. 32. (Antushevich A.A., Antonov V.G., Grebenyuk A.N., Antushevich A.E., Ladanova T.V., Burova E.B. 2013. Pathophysiologic rationale of effectiveness of glutoxim supportive therapy add-on to radiotherapy management of oropharyngeal cancer. Vestnik Rossiiskoi Voenno-medicinskoi akademii. V. 3. № 43. P. 32.)
Борисов А.Е, Кожемякин Л.А., Антушевич А.Е, Кетлицкая О.С., Кащенко В.А., Чепур С.В., Кацалуха В.В., Васюкова Е.Л., Новиченков А.О., Мотущук И.Е. 2001. Клинико-экспериментальное обоснование регионарного и системного введения препаратов группы тиопоэтинов при циррозе печени. Вестник хирургии им. И.И. Грекова. Т. 4. № 2. С. 32. (Borisov A.E., Kozhemyakin L.A., Antushevich A.E., Ketliskaya O.S., Kashchenko V.A., Chepur S.V., Katsalucha V.V., Vasyukova E.L., Novichenkov A.O., Motushchuk I.E. 2001. Clinical and experimental grounds of the regional and systemic administration of the thiopoetin group medicines for cirrhosis of the liver. First communication. Vestnic hirurgii im. I.I. Grekova. V. 4. № 2. P. 32.)
Курилова Л.С., Крутецкая З.И., Лебедев О.Е., Антонов В.Г. 2008. Влияние окисленного глутатиона и его фармакологического аналога препарата глутоксим на внутриклеточную концентрацию Са2+ в макрофагах. Цитология. Т. 50. № 5. С. 452. (Kurilova L.S., Krutetskaya Z.I., Lebedev O.E., Antonov V.G. 2008. The effect of oxidized glutathione and its pharmacological analogue glutoxim on intracellular Са2+ concentration in macrophages. Cell Tiss. Biol. (Tsitologiya). V. 2. P. 322.)
Курилова Л.С., Крутецкая З.И., Лебедев О.Е., Крутецкая Н.И., Антонов В.Г. 2012. Участие актинового цитоскелета в действии препаратов глутоксим и моликсан на внутриклеточную концентрацию Са2+ в макрофагах. Цитология. Т. 54. № 2. С. 135. (Kurilova L.S., Krutetskaya Z.I., Lebedev O.E., Krutetskaya N.I., Antonov V.G. 2012. The involvement of actin cytoskeleton in glutoxim and molixan effect on intracellular Ca2+ concentration in macrophages. Cell Tiss. Biol. (Tsitologiya). V. 6. № 3. P. 240.)
Соколова Г.Б., Синицын М.В., Кожемякин Л.А., Перельман М.И. 2002. Глутоксим в комплексной терапии туберкулеза. Антибиотики и химиотерапия. Т. 2. С. 20. (Sokolova G.B., Sinitsyn M.V., Kozhemiakin L.A., Perel’man M.I. 2002. Glutoxim in the complex treatment of tuberculosis. Antibiot. Khimioter. V. 2. P. 20.)
Толстой О.А., Цыган В.Н., Климов А.Г., Степанов А.В., Антушевич А.Е. 2019. Экспериментальная оценка эффективности препарата моликсан по восстановлению работоспособности вирусинфицированных лабораторных животных. Известия Рос. Военно-Мед. Акад. Т. 38. № 1. С. 271. (Tolstoy O.A., Tsygan V.N., Klimov A.G., Stepanov A.V., Antushevich A.E. 2019. Experimental evaluation of the efficiency of the drug molixan on restoring the operation of virus-infected laboratory animals. Bull. Russ. Military Med. Acad. V. 38. № 1. P. 271.)
Albarran L., Lopez J.J., Salido G.M., Rosado J.A. 2016. Histo-rical overview of store-operated Ca2+ entry. In: Calcium entry pathways in non-excitable cells. Wien: Springer-Verlag. P. 3.
Azimi I., Flanagan J.U., Stevenson R.J., Inserra M., Vetter I., Monteith G.R., Denny W.A. 2017. Evaluation of known and novel inhibitors of Orai1-mediated store-operated Ca2+ entry in MDA-MB-231 breast cancer cells using a Fluorescence Imaging Plate Reader assay. Bioorg. Med. Chem. V. 25. P. 440.
Berridge M.J., Bootman M.D., Lipp P. 1998. Calcium, a life and death signal. Nature. V. 395. P. 645.
Berridge M.J., Bootman M.D., Roderick H.L. 2003. Calcium signalling: dynamics, homeostasis and remodeling. Nat. Rev. Mol. Cell Biol. V. 4. P. 517.
Berridge M.J., Lipp P., Bootman M.D. 2000. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. V. 1. P. 11.
Bird G. S., Putney J.W. 2018. Pharmacology of store-operated calcium entry channels. In: Calcium entry channels in non-excitable cells. CRC Press. P. 311.
Bogeski I., Al-Ansary D., Qu B., Niemeyer B.A., Hoth M., Peinelt Ch. 2010. Pharmacology of ORAI channels as a tool to understand their physiological functions. Expert. Rev. Clin. Pharmacol. V. 3. P. 291.
Bruce J.I.E., Elliott A.C. 2000. Pharmacological evaluation of the role of cytochrome P450 in intracellular calcium signaling in rat pancreatic acinar cells. Brit. J. Physiol. V. 131. P. 761.
Conrad R. E. 1981. Induction and collection of peritoneal exudate macrophages. In: Manual of macrophages methodo-logy. N.Y.: Marcell Dekker. P. 5.
Derler I., Schindl R., Fritsch R., Heftberger P., Riedl M. Ch., Begg M., House D., Romanin Ch. 2013. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium. V. 53. P. 139.
Djuric S.W., BaMaung N.Y., Basha A., Liu H., Luly J.R., Madar D.J., Sciotti R.J., Tu N.P., Wagenaar F.L., Wiedeman P.E., Zhou X., Ballaron S., Bauch J., Chen Y.W., Chiou X.G., Fey T., Gauvin D., Gubbins E., Hsieh G.C., Marsh K.C., Mollison K.W., Pong M., Shaughnessy T.K., Sheets M.P., Smith M., Trevillyan J.M., Warrior U., Wegner C.D., Carter G.W. 2000. 3,5-Bis(trifluoromethyl)pyrazoles: a novel class of NFAT transcription factor regulator. J. Med. Chem. V. 43. P. 2975.
Feske St. 2019. CRAC channels and disease – From human CRAC channelopathies and animal models to novel drugs. Cell Calcium. V. 80. P. 112.
Gao R., Gao X., Xiaa J., Tiana Y., Barretta J.E., Daib Y., Hu H. 2013. Potent analgesic effects of a store-operated calcium channel inhibitor. Pain. V. 154. P. 2034.
Gao X.H., Gao R., Tian Y.Z., McGonigle P., Barrett J.E., Dai Y., Hu H. 2015. A store-operated calcium channel inhibitor attenuates collagen-induced arthritis. Br. J. Pharmacol. V. 172. P. 2991.
Goeger D.E., Riley R.T., Dorner J.W., Cole R.J. 1988. Cyclopiazonic acid inhibition of the Ca2+ transport ATPase in rat skeletal muscle sarcoplasmic reticulum vesicles. Biochem. Pharmacol. V. 37. P. 978.
Grynkiewicz G., Poenie M., Tsien R.Y. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence pro-perties. J. Biol. Chem. V. 260. P. 3440.
He L.-P., Hewavitharana T., Soboloff J., Spassova M. A., Gill D.L. 2005. A functional link between store-operated and TRPC channels revealed by the 3, 5-Bis(trifluoromethyl)pyrazole derivative, BTP2. J. Biol. Chem. V. 280. P. 10 997.
Ishikawa J., Ohga K., Yoshino T., Takezawa R., Ichikawa A., Kubota H., Yamada T. 2003. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J. Immunol. V. 170. P. 4441.
Jairaman A., Prakriya M. 2013. Molecular pharmacology of store-operated CRAC channels. Channels. V. 7. P. 402.
Kimura M., Nishi K., Higashikawa A., Ohyama S., Sakurai K., Tazaki M., Shibukawa Y. 2018. High pH-sensitive store-op-erated Ca2+ entry mediated by Ca2+ release-activated Ca2+ channels in rat odontoblasts. Front. Physiol. 9:443. https://doi.org/10.3389/fphys.2018.00443
Lacruz R.S., Feske St. 2015. Diseases caused by mutations in ORAI1 and STIM1. Ann. N.Y. Acad. Sci. V. 1356. P. 45.
Law M., Morales J.L., Mottram L.F., Iyer A., Peterson B.R., August A. 2011. Structural requirements for the inhibition of calcium mobilization and mast cell activation by the pyrazole derivative BTP2. J. Biochem. Cell Biol. V. 43. P. 1228.
Lewis R.S. 2020. Store-operated calcium channels: from function to structure and back again. Cold Spring Harb. Perspect. Biol. 12(5). https://doi.org/10.1101/cshperspect.a035055
Liu S., Hasegawa H., Takemasa E., Suzuki Y., Oka K., Kiyoi T., Takeda H., Ogasawara T., Sawasaki T., Yasukawa M., Maeyama K. 2017. Efficiency and safety of CRAC inhibitors in human rheumatoid arthritis xenograft models. J. Immunol. V. 199. P. 1584.
Lunz V., Romanin C., Frischauf I. 2019. STIM1 activation of Orai1. Cell Calcium. V. 77. P. 29.
Monahan R.A., Dvorak H.F., Dvorak A.M. 1981. Ultrastructural localization of nonspecific esterase activity in guinea pig and human monocytes, macrophages and lymphocytes. Blood. V. 58. P. 1089.
Moreno C., Vaca L. 2012. Microdomain organization of SOCE signaling. In: Store-operated Ca2+ entry (SOCE) pathways. Wien: Springer-Verlag. P. 93.
Miyoshi M., Liu Sh., Morizane A., Takemasa E., Suzuki Y., Kiyoi T., Maeyama K., Mogi M. 2018. Efficacy of constant long-term delivery of YM-58483 for the treatment of rheumatoid arthritis. Eur. J.Pharmacol. V. 824. P. 89.
Nguyen N.T., Han W., Cao W.-M., Wang Y., Wen S., Huang Y., Li M., Du L., Zhou Y. 2018. Store-operated calcium entry mediated by ORAI and STIM. Compr. Physiol. V. 8. P. 981.
Nurbaeva M. K., Eckstein M., Snead M.L., Feske S., Lacruz R.S. 2015. Store-operated Ca2+ entry modulates the expression of enamel genes. J. Dent. Res. V. 94. P. 1471.
Nwokonko R.M., Cai X., Loktionova N.A., Wang Y., Zhou Y., Gill D.L. 2017. The STIM-Orai pathway: conformational coupling between STIM and Orai in the activation of store-operated Ca2+ entry. Adv. Exp. Med. Biol. V. 993. P. 83.
Prakriya M., Lewis R.S. 2015. Store-operated calcium channels. Physiol. Rev. V. 95. P. 1383.
Putney J.W. 1990. Capacitative calcium entry revisited. Cell Calcium. V. 11. P. 611.
Putney J.W. 2001. Pharmacology of capacitative calcium entry. Mol. Interv. V. 1. P. 84.
Putney J.W. 2010. Pharmacology of store-operated calcium channels. Mol. Interv. V. 10. P. 209.
Putney J. W. 2011. The physiological function of store-operated calcium entry. Neurochem. Res. V. 36. P. 1157.
Putney J.W. 2017. Store-operated calcium entry: an historical overview. Adv. Exp. Med. Biol. V. 981. P. 205.
Rahman S., Rahman T. 2017. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca2+ entry pathway. Sci. Rep. 7:12881. https://doi.org/10.1038/s41598-017-13343-x
Randriamampita C., Trautmann A. 1987. Ionic channels in murine macrophages. Cell. Biol. V. 105. P. 761.
Sabourin J., Gal L.L., Saurwein L., Haefliger J.-A., Raddatz E., Allagnat F. 2015. Store-operated Ca2+ entry mediated by Orai1 and TRPC1 participates to insulin secretion in rat beta-cells. J. Biol. Chem. V. 290. P. 30 530.
Sadaghiani A.M., Lee S.M., Odegaard J.I., Leveson-Gower D.B., McPherson O.M., Novick P., Kim M.R., Koehler A.N., Negrin R., Dolmetsch R.E., Park Ch.Y. 2014. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem. Biol. V. 21. P. 1278.
Shaw P.J., Feske St. 2013. Physiological and pathophysiological functions of SOCE in the immune system. Front. Biosci. V. 4. P. 2253.
Stauderman K.A. 2018. CRAC channels as targets for drug discovery and development. Cell Calcium. V. 74. P. 147.
Sweeney Z.K., Minatti A., Button D.C., Patrick S. 2009. Small-molecule inhibitors of store-operated calcium entry. Chem. Med. Chem. V. 4. P. 706.
Takezawa R., Cheng H., Beck A., Ishikawa J., Launay P., Kubota H., Kinet J.P., Fleig A., Yamada T., Penner R. 2006. A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol. Pharmacol. V. 69. P. 1413.
Thastrup O., Dawson A.P., Scharff O., Foder B., Cullen P.J., Drobak B.K., Bjerrum P.J., Christensen S.B., Hanley M.R. 1989. Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions. V. 27. P. 17.
Tian Ch., Du L., Zhou Y., Li M. 2016. Store-operated CRAC channel inhibitors: opportunities and challenges. Future Med. Chem. V. 8. P. 817.
Trebak M., Kinet J.-P. 2019. Calcium signalling in T cells. Nat. Rev. Immunol. V. 19. P. 154.
Trevillyan J.M., Chiou X.G., Chen Y.W., Ballaron S.J., Sheets M.P., Smith M.L., Wiedeman P.E., Warrior U., Wilkins J., Gubbins E.J., Gagne G.D., Fagerland J., Carter G.W., Luly J.R., Mollison K.W., Djuric S.W. 2001. Potent inhibition of NFAT activation and T cell cytokine production by novel low molecular weight pyrazole compounds. J. Biol. Chem. V. 276. P. 48118.
Vaca L. 2010. SOCIC: the store-operated calcium influx complex. Cell Calcium. V. 47. P. 199.
Vaeth M., Feske S. 2018. Ion channelopathies of the immune system. Curr. Opin. Immunol. V. 52. P. 39.
Vig M., Kinet J.-P. 2009. Calcium signaling in immune cells. Nat. Immunol. V. 10. P. 21.
Wei D., Mei Y., Xia J., Hu H. 2017. Orai1 and Orai3 mediate store-operated calcium entry contributing to neuronal excitability in dorsal root ganglion neurons. Front. Cell. Neurosci. 11:400. https://doi.org/10.3389/fncel.2017.00400
Waldron R.T., Chen Y., Pham H., Go A., Su H.-Y., Hu Ch., Wen L., Husain S.Z., Sugar C.A., Roos J., Ramos S., Lugea A., Dunn M., Stauderman K., Pandol S.J. 2019. The Orai Ca2+ channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. J. Physiol. V. 597.12. P. 3085.
Xia J., Pan R., Gao X., Meucci O., Hu H. 2014. Native store-op-erated calcium channels are functionally expressed in mouse spinal cord dorsal horn neurons and regulate resting calcium homeostasis. J. Physiol. V. 592.16. P. 3443.
Xie Q., Zhang Y., Zhai C., Bonanno J. A. 2002. Calcium influx factor from cytochrome P-450 metabolism and secretion-like coupling mechanisms for capacitative calcium entry in corneal endothelial cells. J. Biol. Chem. V. 277. P. 16 559.
Zeng B., Chen G.-L., Garcia-Vaz E., Bhandari S., Daskoulidou N., Berglund L.M., Jiang H., Hallett T., Zhou L.-P., Huang L., Xu Z.-H., Nair V., Nelson R. G., Ju W., Kretzler M., Atkin S.L., Gomez M.F., Xu Sh.-Z. 2017. ORAI channels are critical for receptor-mediated endocytosis of albumin. Nat. Commun. 8:1920. https://doi.org/10.1038/s41467-017-02094-y
Zitt C., Strauss B., Schwarz E.C., Spaeth N., Rast G., Hatzelmann A., Hoth M. 2004. Potent inhibition of Ca2+-release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J. Biol. Chem. V. 279. P. 12 427.
Дополнительные материалы отсутствуют.