

Цитология, 2021, T. 63, № 6, стр. 513-530
Межклеточный транспорт митохондрий: молекулярные механизмы и роль в поддержании энергетического гомеостаза в тканях
Ю. А. Успенская 1, 2, *, Н. А. Малиновская 1, А. Б. Салмина 1, 3
1 Научно-исследоватeльский институт молекулярной медицины и патобиохимии Красноярского государственного медицинского университета им. проф. В.Ф. Войно-Ясенецкого Министерства здравоохранения РФ
660022 Красноярск, Россия
2 Красноярский государственный аграрный университет
660049 Красноярск, Россия
3 Отдел исследований мозга Научного центра неврологии
125367 Москва, Россия
* E-mail: yulia.uspenskaya@mail.ru
Поступила в редакцию 16.08.2021
После доработки 26.08.2021
Принята к публикации 30.08.2021
Аннотация
Митохондрии, которые определяют метаболические процессы в клетках и клеточную выживаемость, часто претерпевают структурно-динамические изменения при различных видах стресса и нарушениях энергетического гомеостаза. Помимо внутриклеточного перемещения митохондрий, большое значение имеет их межклеточный транспорт. Межклеточный трансфер происходит как в физиологических условиях, так и при различных патологиях, который сопровождается устранением повреждений стрессированных клеток и восстановлением структурно-функциональных нарушений тканей, вызванных митохондриальной дисфункцией. Настоящий обзор суммирует последние данные, полученные исследователями в этой области, и дает общее представление о молекулярных механизмах межклеточного транспорта митохондрий и его роли в поддержании энергетического гомеостаза в тканях. Отдельно обсуждаются перспективные пути изучения передачи митохондрий для таргетной терапии многих заболеваний.
Митохондрии являются одной из самых сложных и важных органелл в клетках эукариот, обеспечивая их необходимой для жизнедеятельности энергией. Митохондриальная дисфункция ассоциирована с большим количеством патологических нарушений и заболеваний (Ballinger, 2005; Gorman et al., 2009). Энергопотребляющие ткани и ткани, подверженные гипоксически-ишемическому повреждению, наиболее уязвимы в плане энергетического истощения из-за дисфункции митохондрий. Таким образом, поддержание количества и качества митохондрий имеет решающее значение для гомеостаза тканей и выживания клеток.
Долгое время считали, что митохондрии удерживаются в цитоплазме и подвергаются частому перепрограммированию и внутриклеточному перемещению (Rafelski, 2013). Широко исследовался двунаправленный (антероградный и ретроградный) внутриклеточный аксональный транспорт митохондрий из-за его значительного влияния на митохондриальный гомеостаз в нейронах (Morris, Hollenbeck, 1993). В настоящее время большой интерес вызывает роль межклеточного транспорта митохондрий в поддержании тканевого гомеостаза (Mittelbrunn, Sanchez-Madrid, 2012; Murray, Krasnodembskaya, 2019). В 2004 г. впервые обнаружено перемещение органелл между клетками млекопитающих через туннелирующие нанотрубочки (ТНТ) (Rustom et al., 2004), а в 2006 г. продемонстрировали межклеточный транспорт митохондрий из мезенхимных стволовых клеток животных (МСК) в клетки с поврежденными митохондриями (Spees et al., 2006). С тех пор накопленные к настоящему времени данные о межклеточном трансфере митохондрий свидетельствуют о более высокой активности митохондрий, чем это предполагали ранее (Spees et al., 2006; Islam et al., 2012; Hayakawa et al., 2016), а перенос митохондрий от клеток-доноров к клеткам-реципиентам представляется многообещающим подходом для поддержания межклеточного энергетического гомеостаза (Vignais et al., 2017; Rodriguez et al., 2018).
Во время митохондриального аэробного дыхания в результате утечки электронов из электрон-транспортной цепи образуются активные формы кислорода (АФК). Обычно количество электронов, выходящих из цепи переноса, минимально, и уровень АФК может контролироваться путем утилизации АФК митохондриями (Murphy, 2009). Однако при стрессовых воздействиях на клетки, таких как ишемия (гипоксия), химическое воздействие и делеция в митохондриальной ДНК (мтДНК), в митохондриях накапливается большое количество АФК в результате усиленной утечки электронов (Angelova, Abramov, 2016). Быстрое повышение уровня АФК вызывает деполяризацию мембранного потенциала митохондрий и впоследствии инициирует митофагию, которая представляет собой селективное разрушение поврежденных митохондрий путем аутофагии (Mohammadalipour et al., 2020). Клетки не могут выжить без этого источника энергии, поэтому замена митохондрий, несомненно, является эффективным способом восстановить истощенные клетки. Интересно, что имеется достаточное количество подтверждений тому, что митохондриальный транспорт происходит не только при восстановлении жизнедеятельности клеток, но и в других ситуациях.
Примечательно, что спонтанный транспорт митохондрий между клетками происходит и в физиологических условиях (Torralba et al., 2016; Liu et al., 2021). С другой стороны, межклеточная передача митохондрий, по-видимому, не только предотвращает тканевое повреждение, часто возникающее при патологии центральной нервной, сердечно-сосудистой и дыхательной систем, но также опосредует клеточную активность, и тем самым оказывает влияние на процессы резистентности к опухолевой терапии и регуляции воспаления. Более того, изучение трансцеллюлярной деградации поврежденных митохондрий из клеток, подвергшихся воздействию стресса, расширяет и понимание митофагии (Davis et al., 2014). Следует отметить, что стволовые клетки являются наиболее популярными донорскими клетками для передачи митохондрий, что указывает на то, что донорство митохондрий может играть ключевую роль в терапии стволовыми клетками.
Настоящий обзор ставит целью не только дать общее представление о межклеточном транспорте митохондрий как фундаментальных органелл, определяющих метаболические процессы в клетках и клеточную выживаемость в физиологических условиях, но и собрать новейшие данные о молекулярных и сигнальных механизмах митохондриального трансфера в патологических условиях. Прогресс в изучении межклеточного транспорта митохондрий открывает перспективы для таргетной терапии многих заболеваний.
ТРАНСПОРТ МИТОХОНДРИЙ В ФИЗИОЛОГИЧЕСКИХ УСЛОВИЯХ ДЛЯ ПОДДЕРЖАНИЯ ГОМЕОСТАЗА И РАЗВИТИЯ ТКАНЕЙ
Клеточная терапия, особенно основанная на использовании стволовых клеток, считается перспективным подходом к лечению сердечных заболеваний (Payne et al., 2005), но специфические механизмы межклеточной передачи сигналов полностью не изучены. Для дальнейшего изучения влияния перекрестных связей между клетками, культивировали дифференцированные мышиные кардиомиоциты с мультипотентными стволовыми клетками человека, полученными из жировой ткани (Acquistapace et al., 2011), и впервые выявили важную роль транспорта митохондрий из стволовых клеток в кардиомиоциты мышей в перепрограммировании клеток. Доля клеток-предшественников кардиомиоцитов резко снизилась после уменьшения количества мтДНК в стволовых клетках. Учитывая, что мышиные кардиомиоциты демонстрируют небольшую гетерогенность, другие авторы (Sinclair et al., 2016) сравнили эффективность транспорта митохондрий между костномозговыми мезенхимными стволовыми клетками (КМ-МСК) и двумя другими популяциями мезенхимных стволовых клеток, полученными из здоровой легочной ткани и жидкости бронхоальвеолярного лаважа от реципиентов после трансплантации легких in vitro. Результаты показали, что МСК из здоровой легочной ткани и жидкости бронхоальвеолярного лаважа от реципиентов после трансплантации легких могут передавать цитоплазматическое содержимое и митохондрии здоровым эпителиальным клеткам бронхов человека с аналогичной эффективностью посредством однонаправленного переноса.
Примечательно, что несколько исследований in vitro показали, что этот спонтанный межклеточный транспорт митохондрий также может быть двунаправленным. Межклеточный обмен цитоплазмой и митохондриями между эпителиоцитами почечных канальцев и мультипотентными мезенхимными стволовыми (стромальными) клетками (ММСК) был обнаружен при их совместном культивировании, и он также был двунаправленным, хотя преобладал транспорт в ММСК (Plotnikov et al., 2010). Вероятно, что двунаправленный обмен клеточных компонентов способствует дифференцировке ММСК, поскольку в них наблюдали появление белков, специфичных для почечных канальцев (Plotnikov et al., 2010). Аналогичным образом, двунаправленный обмен митохондриями был обнаружен в физиологических условиях культивирования гладкомышечных клеток сосудов человека и КМ-МСК, и этот процесс способствовал пролиферации, но не дифференцировке МСК (Vallabhaneni et al., 2012). С другой стороны, спонтанный двунаправленный перенос митохондрий происходит между соматическими клетками через нанотрубочки, о чем свидетельствуют межклеточные взаимодействия между мышиными кардиомиоцитами и кардиальными фибробластами, обеспечивающие структурные и функциональные связи для поддержания межклеточного гомеостаза в миокарде (He et al., 2011). Несмотря на то, что исследования межклеточного транспорта митохондрий в отсутствие стрессовых факторов ограничены, они также представляют собой значимый пул, необходимый для изучения потенциальной роли митохондриального транспорта в поддержании тканевого гомеостаза.
ТРАНСПОРТ МИТОХОНДРИЙ В УСЛОВИЯХ ПАТОЛОГИИ
Транспорт митохондрий в ЦНС. Двунаправленный аксональный транспорт митохондрий – это особый вид внутриклеточной активности, необходимый для восполнения энергозатрат в различных областях нейронов (Morris, Hollenbeck, 1993). Недавно было показано, что межклеточный митохондриальный транспорт является неотъемлемым биологическим событием в ЦНС, который, как полагают, играет решающую роль в устранении ишемических и геморрагических повреждений (Babenko et al., 2015, 2018; Morancho et al., 2015; Hayakawa et al., 2016, 2018; Chou et al., 2017; Huang et al., 2020), восстановлении спинного мозга после травм (Gollihue et al., 2018; Li et al., 2019), защите нейронов от нейротоксического действия, индуцированного химиотерапией (Boukelmoune et al., 2018; English et al., 2020), и нейродегенерации (Rostami et al., 2017; Nitzan et al., 2019; Valdinocci et al., 2019).
Эксперимент на мышиной модели инсульта подтвердил, что нормально функционирующие митохондрии в астроцитах могут транспортироваться к поврежденным нейронам для устранения ишемического повреждения и восстановления нервной системы (Hayakawa et al., 2016). При этом межклеточный транспорт митохондрий, вероятно, опосредуется кальций-зависимым механизмом, включающим CD38-сигнальный путь, и подавление CD38-сигнальной системы может привести к снижению транспорта митохондрий, жизнеспособности клеток и нарушению постишемического восстановления (Hayakawa et al., 2016). Бабенко с коллегами (Babenko et al., 2015, 2018) показали, что митохондрии из мультипотентных МСК могут передаваться в нейроны или астроциты, что приводит к восстановлению клеточного дыхания в клетках-реципиентах и уменьшению ишемического повреждения. Помимо МСК, эндотелиальные клетки-предшественники (ПЭК) также использовались для клеточной терапии благодаря их способности регулировать ангиогенез и васкулогенез (Morancho et al., 2015). Подтверждено (Hayakawa et al., 2018), что митохондрии из ПЭК могут доставляться в поврежденные эндотелиальные клетки головного мозга. Результаты этих авторов показали, что уровни митохондриального белка TOM40, количество копий мтДНК и продукция АТФ были повышены в поврежденных эндотелиоцитах головного мозга. Плотность эндотелия восстанавливалась после обработки митохондриальными частицами, полученными из ПЭК, что демонстрирует способность митохондрий из ПЭК поддерживать функционирование церебральных эндотелиоцитов (Hayakawa et al., 2018).
В другом исследовании сообщалось о транспорте митохондрий после субарахноидального кровоизлияния и травмы спинного мозга. Авторы в экспериментах на крысах и у пациентов (Chou et al., 2017) показали, что митохондрии из астроцитов могут переноситься в спинномозговую жидкость после субарахноидального кровоизлияния. Более того, митохондриальный мембранный потенциал уменьшался в течение первых 72 ч после кровоизлияния и далее начинал расти, что коррелировало, соответственно, с неблагоприятным течением в начале, но в итоге с благоприятным исходом после субарахноидального кровоизлияния. Другой эксперимент, связанный с травмой спинного мозга, продемонстрировал, что экзогенные митохондрии могут быть трансплантированы в поврежденный спинной мозг крысы и способствовать поддержанию энергетического уровня, а также функциональному восстановлению после травмы (Gollihue et al., 2018).
Аналогичные исследования при сокультивировании КМ-МСК и нейронов в условиях глюкозо-кислородного голодания также выявили направленный транспорт митохондрий по пути нейронов и повышение их выживаемости, что иллюстрирует потенциальный терапевтический эффект митохондрий при травмах спинного мозга (Li et al., 2019). Дальнейшие исследования показали, что трансплантация как КМ-МСК, так и митохондрий, полученных из КМ-МСК, может уменьшать апоптоз нейронов и способствовать восстановлению локомоторных функций у крыс после травмы спинного мозга, доказывая, что транспорт митохондрий может рассматриваться как потенциальный механизм терапии стволовыми клетками при спинномозговых травмах (Li et al., 2019).
Когнитивные нарушения, вызванные химиотерапией, являются одной из критических проблем при лечении рака (Wefel, Schagen, 2012). Было продемонстрировано, что цисплатин (химиотерапевтический препарат на основе платины) может нарушать функцию внутрисинаптосомальных митохондрий и изменять морфологию митохондрий нейронов у мышей (Chiu et al., 2017). Недавно появилось сообщение о защитных эффектах межклеточного транспорта митохондрий при их дисфункции в условиях цисплатин-индуцированной нейротоксичности (Boukelmoune et al., 2018; English et al., 2020). При сокультивировании МСК и обработанных цисплатином нейральных стволовых клеток (НСК) МСК транспортировали свои функционирующие митохондрии в НСК, что сопровождалось снижением гибели НСК и восстановлением в них митохондриального мембранного потенциала (Boukelmoune et al., 2018). Кроме того, установлено, что здоровые митохондрии могут передаваться из астроцитов в поврежденные in vitro цисплатином нейроны, увеличивая их выживаемость и восстанавливая в них кальциевую динамику (English et al., 2020). Интересно, что та же доза цисплатина в астроцитах не влияла на их жизнеспособность (English et al., 2020). Результаты показали, что астроциты могут защищать нейроны от проявлений нейротоксичности, индуцированной химиотерапией in vivo, за счет передачи своих функционирующих митохондрий поврежденным нейронам.
Следует упомянуть о экспериментах с астроцитами, полученными из коры головного мозга крыс (Hayakawa et al., 2016). Исследователи выяснили, что астроциты действительно выделяют митохондрии в окружающую среду. С помощью метода CRISPR/Cas9 были получены и генномодифицированные клетки с усиленным производством белка CD38, который катализирует синтез циклической АДФ-рибозы, сигнальной молекулы, необходимой для работы кальциевых мембранных каналов, в том числе и в митохондриях. Как и ожидали авторы, генномодифицированные клетки с усиленной экспрессией CD38 производили и выделяли в среду намного больше митохондрий (Hayakawa et al., 2016). Ученые предположили, что этот процесс может развиваться при повреждении нейронов, помогая их восстановлению.
Митохондриальная дисфункция является важным следствием нейродегенеративных заболеваний, таких как болезнь Альцгеймера (БА) и болезнь Паркинсона (Nitzan et al., 2019; Valdinocci et al., 2019). Показали, что мыши с экспериментальной БА, получавшие внутривенно свежевыделенные митохондрии человека, проявляли лучшие когнитивные способности, чем мыши в контрольной группе, и что у мышей, получавших лизат митохондрий, существенно уменьшались потери нейронов и подавлялись процессы глиоза по сравнению с мышами с БА, которым не вводили митохондрии (Nitzan et al., 2019). Хотя вопросы эффективности и безопасности вмешательства требуют дальнейшего рассмотрения, это исследование наглядно демонстрирует необходимость улучшения митохондриального биогенеза у пациентов с БА.
Прогрессирование болезни Паркинсона связано с агрегацией патологического α-синуклеина (α-syn). Недавние исследования показали, что белковые агрегаты α-syn могут связываться с митохондриями, вызывая их повреждение и фрагментацию (Grassi et al., 2018). Доказано, что α-syn может накапливаться в астроцитах, подвергнутых стрессовому воздействию, следствием которого является набухание эндоплазматического ретикулума и митохондриальная дисфункция (Rostami et al., 2017). Кроме того, накопленный α-syn из поврежденных астроцитов доставляется к соседним здоровым клеткам посредством прямого контакта или ТНТ, что, в свою очередь, индуцировало транспорт митохондрий от здоровых астроцитов к астроцитам, претерпевшим стресс (Rostami et al., 2017). Предполагается, что передача патологического α-syn между клетками и межклеточный транспорт митохондрий при прогрессировании болезни Паркинсона может представлять терапевтическую мишень для ее лечения.
Транспорт митохондрий в сердечно-сосудистой системе. Сердце – энергоемкий и автономный орган, которому требуется постоянное поступление кислорода для поддержания его физиологической функции. Митохондрии обеспечивают сердце первичной энергией за счет аэробного дыхания и составляют 30% объема кардиомиоцитов. Таким образом, митохондриальная дисфункция при кардиоваскулярной патологии или мутации мтДНК, вызванные окислительным стрессом, тесно связаны с сердечно-сосудистыми заболеваниями (Ballinger, 2005).
Ишемия является основной причиной повреждения миокарда и апоптоза, поскольку блокирование поступления кислорода к кардиомиоцитам обычно приводит к дисфункции митохондрий (Liu et al., 2021). Было показано, что трансплантация функционирующих митохондрий в ишемизированную ткань миокарда кроликов сопровождается кардиопротекторным действием и значительно уменьшает размер инфаркта сердца через 4 нед. после выздоровления (Masuzawa et al., 2013). С помощью флуоресцентной микроскопии было обнаружено, что митохондрии частично интернализуются кардиомиоцитами через 2–8 ч после трансплантации. Хотя конкретный механизм интернализации митохондрий не выявлен, результаты показали, что трансплантированные митохондрии могут увеличивать потребление кислорода, продукцию АТФ и секрецию хемокинов в ишемизированной ткани миокарда, а также способствовать синтезу кардиоспецифичных белков, необходимых для поддержания энергетического гомеостаза миокарда (Masuzawa et al., 2013).
Помимо прямой трансплантации митохондрий, МСК в экспериментах in vitro способны восстанавливать жизнеспособность кардиомиобластов, подвергшихся ишемии, за счет транспорта функциональных митохондрий из донорских клеток (Cselenyak et al., 2010). На примере модели гипоксии/реоксигенации кардиомиоцитов установлено, что однонаправленный транспорт митохондрий как от интактнах, так и от индуцированных гипоксией/реоксигенацией миофибробластов по пути поврежденных кардиомиоцитов сопровождался подавлением в них апоптоза (Shen et al., 2018). Полученные результаты обновили существовавшие ранее данные о межклеточном трансфере митохондрий, выявив двунаправленный транспорт митохондрий между кардиофибробластами и кардиомиоцитами в условиях нормоксии (He et al., 2011). Кроме того, поврежденные кардиомиоциты, индуцированные липополисахаридом (Yang et al., 2016) или антрациклином (Zhang et al., 2016), также демонстрировали способность к восстановлению за счет функционирующих митохондрий, полученных из МСК.
Транспорт митохондрий в другие тканевые системы. МСК могут происходить из разных источников, поэтому разные типы МСК обладают разной способностью к дифференцировке и сохранению жизнеспособности других клеток. Так, установлено, что помимо восстановления жизнедеятельности наиболее уязвимых органов, МСК также транспортируют митохондрии в клетки, подвергшиеся стрессорному воздействию, и восстанавливают их аэробное дыхание. Эндотелиальные клетки пупочной вены человека, подвергнутые глюкозо-кислородной депривации и реоксигенации, продемонстрировали восстановление при совместном культивировании с КМ-МСК человека, и этот эффект был опосредован однонаправленным переносом здоровых митохондрий от КМ-МСК к поврежденным эндотелиоцитам (Liu et al., 2014).
В другой совместной культуре в физиологических условиях происходил транспорт митохондрий из МСК, полученных из индуцированных плюрипотентных стволовых клеток человека, к эпителиальным клеткам роговицы кролика, при этом в условиях окислительного стресса, вызванного действием ротенона, трансфер митохондрий в клетки эпителия роговицы значительно возрастал (Jiang et al., 2016). Трансплантация скаффолдов с МСК способствовала заживлению ран роговицы, полученных на кроличьих моделях с использованием щелочи, в результате передачи митохондрий из МСК в клетки эпителия роговицы (Jiang et al., 2016). Перенос митохондрий также происходил между МСК человека, когда МСК реципиента подвергались H2O2-индуцированному окислительному стрессу, а донорские МСК были предварительно обработаны N-ацетил-L-цистеином и L-аскорбат-2-фосфатом, которые благоприятно влияли на митохондриальный биогенез в MСК (Li et al., 2017). Таким образом, происходило подавление окислительного стресса в МСК после их обработки перексидом водорода, а также уменьшение фрагментации поврежденных митохондрий (Li et al., 2017).
Недавно было показано, что МСК, полученные из Вартонова студня пупочного канатика человека (WJ-MSCs), являются еще одним хорошим источником донорских митохондрий (Lin et al., 2015). Миоклоническая эпилепсия с рваными мышечными волокнами (синдром MERRF) – редкое митохондриальное заболевание, которое приводит к нервно-мышечным расстройствам. Совместное культивирование клеток WJ-MSC с цитоплазматическими гибридами MERRF, которые демонстрируют высокий коэффициент митохондриальных мутаций, сопровождалось повышением митохондриальной биоэнергетики и жизнеспособности цитоплазматических гибридов MERRF (Chuang et al., 2017). При этом было показано, что положительные эффекты связаны именно с транспортом функционирующих митохондрий от WJ-MSC к клеткам MERRF.
Транспорт митохондрий для их деградации. Другая сторона межклеточного транспорта митохондрий заключается в том, что поврежденные митохондрии могут быть доставлены в другие клетки для деградации, повторного использования или даже для оказания положительного действия на передачу сигнала. Так, Девис с коллегами (Davis et al., 2014) впервые описали трансцеллюлярную деградацию митохондрий с использованием введенного с помощью вируса специфического белка, меченного флуорофорной группой. Было показано, что поврежденные митохондрии в ганглионарных клетках сетчатки в области головки зрительного нерва переносятся в соседние астроциты и деградируют внутри лизосом. Выявленный процесс деградации митохондрий в ганглионарных клетках сетчатки получил название трансмитофагии в отличие от известной митофагии. Интересно, что в отличие от переноса митохондрий из нормально функционирующих МСК к макрофагам, наблюдаемого при восстановлении структурно-функциональных нарушений тканей (Morrison et al., 2017), поврежденные в условиях окислительного стресса МСК также передавали свои частично деполяризованные митохондрии в макрофаги в составе микровезикул в экспериментальной системе совместного культивирования in vitro (Phinney et al., 2015).
Этот процесс рассматривается как форма митофагии, помогающая МСК выживать при окислительном стрессе. Примечательно, что транспортированные деполяризованные митохондрии повторно использовались посредством слияния митохондрий в макрофагах клеток реципиента для увеличения биоэнергетической эффективности (Phinney et al., 2015). Хотя поврежденные митохондрии были частично деполяризованы, структура их митохондриальной мембраны не была нарушена, что способствовало слиянию с мембранами функционирующих митохондрий в макрофагах клеток реципиента (Phinney et al., 2015). Опухолевые клетки также могут транспортировать нефункциональные митохондрии в нормальные клетки в опухолевом микроокружении для адаптации к различным нарушениям, тем самым способствуя прогрессированию опухоли. Так, обнаружен (Wang et al., 2018) двунаправленный транспорт митохондрий через ТНТ между КМ-МСК и T-лимфоидными клетками (T-ALL-клетками), полученными от больных с Т-клеточным острым лимфобластным лейкозом. Авторы показали, что T-ALL-клетки после химиотерапевтического воздействия транспортировали гораздо больше митохондрий в МСК, чем сами КМ-МСК, что подавляло продукцию митохондриальных АФК в T-ALL-клетках и повышало их химиорезистентность (Wang et al., 2018). Паттерн-распознающие молекулы, ассоциированные с митохондриальным повреждением (DAMP), такие как мтДНК, N-формил-пептиды и митохондриальные белки, высвобождаются из поврежденных клеток, распознаются иммунными клетками (нейтрофилами и моноцитами или макрофагами) и вызывают иммунный ответ (Krysko et al., 2011).
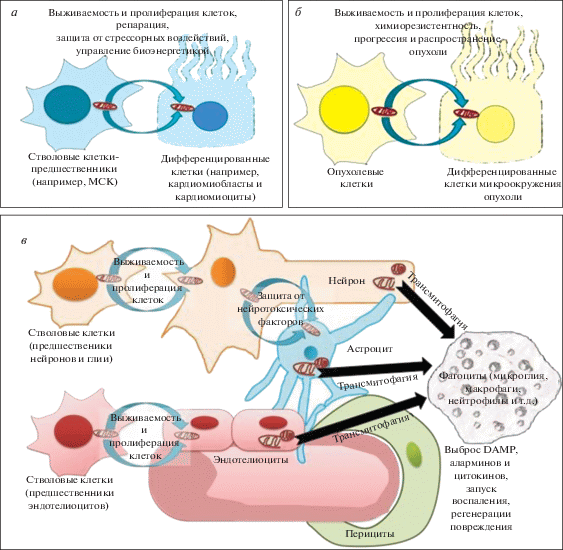
Таким образом, митохондрии можно рассматривать не только как сенсоры клеточного стресса, но и как регуляторы передачи сигналов опасности, выполняющие роль предупредительного знака для клетки или ткани и их своевременной реакции (Galluzzi et al., 2012) (рис. 1). Так, недавние открытия показали, что нормально функционирующие митохондрии могут высвобождаться из поврежденных клеток и действовать как специальные молекулы, при высвобождении которых запускаются воспалительные реакции (Maeda, Fadeel, 2014) или процессы восстановления повреждений тканей (Mahrouf-Yorgov et al., 2017). В экспериментах in vitro с сокультивируемыми клетками митохондрии, высвобождаемые из поврежденных соматических клеток (кардиомиоцитов или эндотелиальных), транспортировались в МСК и подвергались в них деградации через активацию сигнального пути гемоксигеназы-1 (HO-1), которая сопровождалась стимуляцией митохондриального биогенеза в МСК (Mahrouf-Yorgov et al., 2017). Активированные в МСК митохондрии затем передавались в соматические клетки, подвергшиеся воздействию пероксида водорода, и понижали в них уровень апоптоза (Mahrouf-Yorgov et al., 2017). Было обнаружено, что накопление в стрессированных клетках АФК, выступающих в роли триггера аутофагии (Bartz et al., 2015), является пусковым сигналом, необходимым для стимуляции HO-1-опосредованного антиапоптотического ответа. Низкий уровень АФК в стрессированных соматических клетках уменьшал высвобождение митохондрий из МСК (Mahrouf-Yorgov et al., 2017).
Рис. 1.
Роль транспорта митохондрий для функционирования стволовых клеток (а), опухолевых клеток при злокачественном росте (б) и клеток нейроваскулярной единицы (в) в норме и при патологии. DAMP – паттерн-распознающие молекулы, ассоциированные с митохондриальным повреждением.
Конкретные механизмы трансфера митохондрий между клетками изучены пока недостаточно, поэтому для определения возможности использовать такие процессы, как транспорт поврежденных митохондрий из соматических клеток в МСК для последующей деградации, а также транспорт функциональных митохондрий из МСК в поврежденные клетки, требуется дальнейшее изучение механизмов транспорта митохондрий между клетками.
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ МЕЖКЛЕТОЧНОГО ТРАНСПОРТА МИТОХОНДРИЙ
В настоящее время молекулярные и сигнальные механизмы митохондриального транспорта изучены пока недостаточно. Большинство исследований показали, что ТНТ и микровезикулы являются наиболее распространенными путями межклеточного трансфера митохондрий (рис. 2). Однако факторы, активирующие транспорт митохондрий в клетках-донорах и клетках-реципиентах, остаются менее изученными.
Рис. 2.
Схема возможных путей и наиболее важных молекулярных механизмов межклеточного транспорта митохондрий. На рисунке показаны целые (неповрежденные функционально активные) и “усеченные” и фрагментированные (поврежденные нефункциональные) митохондрии. АФК – активные формы кислорода, ЭПР – эндоплазматический ретикулум, Сх43 – коннексин 43, EGFR – рецептор эпидермального фактора роста, PI3K, AKT, mTOR – киназы, M-Sec – белок, известный как TNFαip2, Miro1 – GTPаза. См. объяснения в тексте.
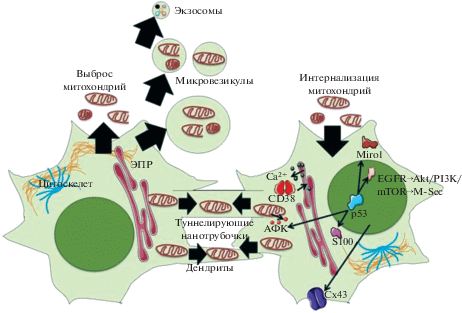
ПУТИ МЕЖКЛЕТОЧНОГО ТРАНСПОРТА МИТОХОНДРИЙ
Туннелирующие нанотрубочки. В 2004 г. было впервые описано образование ТНТ между крысиными клетками феохромоцитомы PC12 (Rustom et al., 2004) и иммунными клетками (Onfelt et al., 2004). Эти нанотрубочки представляют собой мембранные трубчатые выступы, отходящие от плазматической мембраны и позволяющие транспортировать различные клеточные компоненты или органеллы, что в значительной степени способствует межклеточному взаимодействию для поддержания тканевого гомеостаза (Vignais et al., 2017). Диаметр этих структур составляет от 50 до 1500 нм, а длина – от нескольких десятков до сотен микрон (Austefjord et al., 2014). Среди всех транспортируемых органелл основными считаются митохондрии, для которых описан как однонаправленный, так и двунаправленный транспорт через ТНТ. Кроме того, было подтверждено, что такие ТНТ-подобные мембранные структуры, как отростки остеоцитов, также выступают в роли переносчиков митохондрий внутри сети остеоцитов (Gao et al., 2019). При блокировании образования ТНТ актин-связывающим токсином цитохалазином B в наномолярной концентрации (350 нМ), который оказывал незначительное влияние на процессы эндоцитоза и фагоцитоза, транспорт органелл между клетками значительно снижался (Bukoreshtliev et al., 2009).
Известно, что различные стрессовые факторы, вызывающие повреждение митохондрий, способствуют образованию ТНТ и последующему транспорту по ним митохондрий, однако не так много исследований сосредоточено на механизме инициации и регуляции образования этих мембранных структур. Сообщалось, что активация фактора p53 является важным событием, запускающим образование ТНТ в ответ на клеточный стресс (Wang et al., 2011). В подвергшихся стрессу клетках активация p53 опосредовала регуляцию экспрессии рецептора эпидермального фактора роста (EGFR) и активируемого им сигнального каскада Akt/PI3K/mTOR, что приводило к сверпродукции M-Sec (TNFαip2) (Wang et al., 2011), способствующего полимеризации актина и формированию ТНТ путем взаимодействия с RalA (Hase et al., 2009). Показано также, что образование ТНТ между обработанными ротеноном эпителиальными клетками роговицы и МСК регулировалось сигнальным путем NF-κB/TNFαip2, активированным ротенон-индуцированными АФК (Jiang et al., 2016). Кроме того, процесс запуска формирования ТНТ может быть связан с взаимодействием p53 и АФК (Chen et al., 2018), в частности, с активацией p53 под действием АФК в условиях окислительного стресса. Наконец, установлено, что инициирование процесса образования ТНТ ассоциировано с увеличением при активации р53-зависимых сигнальных путей каспаза-3-зависимого расщепления белка S100A4 (Sun et al., 2012) – кальций-связывающего белка семейства S100. Сообщалось, что наличие градиента белка S100A4 способствует росту ТНТ от поврежденных клеток с низкой концентрацией S100A4 к клеткам-мишеням с более высокой концентрацией S100A4 (Sun et al., 2012).
На экспериментальной модели острого липополисахарид-индуцированного повреждения легких у мышей in vivo получили доказательства (Islam et al., 2012) участия трансмембранного белка щелевых контактов коннексина 43 (Сх43) в передаче митохондрий путем стабилизации участков прикрепления костномозговых стволовых клеток (BMSC) к клеткам альвеолярного эпителия, а также стимулирования образования ТНТ и микровезикул. Однако образование ТНТ и микровезикул подавлялось в Cx43-мутантных BMSC в результате нарушения функционирования контактов между BMSC и альвеолами. В таких условиях транспорт митохондрий был значительно подавлен и восстановления после повреждения легких не происходило.
Тем не менее, в некоторых других исследованиях также сообщалось об участии Cx43 в образовании ТНТ (Osswald et al., 2015; Torralba et al., 2016). Было подтверждено (Osswald et al., 2015), что митохондрии быстро перемещаются по мембранным микротрубочкам опухолевых клеток, и что в области пересечения двух разных микротрубочек часто локализован Cx43, способствующий распространению кальция по этим структурам. Потеря Cx43 снижает синхронность распространения межклеточных кальциевых волн и долю клеток астроцитомы с множественными микротрубочками, что указывает на роль Cx43 в стабилизации этих структур в опухолевых клетках. Кроме того, сообщалось об экспрессии Cx43 в остеоцитарной сети, способствующей выживанию остеоцитов (Ma et al., 2019), что свидетельствует о вероятном участии Cx43 в транспорте митохондрий между остеоцитами за счет усиления межклеточных контактов. Хотя механизмы, опосредующие участие белков щелевых контактов в межклеточном транспорте митохондрий, требуют дальнейшего изучения, можно предположить, что Cx43 принимает участие в заякоривании митохондрий с помощью ТНТ.
Известно, что для ТНТ-опосредованного транспорта митохондрий требуется участие Rho-подобных ГТФаз, локализованных во внешней митохондриальной мембране, таких как Miro1 (Ahmad et al., 2014; Babenko et al., 2015, 2018; Zhang et al., 2016). В нейронах Miro1 действует как заякоренный в наружной мембране митохондрии белок, который взаимодействует с митофузином1/2 и кинезином-1 через адаптерный белок milton (TRAK, существующий в нескольких изоформах – TRAK1/2 и OIP106/98), с образованием белкового комплекса, позволяющего митохондриям перемещаться по микротрубочкам (Misko et al., 2010). Показано влияние Miro1 на ТНТ-опосредованный межклеточный транспорт митохондрий от МСК к стрессированным эпителиальным клеткам (Ahmad et al., 2014). Сверхэкспрессия Miro1 в МСК способствовала усилению транспорта митохондрий и уменьшению повреждения эпителиальных клеток, тогда как нокаутирование гена белка Miro1 в МСК подавляло перенос митохондрий и снижало эффективность восстановления.
Интересно, что по сравнению с КМ-МСК, индуцированные плюрипотентные стволовые клетки были более эффективны в роли доноров митохондрий, а также способствовали восстановлению после антрациклин-индуцированной кардиомиопатии благодаря более высокой экспрессии Miro1 (Zhang et al., 2016). Бабенко с коллегами (Babenko et al., 2015) также обнаружили тесную взаимосвязь между Miro1 и транспортом митохондрий, о чем свидетельствовали увеличение количества белка Miro1 в МСК после их сокультивирования с нейронами и эффективность передачи митохондрий в МСК со сверхэкспрессией Miro1 при их трансплантации крысам с экспериментальным ишемическим инсультом (Babenko et al., 2018). Эти результаты доказывают значительный вклад транспорта митохондрий в постишемическую нейропротекцию. Таким образом, возможно, что межклеточный перенос митохондрий через ТНТ имеет схожие механизмы с Miro1-опосредованным аксональным транспортом митохондрий в нейронах.
Микровезикулы. Внеклеточные микровезикулы являются важными посредниками межклеточной коммуникации (Mittelbrunn, Sanchez-Madrid, 2012). В зависимости от их происхождения везикулы можно подразделить на несколько типов с различными размерами, которые включают экзосомы (30–100 нм), микровезикулы (от 100 нм до 1 мкм) и апоптотические тельца (>1 мкм) (Mittelbrunn, Sanchez-Madrid, 2012). В силу небольших размеров, экзосомы, происходящие из мембран эндосом, могут содержать только небольшие белки, липиды и РНК (Kourembanas, 2015), а также мтДНК (Guescini et al., 2010). Микровезикулы, образованные путем блеббинга плазматической мембраны, могут содержать целую органеллу из-за их большего диаметра и участвовать в транспорте митохондрий и мтДНК (Spees et al., 2006; Islam et al., 2012; Phinney et al., 2015; Hayakawa et al., 2016; Morrison et al., 2017). Было обнаружено, что BMSC предоставляют функционирующие митохондрии клеткам альвеолярного эпителия не только через ТНТ, но также и через микровезикулы, для образования которых показана зависимость от экспрессии Cx43(Islam et al., 2012). Наконец, микровезикулы также участвуют в трансмитофагии поврежденных ганглионарных клеток сетчатки (Davis et al., 2014) и стресс-активированных МСК (Phinney et al., 2015), что сопровождается реутилизацией деполяризованных митохондрий.
Выброс и интернализация митохондрий. Как уже говорилось выше, считается, что транспорт митохондрий от клеток-доноров к клеткам-реципиентам в большинстве случаев зависит от наличия таких межклеточных структур, как ТНТ и микровезикулы. Однако в некоторых исследованиях сообщается, что митохондрии или их фрагменты могут высвобождаться из клетки или поступать внутрь нее без формирования межклеточных структур путем выброса из клетки или погружения в клетку в процессе интернализации. Хотя доказательств транспорта митохондрий за счет процессов их выброса и поглощения из донорских клеток в клетки-реципиенты не так много, тем не менее, трансмембранная подвижность митохондрий также может способствовать межклеточной передаче митохондрий.
Предполагается, что выброс из клетки митохондрий необходим в основном для контроля качества митохондрий (Lyamzaev et al., 2008; Nakajima et al., 2008) или передачи стресс-сигналов (Boudreau et al., 2014; Maeda, Fadeel, 2014). Например, клетки HeLa выбрасывали фрагменты митохондрий в процессе элиминации митохондрий при митоптозе в условиях окислительного стресса (Lyamzaev et al., 2008). Другие авторы обнаружили, что цитоплазматические вакуоли, отделившиеся от плазматической мембраны), поглощали поврежденные митохондрии, а затем вытесняли их из погибающих клеток (индуцированных фактором некроза опухоли TNFα) путем повторного слияния с мембраной клеток каспаза-зависимым путем (Nakajima et al., 2008). Интересно, что для осуществления процессов блеббинга и выброса митохондрий требовалось отсутствие повреждений со стороны актинового и тубулинового цитоскелета, поскольку дестабилизация актина или тубулина подавляла образование цитоплазматических вакуолей.
В некоторых случаях выброс митохондрий из стрессированных клеток рассматривали как сигнал для запуска воспалительных реакций (Maeda, Fadeel, 2014). Выброс митохондрий и их фрагментов из клеток был обнаружен также в линиях клеток Jurkat и фибросаркомы мыши L929 с дефицитом Fas-белка, ассоциированного с доменом смерти, подвергнутых воздействию TNFα (Maeda, Fadeel, 2014). Дальнейшие эксперименты in vitro показали, что очищенные митохондрии, высвобождаемые некротизированными клетками, могут поглощаться макрофагами и дендритными клетками человека, что сопровождается модулирующим влиянием на продукцию цитокинов макрофагами и индукцию созревания дендритных клеток (Maeda, Fadeel, 2014). В другом исследовании показано, что функциональные митохондрии могут высвобождаться из активированных тромбоцитов как в виде инкапсулированных в мембрану микрочастиц, так и в виде свободных органелл (Boudreau et al., 2014). В дополнение к исследованиям in vitro, исследование in vivo показало, что внутривенное введение мышам внеклеточных митохондрий ускоряло адгезию нейтрофилов к эндотелию сосудов и что сопровождалось активацией нейтрофилов и воспалительной реакцией (Boudreau et al., 2014).
Интернализация митохондрий была впервые описана в 1982 г. Кларком и Шэем (Clark, Shay, 1982), которые назвали это явление митохондриальной трансформацией. В работе этих авторов было показано, что в чувствительных к хлорамфениколу и эфрапептину клетках млекопитающих происходит интернализация митохондрий, которая приводит к увеличению клеточной выживаемости при воздействии антибиотиков (Clark, Shay, 1982). Полученные результаты предполагают, что явление митохондриальной трансформации может служить терапевтической мишенью для лечения митохондриальных нарушений.
Считается, что трансплантация изолированных функциональных митохондрий и их интернализация в клетках-реципиентах путем макропиноцитоза (Chang et al., 2019) может сопровождаться восстановлением эффективности окислительного фосфорилирования в клетках-реципиентах и их пролиферации in vitro (Hayakawa et al., 2018; Chang et al., 2019). Недавние исследования продемонстрировали, что интернализация митохондрий зависит от целостности внешней митохондриальной мембраны и присутствия на ней белков синцитина-1 и синцитина-2, которые могут выступать в роли лигандов в процессах взаимодействия между митохондриями и клетками-реципиентами (Chang et al., 2019). В дополнение к экспериментам in vitro, некоторые исследования in vivo тоже показали положительное влияние трансплантации митохондрий на восстановление повреждений тканей (Masuzawa et al., 2013; Emani et al., 2017; Gollihue et al., 2018). Так, была проведена серия экспериментов in vivo и клинических исследований по изучению кардиозащитного эффекта трансплантации митохондрий на ишемическое и реперфузионное повреждение миокарда (Masuzawa et al., 2013; Emani et al., 2017). Функциональные аутологичные митохондрии, изолированные из неишемизированных скелетных мышц кролика, вводили непосредственно в ишемизированную зону сердца, где они могли быть интернализованы поврежденными кардиомиоцитами. Это приводило к ограничению размера инфаркта и восстановлению функции миокарда при постинфарктной сердечной недостаточности (Masuzawa et al., 2013). Кроме того, эта группа ученых (Emani et al., 2017) сообщила о первом клиническом применении митохондриальной аутотрансплантации для восстановления функции миокарда у детей, которым проводили экстракорпоральную мембранную оксигенацию (ЭКМО) из-за ишемического/реперфузионного повреждения миокарда (Emani et al., 2017).
Хотя некоторые вопросы, касающиеся дозы и способа митохондриальной трансплантации, еще нуждаются в корректировке, все-таки первичные результаты обнадеживают. Действительно, у четырех из пяти пациентов наблюдали улучшение сократительной функции сердца, что позволило прекратить поддержку ЭКМО (Emani et al., 2017). К настоящему времени механизм захвата митохондрий полностью не изучен, но трансплантация функционально активных митохондрий, по-видимому, является потенциальной терапевтической стратегией для лечения повреждения тканей.
ТРИГГЕРНЫЕ СИГНАЛЫ
По мере накопления доказательств появилось понимание того, что стрессовые сигналы, вызванные дисфункцией митохондрий или окислительным стрессом, могут передаваться от клеток-реципиентов клеткам-донорам и запускать в них транспорт митохондрий. Сообщается, что помимо паттерн-распознающих молекул, ассоциированных с митохондриальным повреждением, и самих поврежденных митохондрий (Krysko et al., 2011; Maeda, Fadeel, 2014; Mahrouf-Yorgov et al., 2017), усиление межклеточного транспорта митохондрий происходит и под действием некоторых других молекул и связанных с ними сигнальных путей. В процессе окислительного фосфорилирования в нормально функционирующих митохондриях небольшая часть электронов выходит из комплексов I и III, увеличивая скорость продукции АФК за счет реакции с O2 (Murphy, 2009).
В физиологических условиях защита клетки от АФК и контроль клеточного гомеостаза осуществляются антиоксидантными ферментами, такими как супероксиддисмутаза, каталаза или глутатионпероксидаза (Murphy, 2009). Однако при различных патологических условиях клетки, подвергшиеся воздействию ишемии (гипоксии), либо химическому воздействию, сопровождающемуся нарушением функций митохондрий, генерируют избыточное количество АФК, которые не могут быть эффективно обезврежены этими антиоксидантными ферментами, что приводит к окислительному повреждению. В клетках, функции которых сопряжены с особенно высокими энергозатратами, усиление продукции АФК при стрессе запускает механизм восстановления функций митохондрий (Ahmad et al., 2014; Mahrouf-Yorgov et al., 2017). И наоборот, МСК, которые обычно выступают в роли доноров митохондрий, поддерживают свои митохондрии в неактивном состоянии и используют для поддержания энергетического баланса аэробный гликолиз (Paliwal et al., 2018), что снижает риск гиперпродукции АФК. Кроме того, МСК характеризуются высоким уровнем экспрессии супероксиддисмутазы, каталазы и глутатионпероксидазы, контролирующих уровень АФК (Valle-Prieto, Conget, 2010). Во время дифференцировки стволовых клеток процессы окислительного фосфорилирования сопровождаются усилением митохондриального биогенеза и изменением морфологии митохондрий для удовлетворения повышенной потребности в энергии (Guo et al., 2020).
Было показано, что в условиях стресса повышенный уровень АФК вызывает фрагментацию митохондрий, их кластеризацию в перинуклеарной области, формирование митоптозных телец и последующий выброс митохондрий из клетки (Lyamzaev et al., 2008). Интересно, что транспорт нефункциональных митохондрий из поврежденных соматических клеток в присутствии антиоксиданта N-ацетил-L-цистеина в МСК был значительно подавлен (Mahrouf-Yorgov et al., 2017). Кроме того, ингибировались процессы активации HO-1 и митохондриального биогенеза в МСК, а также трансфера митохондрий из МСК в соматические клетки (Mahrouf-Yorgov et al., 2017). Поскольку митохондрии участвуют в синтезе гема, было сделано предположение, что АФК-индуцируемая трансмиттофагия поврежденных митохондрий в клетках-реципиентах сопровождается высвобождением гема в МСК, что приводит к активации в них сигнального пути HO-1 (Mahrouf-Yorgov et al., 2017).
Как известно, активация HO-1 стимулирует митохондриальный биогенез (MacGarvey et al., 2012), что сопровождается повышением экспрессии рецептора PPARγ (рецептора, активирующего пролиферацию пероксисом) и митохондриального фактора транскрипции A в МСК. Вероятно, это способствует слиянию митохондрий и их последующему транспорту для устранения повреждений стрессированных соматических клеток (Mahrouf-Yorgov et al., 2017). Кроме того, в недавнем исследовании было подтверждено индуцирующее действие АФК на передачу митохондрий из гемопоэтических стволовых клеток к КМ-МСК (Mistry et al., 2019). Так, накопление АФК в гемопоэтических стволовых клетках, вызванное грамотрицательной бактериальной микрофлорой, активировало PI3K-сигнальный путь и способствовало Cx43-опосредованному транспорту митохондрий из КМ-МСК в гемопоэтические стволовые клетки (Mistry et al., 2019). Интересно, что p53-зависимая активация сигнального пути Akt/PI3K/mTOR также была связана с формированием ТНТ, приводя к гиперпродукции TNFα. Похоже, существует тесная взаимосвязь между АФК и p53: АФК, вероятно, являются индуктором транспорта митохондрий в стрессированных клетках, а PI3K может выступать в роли медиатора, опосредующего межклеточные взаимосвязи и, соответственно, процесс трансфера.
CD38, многофункциональный трансмембранный гликопротеин, известен как катализатор синтеза вторичных посредников Ca2+-зависимого сигналинга, таких как циклическая АДФ-рибоза (Howard et al., 1993) и никотиновая кислота – адениндинуклеотидфосфат (Guse, Lee, 2008) из никотинамидадениндинуклеотида (НАД+) и никотинамидадениндинуклеотидфосфата (НАДФ+) соответственно. Эти реактивные метаболиты необходимы для мобилизации внутриклеточного кальция. Недавно было показано, что CD38 участвует в двух механизмах транспорта митохондрий (Hayakawa et al., 2016; Marlein et al., 2019). Так, в донорских астроцитах белок CD38 способствовал переносу митохондрий в соседние нейроны через микровезикулы (Hayakawa et al., 2016), тогда как в миеломных клетках реципиента CD38 стимулировал транспорт митохондрий из соседних костномозговых стволовых клеток через ТНТ (Marlein et al., 2019).
С одной стороны, экспрессия CD38 в астроцитах опосредуется глутаматом, высвобождаемым нейронами при сокультивировании клеток (Bruzzone et al., 2004), а избыточный глутамат стимулирует генерацию АФК в нейронах (Kritis et al., 2015), и таким образом, вероятно, что эксайтотоксическое воздействие глутамата в ишемизированных нейронах может быть потенциальным триггером транспорта митохондрий из соседних астроцитов. С другой стороны, известно, что CD38 усиливает адгезию клеток (Deaglio et al., 2000), и что экспрессия CD38 в миеломных клетках положительно коррелирует с заякориванием ТНТ в сокультивируемых гемопоэтических стволовых клетках (Marlein et al., 2019), что доказывает взаимосвязь между экспрессией CD38 и прикреплением микротрубочек к мембранам. Несколько исследований продемонстрировали, что липополисахарид может индуцировать транспорт митохондрий (Islam et al., 2012; Yang et al., 2016; Morrison et al., 2017), а также увеличивать экспрессию CD38 (Lee et al., 2012). Таким образом, CD38, вероятно играет роль в запуске митохондриального транспорта, хотя остается еще немало вопросов в отношении специфической функции CD38 в этом процессе, в том числе в отношении его изоформы, присутствующей на митохондриальной мембране.
ЭНДОПЛАЗМАТИЧЕСКИЙ РЕТИКУЛУМ (ЭР) ОПОСРЕДУЕТ ТРАНСПОРТ МИТОХОНДРИЙ
В настоящее время имеются свидетельства в пользу того, что ЭР тесно контактирует с митохондриями и играет критическую роль в регуляции биогенеза митохондрий в результате взаимодействия этих двух органелл (Rowland, Voeltz, 2012). Участки контакта, которые ЭР образует с митохондриями (MAMs), координируют множество процессов в этих органеллах, включая передачу кальциевых сигналов, синтез липидов и транспорт митохондрий внутри клетки (Rowland, Voeltz, 2012). Известно, что белок Mfn2 необходим для аксонального транспорта митохондрий, во время которого он взаимодействует с комплексом Miro/Milton в системе микротрубочек (Misko et al., 2010). Нокаут Mfn2 в нейронах вызывает дефекты аксонального транспорта (Misko et al., 2010). Принимая во внимание роль Mfn2 в прикреплении митохондрий к ЭР, можно предположить, что Mfn2 регулирует аксональный транспорт митохондрий через MAMs. В недавних экспериментах продемонстрирован транпорт митохондрий между остеоцитами через тубулиновые структуры их отростков, и для этого процесса требовались межклеточные контакты (Gao et al., 2019). Кроме того, транспорт митохондрий связан с функционированием ЭР (Gao et al., 2019). Ингибирование Mfn2 или другого белка, присутствующего в области контакта между ЭР и митохондриями – везикуло-ассоциированного мембранного белка В – значительно подавляло транспорт митохондрий в сети остеоцитов, что подтверждает важную роль сайтов контактирования ЭР и митохондрий в механизме переноса митохондрий (Gao et al., 2019).
Как микрофиламенты, так и микротрубочки являются основными компонентами цитоскелета (Vignais et al., 2017) и представляют собой наиболее распространенные пути межклеточного переноса митохондрий. В основе формирования ТНТ лежит процесс полимеризации актина, включающий удлинение микрофиламентов. Для транспорта митохондрий по аксонам нейронов (Misko et al., 2010) или по ТНТ (Ahmad et al., 2014; Babenko et al., 2015, 2018) необходим белок Miro1, который взаимодействует с молекулярным мотором кинезином-1, вызывая перемещение митохондрий вдоль микротрубочек. Другое исследование показало, что ЭР, как и митохондрии, двигаются только по стабильным ацетилированным микротрубочкам (Friedman et al., 2010), при этом места контактов между ЭР и митохондриями сохраняются даже в процессе их морфологических изменений (Friedman et al., 2010). Кроме того, распределение ЭР внутри клеток зависит от расположения микротрубочек (Kimura et al., 2017). Возможно, что митохондрии образуют комплексы с ЭР и микротрубочками в процессе переноса по трубчатым структурам, а микрофиламенты обеспечивают структурную основу для транспорта митохондрий. Таким образом, вполне вероятно, что существует система координации микротрубочек, микрофиламентов и MAMs, облегчающая процесс транспорта митохондрий между клетками через каналоподобные мембранные структуры. Однако механизмы межклеточного трансфера митохондрий могут отличаться в клетках разных типов, поэтому остается пока неясным, являются ли микрофиламенты, микротрубочки и MAMs незаменимыми элементами для различных вариантов митохондриального транспорта.
ПРОБЛЕМЫ И ПУТИ ИЗУЧЕНИЯ ТРАНСПОРТА МИТОХОНДРИЙ
Несмотря на то, что различные механизмы транспорта митохондрий описаны достаточно широко и имеют четкое физиологическое значение (например, регуляция гомеостаза и развития тканей), некоторые важные вопросы все еще остаются без ответа.
Очевидно, что механизм межклеточного транспорта митохондрий не универсален и имеет особенности в различных типах клеточных систем. Однако остается неясным, зависит ли тот или иной вариант митохондриального транспорта от типа клеток-доноров или клеток-реципиентов. Кроме того, непонятен механизм запуска каскада событий, приводящих к формированию ТНТ. В ряде исследований описаны сигнальные механизмы инициации образования ТНТ, исходящие от клеток-реципиентов (Hase et al., 2009; Wang et al., 2011; Sun et al., 2012; Jiang et al., 2016), тогда как другие исследования продемонстрировали, что процесс формирования ТНТ начинается в донорских клетках (Islam et al., 2012; Zhang et al., 2016). Интересно, что в клетках, формирующих синцитий, таких как остеоциты и астроциты, отростки могут рассматриваться как особая форма ТНТ, обеспечивающая транспорт митохондрий в сети. Однако каким образом митохондрии проходят через мембранные барьеры и доставляются к участкам контакта между двумя клетками, полностью не изучено.
Несмотря на имеющиеся сведения об участии белка щелевых контактов Cx43 в передаче митохондрий, маловероятно, что митохондрии могут напрямую проходить через такие узкие щелевые каналы. Скорее всего, Сх43 может выступать в качестве поверхности прикрепления к мембранным структурам двух клеток. Основываясь на описанной выше роли контакта между ЭР и митохондриями в транспорте митохондрий в остеоцитах (Gao et al., 2019), можно предположить, что ЭР также регулирует межклеточные процессы переноса митохондрий в ТНТ-опосредованных моделях, однако подробные механизмы этого феномена пока неясны. Например, неизвестно, как ЭР управляет движением митохондрий по микротрубочкам, что происходит с участком контакта ЭР-митохондрии до того, как митохондрии попадают в клетку-реципиент, участвует ли ЭР в прорыве мембранного барьера в местах контакта клеток и способствует ли движению митохондрий вдоль микротрубочек клеток. Известно, что трубчатая сеть ЭР тесно взаимодействует с плазматической мембраной, и MAMs играют решающую роль в передаче кальциевых сигналов и метаболизме липидов (Wu et al., 2018). Таким образом, весьма вероятно, что ЭР является ключевым медиатором доставки митохондрий из цитоплазмы к плазматической мембране в результате связывания митохондрий с его трубчатой сетью и дальнейшего прохождения через мембранные барьеры в участке контакта ЭР с плазматической мембраной. Эти процессы неизбежно будут включать изменения внутриклеточного ионного гомеостаза (особенно Ca2+) и липидного метаболизма в местах заякоривания митохондрий и ЭР в мембране, однако конкретные механизмы этих событий еще неясны.
Ранее было показано, что деградация подвергшихся стрессу митохондрий, полученных из поврежденных соматических клеток, необходима для активации сигнального пути HO-1 в здоровых МСК и транспорта функциональных митохондрий в стрессированные клетки для стимуляции в них митохондриального биогенеза (Mahrouf-Yorgov et al., 2017). В клетках, подвергшихся стрессу, поврежденные митохондрии подвергаются фрагментации и последующей митофагии (Zhu et al., 2018). Продемонстрировано, что участки контакта ЭР и митохондрий координируют деление митохондрий (Lewis et al., 2016), и можно предположить, что ЭР также участвует в деградации поврежденных митохондрий, однако многие аспекты этого механизма неизвестны. Неясно, например, транспортируется ли ЭР из стрессированных клеток вместе с поврежденными митохондриями вдоль микротрубочек. Поэтому необходимы дальнейшие исследования процессов деградации митохондрий и изучение ее роли в контроле качества митохондрий.
Известно, что подвижность митохондрий обычно коррелирует с изменениями в биогенезе митохондрий, которые приводят к изменениям внутриклеточного дыхательного метаболизма. Хотя усиление митохондриального биогенеза и процесса слияния митохондрий наблюдали в донорских МСК еще до донирования митохондрий (Mahrouf-Yorgov et al., 2017), неясно, является ли такое изменение митохондриального биогенеза обязательным и необходимы для преобразования транспортированных митохондрий в клетках-реципиентах.
Поскольку дисфункция митохондрий лежит в основе гипоксически-ишемического повреждения тканей и ряда наследственных митохондриальных заболеваний, коррекция митохондриальных патологий представляется весьма перспективным направлением для уменьшения митохондриальной (энергетической) недостаточности. Однако эффективность существующих терапевтических стратегий, которые в основном сосредоточены на возобновлении функционирования дыхательной цепи митохондрий и модификации мтДНК, остается недостаточной (Patananan et al., 2016). Принимая во внимание то, что стволовые клетки являются наиболее распространенными кандидатами на донорство митохондрий (и такой механизм, вероятно, отчасти определяет терапевтический эффект трансплантированных стволовых клеток), актуальным является дальнейшее изучение механизмов передачи митохондрий в клетки-реципиенты с целью поиска способов ее усиления, что может явиться новой стратегией митохондриальной терапии. Учитывая, что спонтанный перенос митохондрий происходит и в физиологических условиях, обеспечивая поддержание гомеостаза тканей, усиление межклеточного транспорта митохондрий, вероятно, может лечь в основу лечения дегенеративных заболеваний в процессе старения, таких как болезнь Альцгеймера и остеопороз. Кроме того, процессы выброса и интернализации митохондрий позволяют проводить прямую изоляцию и трансплантацию митохондрий в терапевтических целях (Masuzawa et al., 2013; Caicedo et al., 2017). Тем не менее, остаются еще некоторые вопросы относительно наиболее подходящих типов клеток для донирования митохондрий, а также возможные этические проблемы (Caicedo et al., 2017). Несмотря на определенные трудности, некоторый прогресс был достигнут в повышении эффективности переноса изолированных митохондрий в клетки-реципиенты. Недавно (Maeda et al., 2020) митохондрии впервые были объединены с трансактиватором транскрипционного (TAT) белка из вируса иммунодефицита человека и декстраном, что значительно усилило проникновение изолированных митохондрий в клетки и произвело более выраженный терапевтический эффект от транспорта митохондрий в поврежденные кардиомиоциты. В комплексе ТАТ–декстран пептид ТАТ способствует поглощению митохондрий клетками-реципиентами, а декстран присоединяет комплекс к внешней мембране митохондрий.
В дальнейшем усилия исследователей могут быть сконцентрированы на оптимизации процесса выделения митохондрий и искусственной трансплантации для повышения эффективности митохондриальной терапии.
Протекторное действие межклеточного транспорта митохондрий в опухолевом микроокружении в отношении выживаемости злокачественных клеток, подавление процесса накопления злокачественными клетками функциональных митохондрий из соседних здоровых клеток может стать новой стратегией для борьбы с опухолевой прогрессией и лекарственной устойчивостью. Например, совсем недавно показано, что даратумумаб (моноклональное антитело к CD38) может ингибировать прогрессирование острого миелоидного лейкоза с помощью механизма, включающего блокирование передачи митохондрий от МСК к миелобластам (Mistry et al., 2021). Поскольку клетки злокачественных опухолей характеризуются высоким уровнем метаболических процессов, необходимы дальнейшие исследования для изучения влияния захвата митохондрий на дыхательный метаболизм раковых клеток и разработки методов подавления митохондриального переноса в опухолевом микроокружении.
ЗАКЛЮЧЕНИЕ
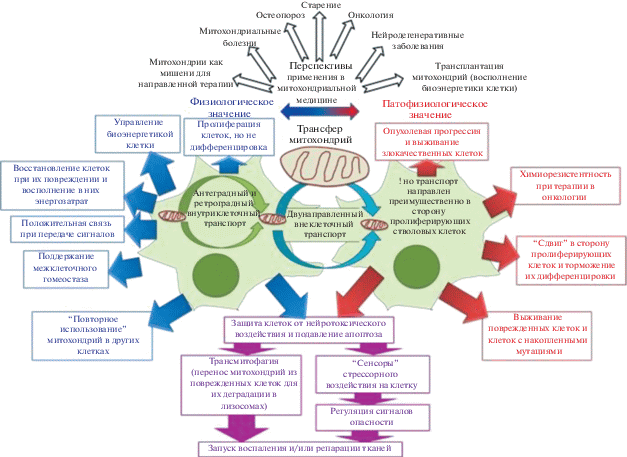
Межклеточный транспорт митохондрий является универсальным биологическим событием, которое происходит как в физиологических, так и патологических условиях для синхронизации энергетического обмена (рис. 3), хотя многие детали этого процесса все еще остаются в значительной степени неясными. С терапевтической точки зрения актуальным является открытие новых аспектов этого механизма с целью усиления или подавления этого вида клеточной коммуникации для восстановления поврежденных тканей и поддержания энергетического гомеостаза. Дальнейшие исследования требуются для изучения характеристик стрессовых факторов, способных инициировать транспорт митохондрий, механизмов формирования межклеточных связей в различных моделях передачи митохондрий, изменений митохондриального биогенеза как в донорских, так и в реципиентных клетках, а также роли иных клеточных органелл и компартментов в межклеточном транспорте митохондрий. Лучшее понимание оптимальных условий для трансфера митохондрий открывает принципиально новые возможности как для фундаментальных исследований в области молекулярной биологии митохондриального транспорта, так и для научно-прикладных исследований по разработке подходов и методов трансплантации митохондрий для управления метаболизмом и адаптацией клеток.
Список литературы
Acquistapace A., Bru T., Lesault P.-F., Figeac F., Coudert A.E., Coz O., Christov C., Baudin X., Auber F., Yiou R., Dubois-Randé J.-L., Rodriguez A.-M. 2011. Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells. V. 29. P. 812.
Ahmad T., Mukherjee S., Pattnaik B., Kumar M., Singh S., Kumar M., Rehman R., Tiwari B.K., Jha K.A., Barhanpurkar A.P., Wani M.R., Roy S.S., Mabalirajan U., Ghosh B., Agrawal A. 2014. Miro1 regulates intercellular mitochondrial transport and enhances mesenchymal stem cell rescue efficacy. EMBO J. V. 33. P. 994.
Angelova P.R., Abramov A.Y. 2016. Functional role of mitochondrial reactive oxygen species in physiology. Free Rad. Biol. Med. V. 100. P. 81.
Austefjord M.W., Gerdes H.H., Wang X. 2014. Tunneling nanotubes: Diversity in morphology and structure. Commun. Integr. Biol. V. 7. P. e27934. https://doi.org/10.4161/cib.27934
Babenko V.A., Silachev D.N., Popkov V.A., Zorova L.D., Pevzner I.B., Plotnikov E.Y., Sukhikh G.T., Zorov D.B. 2018. Miro1 enhances mitochondria transfer from multipotent mesenchymal stem cells (MMSC) to neural cells and improves the efficacy of cell recovery. Molecules. V. 23. P. 687.
Babenko V.A., Silachev D.N., Zorova L.D., Pevzner I.B., Khutornenko A.A., Plotnikov E.Y., Sukhikh G.T., Zorov D.B. 2015. Improving the post-stroke therapeutic potency of mesenchymal multipotent stromal cells by cocultivation with cortical neurons: The role of crosstalk between cells. Stem Cells Transl. Med. V. 4. P. 1011.
Ballinger S.W. 2005. Mitochondrial dysfunction in cardiovascular disease. Free Rad. Biol. Med. V. 38. P. 1278.
Bartz R.R., Suliman H.B., Piantadosi C.A. 2015. Redox mechanisms of cardiomyocyte mitochondrial protection. Front. Physiol. V. 6. P. 291.
Boudreau L.H., Duchez A.-C., Cloutier N., Soulet D., Martin N., Bollinger J., Paré A., Rousseau M., Naika G.S., Lévesque T., Laflamme C., Marcoux G., Lambeau G., Farndale R.W., Pouliot M., et al. 2014. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. V. 124. P. 2173.
Boukelmoune N., Chiu G.S., Kavelaars A., Heijnen C.J. 2018. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol. Commun. V. 6. P. 139.
Bruzzone S., Verderio C., Schenk U., Fedele E., Zocchi E., Matteoli M., De Flora A. 2004. Glutamate-mediated overexpression of CD38 in astrocytes cultured with neurones. J. Neurochem. V. 89. P. 264.
Bukoreshtliev N.V., Wang X., Hodneland E., Gurke S., Barroso J.F.V., Gerdes H.-H. 2009. Selective block of tunneling nanotube (TNT) formation inhibits intercellular organelle transfer between PC12 cells. FEBS Lett. V. 583. P. 1481.
Caicedo A., Aponte P.M., Cabrera F., Hidalgo C., Khoury M. 2017. Artificial mitochondria transfer current challenges, advances, and future applications. Stem Cells Int. V. 2017. P. 7 610 414. https://doi.org/10.1155/2017/7610414
Chang C.-Y., Liang M.-Z., Chen L. 2019. Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl. Neurodegener. V. 8. P. 17.
Chen Y., Liu K., Shi Y., Shao C. 2018. The tango of ROS and p53 in tissue stem cells. Cell Death Differ. V. 25. P. 637.
Chiu G.S., Maj M.A., Rizvi S., Dantzer R., Vichaya E.G., Laumet G., Kavelaars A., Heijnen C.J. 2017. Pifithrin-μ prevents cisplatin-induced chemobrain by preserving neuronal mitochondrial function. Cancer Res. V. 77. P. 742.
Chou S.H., Lan J., Esposito E., Ning M., Balaj L., Ji X., Lo E.H., Hayakawa K. 2017. Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke. V. 48. P. 2231.
Chuang Y.-C., Liou C.-W., Chen S.-D., Wang P.-W., Chuang J.-H., Tiao M.-M., Hsu T.-Y., Lin H.-Y., Lin T.-K. 2017. Mitochondrial transfer from Wharton’s jelly mesenchymal stem cell to MERRF cybrid reduces oxidative stress and improves mitochondrial bioenergetics. Oxid. Med. Cell. Longev. V. 2017. P. 5691215. https://doi.org/10.1155/2017/5691215
Clark M.A., Shay J.W. 1982. Mitochondrial transformation of mammalian cells. Nature. V. 295. P. 605.
Cselenyak A., Pankotai E., Horvath E.M., Kiss L., Lacza Z. 2010. Mesenchymal stem cells rescue cardiomyoblasts from cell death in an in vitro ischemia model via direct cell-to-cell connections. BMC Cell Biol. V. 11. P. 29.
Davis C.O., Kim K.-Y., Bushong E.A., Mills E.A., Boassa D., Shih T., Kinebuchi M., Phan S., Zhou Y., Bihlmeyer N.A., Nguyen J.V., Jin Y., Ellisman M.H., Marsh-Armstrong N. 2014. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA. V. 111. P. 9633.
Deaglio S., Mallone R., Baj G., Arnulfo A., Surico N., Dianzani U., Mehta K., Malavasi F. 2000. CD38/CD31, a receptor/ligand system ruling adhesion and signaling in human leukocytes. Chem. Immunol. V. 75. P. 99.
Emani S.M., Piekarski B.L., Harrild D., Del Nido P.J., McCully J.D. 2017. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. V. 154. P. 286.
English K., Shepherd A., Uzor N.-E., Trinh R., Kavelaars A., Heijnen C.J. 2020. Astrocytes rescue neuronal health after cisplatin treatment through mitochondrial transfer. Acta Neuropathol. Commun. V. 8. P. 36.
Friedman J.R., Webster B.M., Mastronarde D.N., Verhey K.J., Voeltz G.K. 2010. ER sliding dynamics and ER–mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. V. 190. P. 363.
Galluzzi L., Kepp O., Kroemer G. 2012. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. V. 13. P. 780.
Gao J., Qin A., Liu D., Ruan R., Wang Q., Yuan J., Cheng T.S., Filipovska A., Papadimitriou J.M., Dai K., Jiang Q., Gao X., Feng J.Q., Takayanagi H., Zhang C., Zheng M.H. 2019. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci. Adv. V. 5. P. eaaw7215. https://doi.org/10.1126/sciadv.aaw7215
Gollihue J.L, Patel S.P., Eldahan K.C., Cox D.H., Donahue R.R., Taylor B.K., Sullivan P.G., Rabchevsky A.G. 2018. Effects of mitochondrial transplantation on bioenergetics, cellular incorporation, and functional recovery after spinal cord injury. J. Neurotrauma. V. 35. P. 1800.
Gorman G.S., Chinnery P.F., DiMauro S., Hirano M., Koga Y., McFarland R., Suomalainen A., Thorburn D.R., Zeviani M., Turnbull D.M. 2009. Mitochondrial diseases. Nat. Rev. Dis. Prim. V. 2. P. 16080. https://doi.org/10.1038/nrdp.2016.80
Grassi D., Howard S., Zhou M., Diaz-Perez N., Urban N.T., Guerrero-Given D., Kamasawa N., Volpicelli-Daley L.A., LoGrasso P., Lasmézas C.I. 2018. Identification of a highly neurotoxic alpha-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci. USA. V. 115. P. E2634.
Guescini M., Genedani S., Stocchi V., Agnati L.F. 2010. Astrocytes and glioblastoma cells release exosomes carrying mtDNA. J. Neural. Transm. V. 117. P. 1.
Guo Y., Chi X., Wang Y., Heng B.C., Wei Y., Zhang X., Zhao H., Yin Y., Deng X. 2020. Mitochondria transfer enhances proliferation, migration, and osteogenic differentiation of bone marrow mesenchymal stem cell and promotes bone defect healing. Stem Cell Res. Ther. V. 11. P. 245.
Guse A.H., Lee H.C. 2008. NAADP: A universal Ca2+ trigger. Sci. Signal. V. 1. P. re10.
Hase K., Kimura S., Takatsu H., Ohmae M., Kawano S., Kitamura H., Ito M., Watarai H., Hazelett C.C., Yeaman C., Ohno H. 2009. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. V. 11. P. 1427.
Hayakawa K., Chan S.J., Mandeville E.T., Park J.H., Bruzzese M., Montaner J., Arai K., Rosell A., Lo E.H. 2018. Protective effects of endothelial progenitor cell-derived extracellular mitochondria in brain endothelium. Stem Cells. V. 36. P. 1404.
Hayakawa K., Esposito E., Wang X., Terasaki Y., Liu Y., Xing Ch., Ji X., Lo E.H. 2016. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. V. 535. P. 551.
He K., Shi X., Zhang X., Dang S., Ma X., Liu F., Xu M., Lv Z., Han D., Fang X., Zhang Y. 2011. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc. Res. V. 92. P. 39.
Howard M., Grimaldi J.C., Bazan J.F., Lund F.E., Santos-Argumedo L., Parkhouse R.M., Walseth T.F., Lee H.C. 1993. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science. V. 262. P. 1056.
Huang L., Reis C., Boling W.W., Zhang J.H. 2020. Stem cell therapy in brain ischemia: The role of mitochondrial transfer. Stem Cells Dev. V. 29. P. 555.
Islam M.N., Das S.R., Emin M.T., Wei M., Sun L., Westphalen K., Rowlands D.J., Quadri S.K., Bhattacharya S., Bhattacharya J. 2012. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. V. 18. P. 759.
Jiang D., Gao F., Zhang Y., Wong D.S.H., Li Q., Tse H.-F., Xu G., Yu Z., Lian Q. 2016. Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. V. 7. P. e2467. https://doi.org/10.1038/cddis.2016.358
Kimura K., Mamane A., Sasaki T., Sato K., Takagi J., Niwayama R., Hufnagel L., Shimamoto Y., Joanny J.-F., Uchida S., Kimura A. 2017. Endoplasmic-reticulum-mediated microtubule alignment governs cytoplasmic streaming. Nat. Cell Biol. V. 19. P. 399.
Kourembanas S. 2015. Exosomes: Vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu. Rev. Physiol. V. 77. P. 13.
Kritis A.A., Stamoula E.G., Paniskaki K.A., Vavilis T.D. 2015. Researching glutamate-induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell Neurosci. V. 9. P. 91.
Krysko D.V., Agostinis P., Krysko O., Garg A.D., Bachert C., Lambrecht B.N., Vandenabeele P. 2011. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. V. 32. P. 157.
Lee C.U., Song E.K., Yoo C.H., Kwak Y.K., Han M.K. 2012. Lipopolysaccharide induces CD38 expression and solubilization in J774 macrophage cells. Mol. Cells. V. 34. P. 573.
Lewis S.C., Uchiyama L.F., Nunnari J. 2016. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science. V. 353. P. aaf5549.
Li C.J., Chen P.K., Sun L.Y., Pang C.Y. 2017. Enhancement of mitochondrial transfer by antioxidants in human mesenchymal stem cells. Oxid. Med. Cell. Longev. V. 2017. P. 8 510 805. https://doi.org/10.1155/2017/8510805
Li H., Wang C., He T., Zhao T., Chen Y.-Y., Shen Y.-L., Zhang X., Wang L.-L. 2019. Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via gap junction. Theranostics. V. 9. P. 2017.
Lin H.-Y., Liou C.-W., Chen S.-D., Hsu T.-Y., Chuang J.-H., Wang P.-W., Huang S.-T., Tiao M.-M., Chen J.-B., Lin T.-K., Chuang Y.-C. 2015. Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion. V. 22. P. 31.
Liu D., Gao Y., Liu J., Huang Y., Yin J., Feng Y., Shi L., Meloni B.P., Zhang C., Zheng M., Gao J. 2021. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct. Target Ther. V. 6. P. 65.
Liu K., Ji K., Guo L., Wu W., Lu H., Shan P., Yan C. 2014. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. V. 92. P. 10.
Lyamzaev K.G., Nepryakhina O.K., Saprunova V.B., Bakeeva L.E., Pletjushkina O.Yu., Chernyak B.V., Skulachev V.P. 2008. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta. V. 1777. P. 817.
Ma L., Hua R., Tian Y., Cheng H., Fajardo R.J., Pearson J.J., Guda T., Shropshire D.B., Gu S., Jiang J.X. 2019. Connexin 43 hemichannels protect bone loss during estrogen deficiency. Bone Res. V. 7. P. 11.
MacGarvey N.C., Suliman H.B., Bartz R.R., Fu P., Withers C.M., Welty-Wolf K.E., Piantadosi C.A. 2012. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am. J. Resp. Crit. Care Med. V. 185. P. 851.
Maeda A., Fadeel B. 2014. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. V. 5. P. e1312. https://doi.org/10.1038/cddis.2014.277
Maeda H., Kami D., Maeda R., Murata Y., Jo J.-I., Kitani T., Tabata Y., Matoba S., Gojo S. 2020. TAT-dextran-mediated mitochondrial transfer enhances recovery from models of reperfusion injury in cultured cardiomyocytes. J. Cell. Mol. Med. V. 24. P. 5007.
Mahrouf-Yorgov M., Augeul L., Crola Da Silva C., Jourdan M., Rigolet M., Manin S., Ferrera R., Ovize M., Henry A., Guguin A., Meningaud J.-P., Dubois-Randé J.-L., Motterlini R., Foresti R., Rodriguez A.-M. 2017. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. V. 24. P. 1224.
Marlein C.R., Piddock R.E., Mistry J.J., Zaitseva L., Hellmich C., Horton R.H., Zhou Z., Auger M.J., Bowles K.M., Rushworth S.A. 2019. CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. V. 79. P. 2285.
Masuzawa A., Black K.M., Pacak C.A., Ericsson M., Barnett R.J., Drumm C., Seth P., Bloch D.B., Levitsky S., Cowan D.B., McCully J.D. 2013. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. V. 304. P. H966.
Misko A., Jiang S., Wegorzewska I., Milbrandt J., Baloh R.H. 2010. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. V. 30. P. 4232.
Mistry J.J., Marlein C.R., Moore J.A., Hellmich C., Wojtowicz E.E., Smith J.G.W., Macaulay I., Sun Y., Morfakis A., Patterson A., Horton R.H., Divekar D., Morris C.J., Haestier A., Di Palma F., et al. 2019. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc. Natl Acad. Sci. USA. V. 116. P. 24610.
Mistry J.J., Moore J.A., Kumar P., Marlein C.R., Hellmich C., Pillinger G., Jibril A., Di Palma F., Collins A., Bowles K.M., Rushworth S.A. 2021. Daratumumab inhibits acute myeloid leukaemia metabolic capacity by blocking mitochondrial transfer from mesenchymal stromal cells. Haematologica. V. 106. P. 589.
Mittelbrunn M., Sanchez-Madrid F. 2012. Intercellular communication: diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell. Biol. V. 13. P. 328.
Mohammadalipour A., Dumbali S.P., Wenzel P.L. 2020. Mitochondrial transfer and regulators of mesenchymal stromal cell function and therapeutic efficacy. Front. Cell Dev. Biol. V. 8. P. 603292. https://doi.org/10.3389/fcell.2020.603292
Morancho A., Ma F., Barceló V., Giralt D., Montaner J., Rosell A. 2015. Impaired vascular remodeling after endothelial progenitor cell transplantation in MMP9-deficient mice suffering cortical cerebral ischemia. J. Cereb. Blood Flow Metab. V. 35. P. 1547.
Morris R.L., Hollenbeck P.J. 1993. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. V. 104. P. 917.
Morrison T.J., Jackson M.V., Cunningham E.K., Kissenpfennig A., McAuley D.F., O’Kane C.M., Krasnodembskaya A.D. 2017. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am. J. Respir. Crit. Care Med. V. 196. P. 1275.
Murphy M.P. 2009. How mitochondria produce reactive oxygen species. Biochem. J. V. 417. P. 1.
Murray L.M.A., Krasnodembskaya A.D. 2019. Concise review: Intercellular communication via organelle transfer in the biology and therapeutic applications of stem cells. Stem Cells. V. 37. P. 14.
Nakajima A., Kurihara H., Yagita H., Okumura K., Nakano H. 2008. Mitochondrial extrusion through the cytoplasmic vacuoles during cell death. J. Biol. Chem. V. 283. P. 24128.
Nitzan K., Benhamron S., Valitsky M., Kesner E.E., Lichtenstein M., Ben-Zvi A., Ella E., Segalstein Y., Saada A., Lorberboum-Galski H., Rosenmann H. 2019. Mitochondrial transfer ameliorates cognitive deficits, neuronal loss, and gliosis in Alzheimer’s disease mice. J. Alzheimers Dis. V. 72. P. 587.
Onfelt B., Nedvetzki S., Yanagi K., Davis D.M. 2004. Cutting edge: membrane nanotubes connect immune cells. J. Immunol. V. 173. P. 1511.
Osswald M., Jung E., Sahm F., Solecki G., Venkataramani V., Blaes J., Weil S., Horstmann H., Wiestler B., Syed M., Huang L., Ratliff M., Karimian Jazi K., Kurz F.T., Schmenger T. et al. 2015. Brain tumour cells interconnect to a functional and resistant network. Nature. V. 528. P. 93.
Paliwal S., Chaudhuri R., Agrawal A., Mohanty S. 2018. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J. Biomed. Sci. V. 25. P. 31.
Patananan A.N., Wu T.H., Chiou P.Y., Teitell M.A. 2016. Modifying the mitochondrial genome. Cell Metab. V. 23. P. 785.
Payne T.R., Oshima H., Sakai T., Ling Y., Gharaibeh B., Cummins J., Huard J. 2005. Regeneration of dystrophin-expressing myocytes in the mdx heart by skeletal muscle stem cells. Gene Ther. V. 12. P. 1264.
Phinney D.G., Di Giuseppe M., Njah J., Sala E., Shiva S., St Croix C.M., Stolz D.B., Watkins S.C., Di Y.P., Leikauf G.D., Kolls J., Riches D.W., Deiuliis G., Kaminski N., Boregowda S.V., McKenna D.H., Ortiz L.A. 2015. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. V. 6. P. 8472.
Plotnikov E.Y., Khryapenkova T.G., Galkina S.I., Sukhikh G.T., Zorov D.B. 2010. Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture. Exp. Cell Res. V. 316. P. 2447.
Rafelski S.M. 2013. Mitochondrial network morphology: building an integrative, geometrical view. BMC Biol. 2013. V. 11. P. 71.
Rodriguez A.-M., Nakhle J., Griessinger E., Vignais M.-L. 2018. Intercellular mitochondria trafficking highlighting the dual role of mesenchymal stem cells as both sensors and rescuers of tissue injury. Cell Cycle. V. 17. P. 712.
Rostami J., Holmqvist S., Lindström V., Sigvardson J., Westermark G.T., Ingelsson M., Bergström J., Roybon L., Erlandsson A. 2017. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. V. 37. P. 11 835.
Rowland A.A., Voeltz G.K. 2012. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. V. 13. P. 607.
Rustom A., Saffrich R., Markovic I., Walther P., Gerdes H.H. 2004. Nanotubular highways for intercellular organelle transport. Science. V. 303. P. 1007.
Shen J., Zhang J.-H., Xiao H., Wu J.-M., He K.-M., Lv Z.-Z., Li Z.-J., Xu M., Zhang Y.-Y. 2018. Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cardiomyocytes from apoptosis. Cell Death Dis. V. 9. P. 81.
Sinclair K.A., Yerkovich S.T., Hopkins P.M., Chambers D.C. 2016. Characterization of intercellular communication and mitochondrial donation by mesenchymal stromal cells derived from the human lung. Stem Cell Res. Ther. V. 7. P. 91.
Spees J.L., Olson S.D., Whitney M.J., Prockop D.J. 2006. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl Acad. Sci. USA. V. 103. P. 1283.
Sun X., Wang Y., Zhang J., Tu J., Wang X.-J., Su X.-D., Wang L., Zhang Y. 2012. Tunneling-nanotube direction determination in neurons and astrocytes. Cell Death Dis. V. 3. P. e438.
Torralba D., Baixauli F., Sánchez-Madrid F. 2016. Mitochondria know no boundaries: mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. V. 4. P. 107.
Valdinocci D., Simões R.F., Kovarova J., Cunha-Oliveira T., Neuzil J., Pountney D.L. 2019. Intracellular and intercellular mitochondrial dynamics in Parkinsonas disease. Front. Neurosci. V. 13. P. 930.
Vallabhaneni K.C., Haller H., Dumler I. 2012. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. V. 21. P. 3104.
Valle-Prieto A., Conget P.A. 2010. Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. V. 19. P. 1885.
Vignais M.L., Caicedo A., Brondello J.M., Jorgensen C. 2017. Cell connections by tunneling nanotubes: Effects of mitochondrial trafficking on target cell metabolism, homeostasis, and response to therapy. Stem Cells Int. V. 2017. P. 6 917 941. https://doi.org/10.1155/2017/6917941
Wang J., Liu X., Qiu Y., Shi Y., Cai J., Wang B., Wei X., Ke Q., Sui X., Wang Y., Huang Y., Li H., Wang T., Lin R., Liu Q., Xiang A.P. 2018. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. V. 11. P. 11.
Wang Y., Cui J., Sun X., Zhang Y. 2011. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. V. 18. P. 732.
Wefel J.S., Schagen S.B. 2012. Chemotherapy-related cognitive dysfunction. Curr. Neurol. Neurosci. Rep. V. 12. P. 267.
Wu H., Carvalho P., Voeltz G.K. 2018. Here, there, and everywhere: the importance of ER membrane contact sites. Science. V. 361. P. eaan5835.
Yang H., Borg T.K., Ma Z., Xu M., Wetzel G., Saraf L.V., Markwald R., Runyan R.B., Gao B.Z. 2016. Biochip-based study of unidirectional mitochondrial transfer from stem cells to myocytes via tunneling nanotubes. Biofabrication. V. 8. P. 015 012. https://doi.org/10.1088/1758-5090/8/1/015012
Zhang Y., Yu Z., Jiang D., Liang X., Liao S., Zhang Z., Yue W., Li X., Chiu S.-M., Chai Y.-H., Liang Y., Chow Y., Han S., Xu A., Tse H.-F., Lian Q. 2016. iPSC-MSCs with high intrinsic MIRO1 and sensitivity to TNF-α yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Rep. V. 7. P. 749.
Zhu T., Chen J.L., Wang Q., Shao W., Qi B. 2018. Modulation of mitochondrial dynamics in neurodegenerative diseases: An insight into prion diseases. Front. Aging Neurosci. V. 10. P. 336.
Дополнительные материалы отсутствуют.
Инструменты
Цитология