

Цитология, 2021, T. 63, № 2, стр. 176-183
Оценка уровня RAGE в клетках гематоэнцефалического барьера при экспериментальной болезни Альцгеймера
Я. В. Горина 1, *, Е. Д. Осипова 1, А. В. Моргун 1, Е. Б. Бойцова 1, О. Л. Лопатина 1, А. Б. Салмина 1
1 НИИ молекулярной медицины и патобиохимии Красноярского государственного медицинского университета
им. проф. В.Ф. Войно-Ясенецкого Министерства здравоохранения РФ
660022 Красноярск, Россия
* E-mail: yana_20@bk.ru
Поступила в редакцию 28.09.2020
После доработки 27.10.2020
Принята к публикации 28.10.2020
Аннотация
Болезнь Альцгеймера (БА) представляет собой прогрессирующее нейродегенеративное расстройство, при котором накопление β-амилоида и нейрофибриллярных клубков является определяющим патологическим признаком. Активация передачи сигналов белков RAGE (рецептора поздних продуктов гликозилирования) играет решающую роль в продукции β-амилоида, образовании нейрофибриллярных клубков и дегенерации нейронов. Цель работы – оценить влияние RAGE и его лиганда β-амилоида (Aβ1-42) на клетки эндотелия церебральных микрососудов в in vitro модели гематоэнцефалического барьера (ГЭБ) при экспериментальной БА. При моделировании экспериментальной БА in vitro выявлено значимое (P ≤ 0.05) повышение уровня RAGE на клетках эндотелия и снижение величины трансэндотелиального электрического сопротивления (ТЭС) как в статической, так и динамической моделях ГЭБ. Однако блокирование активности белка RAGE привело к стойкому и длительному повышению величины ТЭС. При этом лиганд RAGE (Aβ1-42) вызвал уменьшение величины ТЭС. БА сопровождается изменением уровня RAGE на клетках эндотелия, что приводит к нарушению структурно-функциональной целостности ГЭБ. Однако блокирование активности белка RAGE ингибирует развитие эндотелиальной дисфункции и восстанавливает целостность ГЭБ, нарушенную действием β-амилоида.
Болезнь Альцгеймера (БА) представляет собой хроническое нейродегенеративное заболевание, поражающее различные области головного мозга и характеризующееся церебральным накоплением бляшек β-амилоида, которые усиливают образование нейрофибриллярных клубков (Jeong, 2017; Chen, 2018). Это способствует гибели нейронов и нарушению синаптической пластичности, усиливая воспаление, окислительный стресс и развитие церебральных микрососудистых осложнений (Sengupta et al., 2016).
Известно, что увеличение продукции β-амилоида в ткани головного мозга, характерное для большинства случаев БА, может быть вызвано различными причинами, при этом снижение скорости клиренса приводит к его накоплению в паренхиме головного мозга и, в конечном итоге, образованию сенильных бляшек как ключевого признака БА (Oh et al., 2018). Рецептор конечных продуктов гликирования (RAGE), являющийся мультилигандным рецептором семейства иммуноглобулинов, участвует в патогенезе неврологических нарушений при БА, например, за счет участия в развитии нейровоспаления и взаимодействия с молекулой β-амилоида, выступающей в качестве его лиганда (Fang et al., 2010; Успенская и др., 2015). Гиперэкспрессия RAGE запускает каскад патологических событий, которые способствуют развитию окислительного стресса и нейровоспаления за счет повышения уровня ядерного фактора NF-kB и провоспалительных цитокинов, что, в конечном итоге, приводит к дисфункции нервных клеток (Tóbon-Velasco et al., 2014).
Интересно, что повышение уровня RAGE диагностируется уже на ранней стадии развития БА (Takeuchi et al., 2007). Кроме того, RAGE обладает значимой транспортной активностью, что было показано не только для β-амилоида, но и для нейропептидов, в частности, окситоцина (Yamamoto et al., 2019). Экспериментально установлено, что аномальная транслокация β-амилоида через гематоэнцефалический барьер (ГЭБ) играет роль в развитии БА (Mohamed et al., 2017). Транслокация в основном осуществляется через RAGE и белок LRP1 – белок-1, подобный рецептору липопротеинов низкой плотности. При этом RAGE действует как транспортер, регулирующий поступление в ткань головного мозга β-амилоида, тогда как выход β-амилоида в периферический кровоток происходит при непосредственном участии LRP1 (Guo et al., 2016). Примечательно, что клинические исследования продемонстрировали взаимосвязь между патологическими признаками БА и уровнями LRP1 и RAGE, а именно, у пациентов с БА наблюдали низкий уровень LRP1 и высокий уровень RAGE, что в свою очередь вызывало интенсивное отложение церебрального β-амилоида (Donahue et al., 2006).
Клетки церебральных микрососудов являются основным компонентом гематоэнцефалического барьера (ГЭБ) и экспрессируют большое число транспортных молекул. Развитие церебральной амилоидной ангиопатии сопровождается нарушением целостности ГЭБ (Yamada et al., 2008). β-Амилоид оказывает RAGE-опосредованное цитотоксическое действие на белки плотных контактов между эндотелиальными клетками церебральных микрососудов (Wan et al., 2015). Показано, что взаимодействие β-амилоида с RAGE индуцирует повышение проницаемости ГЭБ за счет нарушения экспрессии белков плотных контактов (ZO-1) в плазматической мембране, увеличения секреции внутриклеточного кальция и секреции матриксной металлопротеиназы в культивируемых эндотелиальных клетках, что указывает на потенциальный молекулярный путь, лежащий в основе β-амилоид-RAGE-индуцированного нарушения целостности ГЭБ. При этом блокирование активности RAGE, ингибирование матриксной металлопротеиназы предотвращало вызванное β-амилоидом патологическое изменение содержания ZO-1, тем самым снижая негативное влияние β-амилоида на эндотелий микрососудов головного мозга и нейротоксический эффект (Kook et al., 2012).
Несмотря на значительный интерес к механизмам транспорта через ГЭБ и токсического действия β-амилоида на клетки эндотелия микрососудов головного мозга, многие стороны этих процессов остаются не выясненными. В связи с этим, цель настоящего исследования заключалась в оценке роли RAGE и его лиганда β-амилоида (Aβ1-42) в клетках эндотелия церебральных микрососудов в модели ГЭБ in vitro при экспериментальной БА.
МАТЕРИАЛ И МЕТОДИКА
Клетки. Материалом для создания моделей ГЭБ in vitro служили первичные культуры клеток астроцитов, нейронов и церебральных эндотелиоцитов, выделенные из головного мозга эмбрионов мыши линии C57BL/6.
Выделение церебральных эндотелиоцитов из коры головного мозга проводили согласно протоколу (Liu et al., 2013). Для этого выделяли кору головного мозга, мелко нарезали, центрифугировали при ускорении 150 g в течение 3 мин при 20°С. Затем добавляли 25% фетальной бычьей сыворотки (FBS, HyClone, Южная Америка), тритурировали, гомогенат центрифугировали при ускорении 600 g в течение 10 мин при 20°С. Забирали нижний слой, тритурировали и центрифугировали 3 раза, затем проводили ферментативную обработку пеллета 0.1%-ным раствором коллагеназы II (ПанЭко, Россия) при 37°С в течение 35 мин с последующим центрифугированием при ускорении 150 g в течение 5 мин. Далее культивировали фрагменты и отдельные эндотелиальные клетки в культуральных флаконах, покрытых желатином (gelatin solution 0.1%; Biological Industries, США) в культуральной среде, состоящей из DMEM (ПанЭко, Россия), 20% FBS (HyClone, Южная Америка), 3 мг/мл глюкозы (Sigma-Aldrich, США), 0.58 мг/мл глутамина (Sigma-Aldrich, США), 100 ЕД/мл пенициллина и 100 мг/мл стрептомицина (HyClone, Южная Америка), при 5% CO2 и 37°С.
Для культивирования нейросфер из головного мозга выделяли гиппокамп и стенки боковых желудочков, иссекали, помещали в 2%-ный раствор глюкозы (Sigma-Aldrich, США) в фосфатно-солевом буфере (Sigma-Aldrich, США) на 1 мин, далее удаляли супернатант, а оставшуюся ткань ресуспензировали в 1 мл культуральной среды NeuroCult NS-A Prolife-ration (StemCell, Канада), тритурировали. К полученной суспензии добавляли 1 мл культуральной среды NeuroCult NS-A Proliferation, собирали супернатант, центрифугировали при ускорении 150 g в течение 5 мин, затем удаляли супернатант и добавляли 1 мл культуральной среды NeuroCult NS-A Proliferation с последующей тритурацией.
С помощью цитометра Scepter Cell Counter (Millipore, США) проводили подсчет числа клеток. Клетки (1.5 × 106 кл./мл расссевали в культуральные флаконы с 25 мл культуральной среды NeuroCult NS-A Proliferation, инкубировали при 5% CO2 и 37°C. Образование нейросфер наблюдали через 24–48 ч.
Получение астроцитов и нейронов осуществляли из нейросфер согласно методике, представленной ранее (Моргун и др., 2013). Для этого использовали культуральную среду NeuroCult NS-A Proliferation Kit (StemCell, Канада), содержащую гепарин, основной фактор роста фибробластов (bFGF) и эпидермальный фактор роста EGF (Goustard-Langelier et al., 2012). Клетки (1.5 × 106 кл./мл) рассевали в культуральные флаконы с 25 мл культуральной среды, затем выполняли дифференцировку нейросфер в астроциты и нейроны в культуральной среде NeuroCult NS-A Differentiation Kit (Моргун и др., 2013).
Моделирование ГЭБ in vitro. После получения культур клеток-компонентов моделировали статическую и динамическую модели ГЭБ. Для создания статической трехкомпонентной модели ГЭБ in vitro на дно 24-луночного планшета (Corning, США) засевали астроциты и нейроны по 1.5 × 105 клеток в каждую лунку. После чего на культуральные вставки добавляли столько же (1.5 × 105) церебральных эндотелиоцитов на каждую вставку. Далее клетки-компоненты ГЭБ сокультивировали в среде DMEM (ПанЭко, Россия) с 10% FBS (HyClone, Южная Америка), 0.58 мг/мл глутамина (Sigma-Aldrich, США), 100 Ед/мл пенициллина и 100 мг/мл стрептомицина (HyClone, Южная Америка) в условиях CO2-инкубатора. Моделирование экспериментальной БА в статической модели ГЭБ in vitro осуществляли путем добавления к клеткам Аβ1-42 (Sigma-Aldrich, США) в конечной концентрации 10 мкМ в питательной среде.
Динамическую модель ГЭБ in vitro использовали в двух вариантах: коммерческая закрытая система для микроперфузии CellASIC ONIX (Merck Millipore, США) и открытая система на основе перфузируемых вставок и лунок 24-луночного культурального планшета.
Для создания динамической модели ГЭБ с использованием системы микроперфузии CellASIC ONIX (Merck Millipore, США) в камеру планшета для микрофлюидной установки засевали клетки-компоненты ГЭБ для формирования трехмерной модели, при этом астроциты и нейроны были отделены от церебральных эндотелиоцитов мембраной рабочей камеры. Соотношение нейронов, астроцитов и эндотелиоцитов составляло 1 : 2 : 1. Клетки культивировали в среде DMEM (ПанЭко, Россия), содержащей 10% FBS (HyClone, Южная Америка), 0.58 мг/мл глутамина (Sigma-Aldrich, США), 100 Ед/мл пенициллина и 100 мг/мл стрептомицина (HyClone, Южная Америка) при скорости потока культуральной среды 1.25 мкл/ч при 5% CO2 и 37°C в течение 48 ч. Моделирование экспериментальной БА в динамической модели ГЭБ in vitro осуществляли путем добавления к клеткам амилоида Аβ1-42 (Sigma-Aldrich, США) в конечной концентрации 10 мкМ в питательной среде.
Динамическая модель ГЭБ на основе перфузируемых вставок и лунок 24-луночного культурального планшета была разработана с возможностью использования шприцевого насоса со стеклянными шприцами типа Hamilton (Швейцария). При присоединении шприцев с обратной стороны каретки насоса была получена система для удаления жидкости из открытой камеры синхронизированно с ее подачей. Апробация системы происходила в течение 2-х сут. Учитывая разницу рабочих объемов лунок в системе CellASIC ONIX (Merck Millipore, США) и 24-луночного планшета, скорость потока среды была увеличена до 1.5 мкл/мин. Использование открытой системы на основе перфузируемых вставок и лунок позволило провести регистрацию ТЭС, что не представляется возможным при использовании закрытых систем.
Иммуногистохимическое исследование. Как в статической, так и динамической моделях ГЭБ in vitro, эндотелиальные клетки фенотипировали с использованием первичных антител против: ZO-1 (разведение 1 : 1000; Santa Cruz, Biotechnology, США), маркера астроцитов GFAP (1 : 1000), маркера нейронов NSE (1 : 1000) и вторичных антител Alexa Fluor 488 (1 : 1000; все от Abcam, Великобритания) согласно протоколу иммуногистохимии (García-Cabezas et al., 2016; Yu et al., 2016).
Для оценки экспрессии RAGE были сформированы 4 экспериментальные группы: контрольная К-I – интактные эндотелиоциты, культивируемые в статической модели ГЭБ; экспериментальная Э-I – эндотелиоциты в присутствии 10 мкМ Аβ1-42, культивируемые в статической модели ГЭБ (моделирование экспериментальной БА); контрольная К-II – интактные эндотелиоциты, культивируемые в динамической модели ГЭБ (закрытая система); экспериментальная Э-II – эндотелиоциты в присутствии 10 мкМ Аβ 1-42, культивируемые в динамической модели ГЭБ (закрытая система; моделирование экспериментальной БА).
Оценку наличия на поверхности целевой молекулы RAGE (Abcam, Великобритания) проводили по стандартному протоколу иммуноокрашивания (García-Cabezas et al., 2016; Yu et al., 2016). Первичные антитела использовали в рабочем разведении 1 : 300. Время инкубации составляло 18 ч при 4°С. Вторичные антитела Alexa Fluor 488 (1 : 1000; Abcam, Великобритания) использовали в разведении 1 : 500, время инкубации составляло 2 ч при 37°С. Микроскопию клеток осуществляли на флуоресцентном микроскопе ZOE (Bio-Rad, США). Считали относительное количество клеток, несущих каждый из видов антигена в образце при анализе не менее 10 полей зрения. При обработке результатов учитывали относительное количество всех клеток, несущих интересующую метку (метки) и выражали в процентах от их общего количества.
Регистрация трансэндотелиального электрического сопротивления (ТЭС). Для непосредственного измерения ТЭС использовали эпителиальный вольтметр EVOM2 и электрод STX2 (World Precision Instruments, США). Регистрацию проводили при формировании модели ГЭБ in vitro в следующих экспериментальных группах: К-I (интактные эндотелиоциты, культивируемые в статической модели ГЭБ); Э-I с моделированием экспериментальной БА (эндотелиоциты в присутствии 10 мкМ Аβ1-42, культивируемые в статической модели ГЭБ); К-III (интактные эндотелиоциты, культивируемые в динамической модели ГЭБ (открытой системе)); Э-III с моделью экспериментальной БА – эндотелиоциты в присутствии 10 мкМ Аβ1-42, культивируемые в динамической модели ГЭБ (открытой системе).
Модуляция активности RAGE и регистрация ТЭС. Для подавления активности RAGE в клетках эндотелия использовали siRNA RAGE (Santa Cruz, США). Трансфекцию siRNA осуществляли методом электропорации с использованием системы трансфекции Nucleofector 2b Device (Lonza, Швейцария) и набора Basic Nucleofector Kit for Primary Mammalian Endothelial Cells (Lonza, Швейцария) по протоколу, рекомендованному фирмой-производителем.
Для химической модуляции активности RAGE в качестве лиганда RAGE использовали Aβ1-42 (Sigma-Aldrich, США) в конечной концентрации 1 мкМ, инкубацию клеток проводили в течение 1 ч.
Регистрацию ТЭС через 1, 2, 4, 6 и 24 ч проводили в контрольной группе К-III и группе Э-III, в каждой из которой было сформировано по 3 подгруппы: интактная – культура эндотелиальных клеток без воздействия siRNA или химических модуляторов; группа siRNA RAGE – эндотелиальные клетки после воздействия siRNA RAGE и группа Aβ1-42 – эндотелиальные клетки после воздействия лиганда RAGE (Aβ1-42).
Статистический анализ. Анализ полученных результатов проводили с помощью программы GraphPad Prizm 8.0.1 (версия 8.0, США). Критерий Колмагорова–Смирнова использовали для оценки нормальности распределения. При отсутствии условий нормальности распределения применяли U-критерий Манна–Уитни. Различия принимали значимыми при P ≤ 0.05. Результаты представлены в виде Me (Q1; Q3), где Ме – медиана, Q1 – нижний квартиль, Q3 – верхний квартиль.
РЕЗУЛЬТАТЫ
При оценке уровня RAGE в интактных эндотелиоцитах, которые культивировали в условиях статической и динамической моделей ГЭБ in vitro, мы не обнаружили значимых различий (P > 0.05). Подобную ситуацию наблюдали и при моделировании экспериментальной БА in vitro (при культивировании клеток с Аβ1-42 в концентрации 10 мкМ) как в статической, так и динамической моделях ГЭБ in vitro (рис. 1а, б).
Рис. 1.
Уровень RAGE на клетках эндотелия при культивировании в статической и динамической моделях ГЭБ in vitro. а – количество RAGE+-клеток интактных эндотелиоцитов, культивируемых в статической (контрольная группа К-I) и динамической (контрольная группа К-II) моделях ГЭБ in vitro (различия статистически не значимы; P > 0.05, U-критерий Манна−Уитни); б – количество RAGE+-клеток эндотелиоцитов в экспериментальных группах при культивировании в присутствии 10 мкМ Аβ1-42 в статической (группа Э-I) и динамической (группа Э-II) моделях ГЭБ in vitro (различия статистически не значимы; P > 0.05, U-критерий Манна−Уитни). Показано количество иммунопозитивных клеток (RAGE+) в % от общего количества клеток в поле зрения. На рис. 1—4 результаты представлены в виде Me (Q1; Q3).

Однако необходимо отметить, что влияние β-амилоида в статических и динамических моделях ГЭБ in vitro вызвало однонаправленные изменения количества эндотелиальных клеток, несущих RAGE (рис. 2а, б) – количество клеток с RAGE возрaстало.
Рис. 2.
Влияние β-амилоида на уровень RAGE на клетках эндотелия при культивировании в статической и динамической моделях ГЭБ in vitro. а – Количество RAGE+-эндотелиоцитов в контрольной (К-I) и экспериментальной (Э-I) группах при культивировании в статической модели ГЭБ in vitro; б – количество RAGE+-эндотелиоцитов в контрольной (К-II) и экспериментальной (Э-II) круппах при культивировании в динамической модели ГЭБ in vitro. Количество положительных клеток представлено в процентах от общего количества клеток в поле зрения. *, #Различия статистически значимы при P < 0.05, U-критерий Манна−Уитни.

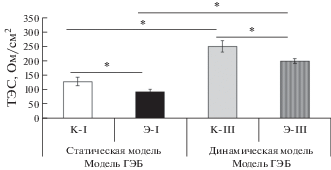
При оценке влияния условий культивирования интактных клеток, а также в экспериментальной модели БА установлено, что присутствие в модели Аβ1-42 приводит к статистически значимому (P ≤ 0.05) снижению величины ТЭС, как в статической, так и динамической моделях ГЭБ in vitro, что соответствует повышению проницаемости ГЭБ, регистрируемому вследствие токсического действия β-амилоида. Также выявлено, что культивирование клеток в динамической модели приводит к статистически значимому (P ≤ 0.05) увеличению величины ТЭС, как в контрольной группе клеток, так и при моделировании БА in vitro (рис. 3).
Рис. 3.
Изменение трансэндотелиального электрического сопротивления (ТЭС) в статической и динамической моделях ГЭБ in vitro в контрольных и экспериментальных группах. *Различия между группами статистически значимы при P < 0.05, U-критерий Манна−Уитни.
При оценке эффектов модуляции RAGE в контрольной культуре эндотелиальных клеток (группа К-III) с использованием Aβ1-42 в качестве лиганда и соответствующей siRNA для блокирования активности RAGE, выявлены изменения величины ТЭС. Блокирование активности RAGE приводит к статически значимому (P ≤ 0.05) увеличению показателей ТЭС в первые часы наблюдения с последующим его уменьшением к концу суток. Культивирование эндотелиальных клеток в присутствии лиганда RAGE (Aβ1-42) практически не оказало влияния на величину ТЭС (рис. 4а).
Рис. 4.
Влияние RAGE-модуляторов на величину ТЭС в динамической модели ГЭБ in vitro. а – В контрольной группе (К-III); б – в экспериментальной группе (Э-III). *Различия между группами статистически значимы при P < 0.05, U-критерий Манна−Уитни.
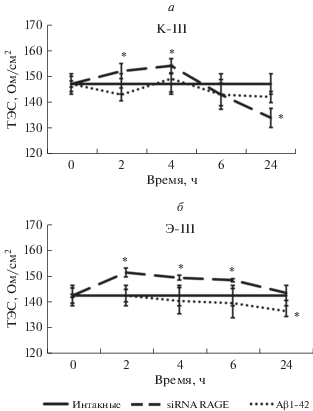
Однако в модели БА in vitro блокирование экспрессии RAGE привело к более стойкому и длительному повышению величины ТЭС. При этом лиганд RAGE вызвал уменьшение величины ТЭС через 24 ч после его добавления к культуре клеток (рис. 4б).
ОБСУЖДЕНИЕ
При развитии нейродегенерации альцгеймеровского типа β-амилоид может накапливаться в ткани мозга за счет усиленной продукции, значительного транспорта из крови и нарушенной элиминации. Как известно, существует три пути элиминации β-амилоида в головном мозге: трансбарьерная доставка в периферичесий кровоток, деградация в ткани мозга и периваскулярный дренаж (Ueno et al., 2014), а соотношение активности RAGE и LRP1 определяет эффективность клиренса β-амилоида (Deane et al., 2004; Gool et al., 2019), которые рассматриваются в качестве терапевтических мишеней при лечении БА (Jedlitschky et al., 2014). При этом скорость клиренса через ГЭБ в 6 раз выше, чем через общий поток спинномозговой жидкости (Bell et al., 2007) и примерно в 2 раза выше, чем скорость деградации (Qosa et al., 2014). Таким образом, нарушение притока и оттока β-амилоида через ГЭБ, по-видимому, являются основным триггером его накопления в головном мозге (Bloom, 2014; Welikovitch et al., 2018).
Повышение уровня RAGE в головном мозге регистрируется уже на ранней стадии БА (Criscuolo et al., 2017), тем самым ускоряя процесс нейродегенерации (Origlia et al., 2009). Важно отметить и то, что, согласно данным клинических исследований, у пациентов с БА повышенная иммунореактивность RAGE наблюдается в основном в нейронах, микроглии и астроцитах гиппокампа и нижней лобной коре (Lue et al., 2001). Недавние исследования показали, что после добавления к культуре нейрональных клеток гиппокампа Aβ 25–35 наблюдали увеличение содержания RAGE, толл-подобного рецептора 4 (TLR4) и ядерного фактора kB на уровне мРНК, наряду с медиаторами воспаления (HMGB1, IL-1β, IL-6 и TNF-α). Этот факт указывает на то, что нейровоспаление является ключевым фактором в прогрессировании БА посредством активации передачи сигналов RAGE/TLR4 (Nan et al., 2019).
Кроме того, гиперэкспрессия RAGE на эндотелии церебральных микрососудов у пациентов с БА наблюдается уже на ранней стадии и заметно увеличивается по мере прогрессирования заболевания (Miller et al., 2008). Поэтому помимо интенсификации процессов транспорта β-амилоида в ткань головного мозга, повреждающее действие β-амилоида может выражаться в альтерации межклеточных взаимодействий в ГЭБ и нарушении метаболизма клеток эндотелия, нарушая процессы неоангиогенеза и механизмы поддержания целостности барьера.
Мы обнаружили, что in vitro модели ГЭБ (статическая и динамическая конфигурации) пригодны для изучения роли RAGE в клетках церебрального эндотелия в (пато)физиологических условиях. Используя эти модели, мы обнаружили, что в эндотелии увеличивается уровень RAGE, ответственных за транспорт β-амилоида, что способствует увеличению проницаемости ГЭБ, реализуемой за счет парацеллюлярного транспорта и (или) повреждения клеток эндотелия. Подавление экспрессии RAGE предупреждает развитие патологической проницаемости ГЭБ in vitro. Коль скоро повышение ТЭС регистрируется и в интактном эндотелии с подавленной экспрессией RAGE, логично предположить, что физиологический уровень экспрессии RAGE повышает пассивную парацеллюлярную проницаемость ГЭБ. Важно, что как статическая, так и динамическая модели ГЭБ демонстрируют схожие качественные и количественные характеристики не только в физиологических условиях, но и при моделировании эндотелиотоксического эффекта β-амилоида in vitro, что служит основанием для рекомендации их применения для решения задач нейробиологии и нейрофармакологии при изучении нейродегенерации альцгеймеровского типа.
Ранее было показано применительно к RAGE в нейронах, что снижение активности RAGE может иметь цитопротекторный эффект, особенно для сохранения нейрональной функции на ранних стадиях развития заболевания (Arancio et al., 2004). Полученные нами результаты расширяют представления о роли RAGE в регуляции проницаемости ГЭБ в норме и при нейродегенерации альцгеймеровского типа.
Список литературы
Моргун А.В., Кувачева Н.В., Комлева Ю.К., Кутищева И.А., Окунева О.С., Дробушевская А.И., Хилажева Е.Д., Черепанов С.М., Салмина А.Б. 2013. Дифференцировка эмбриональных прогениторных клеток мозга крыс в астроциты и нейроны. Сибирское медицинское обозрение. № 6. С. 9. (Morgun A.V., Kuvacheva N.V., Komleva Yu.K., Kutishcheva I.A., Okuneva O.S., Drobushevskaya A.I., Khilazheva E.D., Cherepanov S.M., Salmina A. B. 2013. Differentiation of rat brain embryonic progenitor cells into astrocytes and neurons. Siberian Med.l Revi. № 6. P. 9.)
Успенская Ю.А., Комлева Ю.К., Пожиленкова Е.А., Салмин В.В., Лопатина О.Л., Фурсов А.А., Лаврентьев П.В., Белова О.А., Салмина А.Б. 2015. Лиганды RAGE-белков: роль в межклеточной коммуникации и патогенезе воспаления. Вестник Российской академии медицинских наук. Т. 70. № 6. С. 704. (Uspenskaya Yu.A., Komleva Yu.K., Pozhilenkova E.A., Salmin V.V., Lopatina O.L., Fursov A.A., Lavrent’ev P.V., Belova O.A., Salmina A.B. 2015. RAGE Protein ligands: Role in cellular communication and inflammation pathogenesis. Bulletin Russ. Acad. Med. Scis. V. 70. № 6. P. 704.)
Arancio O., Zhang H.P., Chen X., Lin C., Trinchese F., Puzzo D., Liu S., Hegde A., Yan F.S., Stern A., Luddy J.S., Lue L.-F., Walker D.G., Roher A., Buttini M. et al. 2004. RAGE Potentiates abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. V. 23. P. 4096. https://doi.org/10.1038/sj.emboj.7600415
Bell R.D., Sagare A.P., Friedman A.E., Bedi G.S., Holtzman D.M., Deane R., Zlokovic B.V. 2007. Transport pathways for clearance of human alzheimer’s amyloid beta-peptide and apolipoproteins e and j in the mouse central nervous system. J. Cereb. Blood Flow Metab. V. 27. P. 909. https://doi.org/10.1038/sj.jcbfm.9600419
Bloom G.S. 2014. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. V. 71. P. 505. https://doi.org/10.1001/jamaneurol.2013.5847
Chen Y.G. 2018. Research progress in the pathogenesis of Alzheimer’s Disease. Chin. Med. J. (Engl). V. 131. P. 624. https://doi.org/10.4103/0366-6999.235112
Criscuolo C., Fontebasso V., Middei S., Stazi M., Ammassari-Teule M., Yan S.S., Origlia N. 2017. Entorhinal cortex dysfunction can be rescued by inhibition of microglial rage in an Alzheimer’s disease mouse model. Sci. Rep. V. 7. P. 42 370. https://doi.org/10.1038/srep42370
Deane R., Wu Z., Zlokovic B.V. 2004. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. V. 35. P. 2628. https://doi.org/10.1161/01.STR.0000143452.85382.d1
Donahue J.E., Flaherty S.L., Johanson C.E., Duncan J.A., Silverberg G.D., Miller M.C., Tavares R., Yang W., Wu Q., Sabo E., Hovanesian V., Stopa E.G. 2006. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. V. 112. P. 405. https://doi.org/10.1007/s00401-006-0115-3
Fang F., Lue L.-F., Yan S., Xu H., Luddy J.S., Chen D., Walker D.G., Stern D.M., Yan S., Schmidt A.M., Chen J.X., Yan S.S. 2010. RAGE-dependent signaling in microglia contributes to neuroinflammation, abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. V. 24. P. 1043. https://doi.org/10.1096/fj.09-139634
García-Cabezas M.Á., John Y.J., Barbas H., Zikopoulos B. 2016. Distinction of neurons, glia and endothelial cells in the cerebral cortex: an algorithm based on cytological features. Front. Neuroanat. V. 10. P. 107. https://doi.org/10.3389/fnana.2016.00107
Gool B.V., Storck S.E., Reekmans S.M., Lechat B., Gordts P.L.S.M., Pradier L., Pietrzik C.U., Roebroek A.J.M. 2019. LRP1 Has a predominant role in production over clearance of Aβ in a mouse model of Alzheimer’s disease. Mol. Neurobiol. V. 56. P. 7234. https://doi.org/10.1007/s12035-019-1594-2
Goustard-Langelier B., Koch M., Lavialle M., Heberden C. 2012. Rat neural stem cell proliferation and differentiation are durably altered by the in utero polyunsaturated fatty acid supply. J. Nutr. Biochem. V. 24. P. 380. https://doi.org/10.1016/j.jnutbio.2012.08.001
Guo Y.-X., He L.-Y., Zhang M., Wang F., Liu F., Peng W.-X. 2016. 1,25-Dihydroxyvitamin D3 regulates expression of LRP1 and RAGE in vitro and in vivo, enhancing Aβ1-40 brain-to-blood efflux and peripheral uptake transport. Neuroscience. V. 322. P. 28. https://doi.org/10.1016/j.neuroscience.2016.01.041
Jedlitschky G., Grube M., Mosyagin I., Kroemer H.K., Vogelgesang S. 2014. Targeting CNS transporters for treatment of neurodegenerative diseases. Curr. Pharm. Des. V. 20. P. 1523. https://doi.org/10.2174/13816128113199990460
Jeong S. 2017. Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Mol. Cells. V. 40. P. 613. https://doi.org/10.14348/molcells.2017.0096
Kook S.-Y., Hong H.S., Moon M., Ha C.M., Chang S., Mook-Jung I. 2012. Aβ1−42-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca2+-calcineurin signaling. J. Neurosci. V. 32. P. 8845. https://doi.org/10.1523/JNEUROSCI.6102-11.2012
Liu Y., Xue Q., Tang Q., Hou M., Qi H., Chen G., Chen W., Zhang J., Chen Y., Xu X. 2013. A simple method for isolating and culturing the rat brain microvascular endothelial cells. Microvasc. Res. V. 90. P. 199. https://doi.org/10.1016/j.mvr.2013.08.004
Lue L.F., Walker D.G., Brachova L., Beach T.G., Rogers J., Schmidt A.M., Stern D.M., Yan S.D. 2001. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: Identification of a cellular activation mechanism. Exper. Neurol. V. 1. P. 29. https://doi.org/10.1006/exnr.2001.7732
Miller M.C., Tavares R., Johanson C.E., Hovanesian V., Donahue J.E., Gonzalez L., Silverberg G.D., Stopa E.G. 2008. Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease. Brain Res. V. 1230. P. 273. https://doi.org/10.1016/j.brainres.2008.06.124
Mohamed L.A., Zhu H., Mousa Y.M., Wang E., Qiu W.Q., Kaddoumi A. 2017. Amylin enhances amyloid-β peptide brain to blood efflux across the blood-brain barrier. J. Alzheimers Dis. V. 56. P. 1087. https://doi.org/10.3233/JAD-160800
Nan K., Han Y., Fang Q., Huang C., Yu L., Ge W., Xiang F., Tao Y.-X., Cao H., Li J. 2019. HMGB1 gene silencing inhibits neuroinflammation via down-regulation of NF-κB signaling in primary hippocampal neurons induced by Aβ 25–35. Int. Immunopharmacol. V. 67. P. 294. https://doi.org/10.1016/j.intimp.2018.12.027
Oh S., Son M., Choi J., Lee S., Byun K. 2018. sRAGE prolonged stem cell survival and suppressed RAGE-related inflammatory cell and T lymphocyte accumulations in an Alzheimer’s disease model. Biochem. Biophys. Res. Commun. V. 495. P. 807. https://doi.org/10.1016/j.bbrc.2017.11.035
Origlia N., Arancio O., Domenici L., Yan S.S. 2009. Beta-Amyloid and synaptic dysfunction: The role of RAGE affiliations expand. Expert Rev. Neurother. V. 9. P. 1635. https://doi.org/10.1586/ern.09.107
Qosa H., Abuasal B.S., Romero I.A., Weksler B., Couraud P.-O., Keller J.N., Kaddoumi A. 2014. Differences in amyloid-β clearance across mouse and human blood-brain barrier models: Kinetic analysis and mechanistic modeling. Neuropharmacol. V. 79. P. 668. https://doi.org/10.1016/j.neuropharm.2014.01.023
Sengupta U., Nilson A.N., Kayed R. 2016. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. V. 6. P. 42. https://doi.org/10.1016/j.ebiom.2016.03.035
Takeuchi M., Sato T., Takino J., Kobayashi Y., Furuno S., Kikuchi S., Yamagishi S. 2007. Diagnostic utility of serum or cerebrospinal fluid levels of toxic advanced glycation end-products (TAGE) in early detection of Alzheimer’s disease. Med. Hypotheses. V. 69. P. 1358. https://doi.org/10.1016/j.mehy.2006.12.017
Tóbon-Velasco J.C., Cuevas E., Torres-Ramos M.A. 2014. Receptor for ages (rage) as mediator of NF-kb pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug. Targets. V. 13. P. 1615. https://doi.org/10.2174/1871527313666140806144831
Ueno M., Chiba Y., Matsumoto K., Nakagawa T., Miyanaka H. 2014. Clearance of beta-amyloid in the brain. Curr. Med. Chem. V. 21. P. 4085. https://doi.org/10.2174/0929867321666141011194256
Wan W., Cao L., Liu L., Zhang C., Kalionis B., Tai X., Li Y., Xia S. 2015. Aβ(1-42) oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J. Neurochem. V. 134. P. 382. https://doi.org/10.1111/jnc.13122
Welikovitch L.A., Carmo S.D., Maglóczky Z., Szocsics P., Lőke J., Freund T., Cuello A.C. 2018. Evidence of intraneuronal Aβ accumulation preceding Tau pathology in the entorhinal cortex. Acta Neuropathol. V. 136. P. 901. https://doi.org/10.1007/s00401-018-1922-z
Yamada K., Hashimoto T., Yabuki C., Nagae Y., Tachikawa M., Strickland D.K., Liu Q., Bu G., Basak J.M., Holtzman D.M., Ohtsuki S., Terasaki T., Iwatsubo T. 2008. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J. Biol. Chem. V. 283. P. 34554. https://doi.org/10.1074/jbc.M801487200
Yamamoto Y., Liang M., Munesue S., Deguchi K., Harashima A., Furuhara K., Yuhi T., Zhong J., Akther S., Goto H., Eguchi Y., Kitao Y., Hori O., Shiraishi Y., Ozaki N. et al. 2019. Vascular RAGE transports oxytocin into the brain to elicit its maternal bonding behaviour in mice. Commun. Biol. V. 2. P. 76. https://doi.org/10.1038/s42003-019-0325-6
Yu Q.C., Song W., Wang D., Zeng Y.A. 2016. Identification of blood vascular endothelial stem cells by the expression of protein C receptor. Cell Res. V. 26. P. 1079. https://doi.org/10.1038/cr.2016.85
Дополнительные материалы отсутствуют.