

Доклады Российской академии наук. Химия, науки о материалах , 2020, T. 490, № 1, стр. 24-28
ИССЛЕДОВАНИЕ ПОЛИМЕРИЗАЦИИ 2,3,4,5,6-ПЕНТАФТОРСТИРОЛА В ПРИСУТСТВИИ ПОЛИ-2-ГИДРОКСИЭТИЛМЕТАКРИЛАТНОГО АГЕНТА ПЕРЕДАЧИ ЦЕПИ
К. Е. Чекуров 1, *, А. И. Барабанова 1, И. В. Благодатских 1, А. В. Муранов 1, Т. В. Лаптинская 2, А. С. Перегудов 1, академик А. Р. Хохлов 1, 2
1 Институт элементоорганических соединений
им. А.Н.Несмеянова Российской академии наук
Москва, Россия
2 Московский государственный университет имени
М.В. Ломоносова. Физический факультет
Москва, Россия
* E-mail: chekurov@polly.phys.msu.ru
Поступила в редакцию 11.12.2019
После доработки 11.12.2019
Принята к публикации 11.12.2019
Аннотация
Радикальная полимеризация 2,3,4,5,6-пентафторстирола (ПФС) в присутствии поли-2-гидроксиэтилметакрилата с дитиобензоатной концевой группой в качестве агента передачи цепи исследована in situ в ДМФА-d7 с помощью ПМР спектроскопии. Установлено, что реакция протекает практически без индукционного периода с более высокой скоростью, чем полимеризация ПФС в присутствии низкомолекулярного агента передачи цепи. Одной из возможных причин наблюдаемых кинетических закономерностей является самоассоциация диблок-сополимеров ПФС и 2-гидроксиэтилметакрилата в процессе полимеризации.
Амфифильные диблок-сополимеры (ДС) на основе 2,3,4,5,6-пентафторстирола (ПФС) и 2-гидроксиэтилметакрилата (ГЭМА) различного состава активно применяются для создания супергидрофобных покрытий [1–3]. Покрытия из амфифильных ДС проявляют супергидрофобные свойства благодаря реализации состояния Касси, которое обеспечивается наношероховатостью поверхности и ее низкой поверхностной энергией [4–6]. Ранее мы показали [3], что превращение гидрофобных покрытий в супергидрофобные зависит от состава ДС и контролируется соотношением между длинами поли-2,3,4,5,6-пентафторстирольного (ППФС) и поли-2-гидроксиэтилметакрилатного блоков (ПГЭМА) ($P_{n}^{{{\text{ПФС}}}}{\text{/}}P_{n}^{{{\text{ГЭМА}}}}$). Увеличение $P_{n}^{{{\text{ПФС}}}}{\text{/}}P_{n}^{{{\text{ГЭМА}}}}$ приводит к существенному повышению краевых углов смачивания водой (${{\theta }^{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$) и дийодметаном (${{\theta }^{{{\text{C}}{{{\text{H}}}_{{\text{2}}}}{{{\text{I}}}_{{\text{2}}}}}}}$), которые достигают предельных значений при $P_{n}^{{{\text{ПФС}}}}{\text{/}}P_{n}^{{{\text{ГЭМА}}}}$ = 3.5 и остаются практически постоянными вплоть до $P_{n}^{{{\text{ПФС}}}}{\text{/}}P_{n}^{{{\text{ГЭМА}}}}$ = 6.2. Наиболее высокие краевые углы ${{\theta }^{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 158 ± 4° и ${{\theta }^{{{\text{C}}{{{\text{H}}}_{{\text{2}}}}{{{\text{I}}}_{{\text{2}}}}}}}$ = 107 ± 3° были достигнуты при нанесении на нейлоновую ткань ДС с $P_{n}^{{{\text{ПФС}}}}{\text{/}}P_{n}^{{{\text{ГЭМА}}}}$ = 6.2. Таким образом, для создания супергидрофобных покрытий нужно научиться направлено получать ДС с требуемым составом, который определяется не только исходной концентрацией реагентов, но и продолжительностью реакции.
Цель настоящей работы – изучение кинетики сополимеризации ПФС в присутствии ПГЭМА ОПЦ-агента с помощью спектроскопии ПМР непосредственно в ходе процесса (in situ). Схема полимеризации ПФС в присутствии ПГЭМА ОПЦ-агента показана ниже.
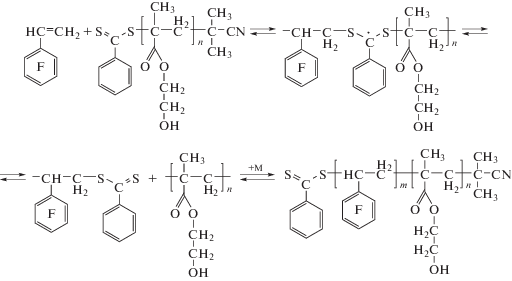
Полимеризацию ПФС ([ПФС] = 2 моль/л) проводили в присутствии ПГЭМА ОПЦ-агента (Mn = 23.5 × 103, Mw/Mn = 1.24 по данным ГПХ) и инициатора – динитрила азоизомасляной кислоты (ДАК) при мольном соотношении [ПГЭМА]/[ДАК] = 4.0. Недавно мы показали, что в этих условиях полимеризация ПФС протекает по механизму ОПЦ полимеризации [7]. Реакцию проводили в ДМФА-d7 в ампуле для измерения спектров ПМР непосредственно в ЯМР-спектрометре, поддерживая постоянную температуру 50°С, и через определенные промежутки времени, вплоть до конверсии ПФС q = 27.2%, записывали спектры реакционных мономерно-полимерных смесей и измеряли интегральную интенсивность соответствующих сигналов. Реакционную смесь перед полимеризацией освобождали от воздуха трехкратным повторением циклов замораживание–оттаивание в вакууме. Количественное определение содержания ПФС в процессе реакции рассчитывали относительно общей интегральной интенсивности всех протонов реакционной смеси. Точность измерения интегральной интенсивности составляла ~5%. Такой подход мы использовали ранее для определения состава сополимеров акриламида с анионоактивными и катионоактивными сомономерами при разных конверсиях [8, 9].
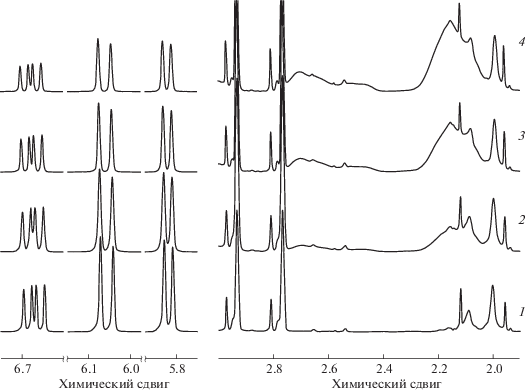
На рис. 1 приведены фрагменты спектров ПМР мономерно-полимерных реакционных смесей, образующихся в ходе полимеризации при различных конверсиях ПФС. В спектре ПМР ПФС в ДМФА-d7 при q = 0% (рис. 1, спектр 1) имеются сигналы с химическими сдвигами при 6.67–5.82 м.д., относящиеся к трем протонам винильной группы (СН=, СН2=). При записи спектров в ДМФА-d7 эти сигналы не перекрываются. Интегральная интенсивность сигналов пропорциональна концентрации ПФС в мономерно-полимерной смеси. Для расчета количества непрореагировавшего ПФС и q можно использовать интегральные интенсивности каждого из сигналов СН2= и СН=, однако для повышения точности мы использовали их сумму. Уширенные сигналы между 2.7 и 2.1 м.д. относятся к ППФС блоку в ДС. Интенсивность этих сигналов растет с увеличением конверсии мономера от 7.8 до 27.2% (рис. 1, спектры 2–4).
Рис. 1.
Спектры ПМР реакционных мономерно-полимерных смесей до полимеризации (1) и после полимеризации ПФС в интервале q от 7.8 до 27.2% (2–4). [ПФС] = 2 моль/л, [ДАК] = 1 × 10–3 моль/л, [ПГЭМА] = 4 × 10–3 моль/л, 50°С, ДМФА-d7.
Полулогарифмическая зависимость ln([М]0/[М]) от времени реакции в присутствии ПГЭМА ОПЦ-агента приведена на рис. 2 (кривая 1). Для сравнения на рис. 2 показана кинетическая кривая (кривая 2) для полимеризации ПФС в присутствии 2-циано-2-пропил дитиобензоата (ЦПТБ) – агента передачи цепи, который применяли для синтеза ПГЭМА ОПЦ-агента. Линейная зависимость ln([М]0/[М]) от времени реакции указывает на первый порядок обеих реакций полимеризации по мономеру. Из рис. 2 можно видеть, что полимеризация ПФС в присутствии ЦПТБ протекает с индукционным периодом (τ ≈ 150 мин), что является вполне обычным для полимеризации различных мономеров в присутствии дитиобензоатов [10]. Появление индукционного периода и замедление скорости реакции объясняют либо медленной фрагментацией радикальных интермедиатов, либо существованием реакций обрыва на радикальных интермедиатах [10].
Рис. 2.
Зависимость конверсии ПФС от времени полимеризации, инициированной ДАК, в присутствии ПГЭМА ОПЦ-агента (1) и ЦПТБ (2). [ПФС] = 2.0 моль/л, [ДАК] = 1.0 × 10–3 моль/л, [ЦПТБ]/[ДАК] = 1.9, [ПГЭМА]/[ДАК] = 4.0, 50°С, ДМФА-d7.
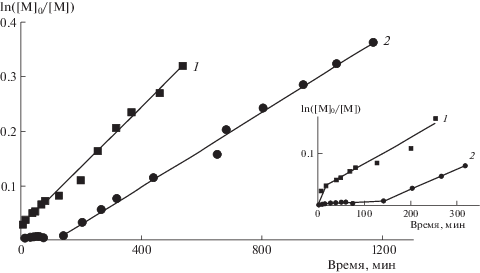
Другая картина наблюдается при полимеризации ПФС в присутствии ПГЭМА ОПЦ-агента. Полимеризация начинается практически сразу при нагревании реакционной смеси и протекает с более высокой скоростью, чем аналогичная реакция в присутствии ЦПТБ, несмотря на более высокую концентрацию ПГЭМА ОПЦ-агента и более низкую концентрацию ДАК ([ПГЭМА]/[ДАК] = = 4.0 по сравнению с [ЦПТБ]/[ДАК] = 1.9).
Одной из возможных причин более высокой скорости полимеризации ПФС в присутствии ПГЭМА ОПЦ-агента может быть ассоциативное поведение диблок-сополимерных макромолекул, формирующихся в ДМФА-d7 во время полимеризации. Очевидно, в ходе реакции при 50°С в ДМФА-d7, селективном растворителе для ПГЭМА, при достижении определенной q диблок-сополимерные растущие радикалы образуют межцепные полимерные ассоциаты (агрегаты), в которых гидрофобные ППФС-блоки в компактизованных конформациях окружены сольватированными ПГЭМА-блоками. Гидрофобный ПФС, вероятно, солюбилизируется внутри таких агрегатов, что приводит к увеличению его локальной концентрации внутри агрегатов и, как следствие, к более высокой скорости полимеризации по сравнению с полимеризацией в присутствии ЦПТБ. Такое поведение является характерным для процессов самоорганизации, вызванной полимеризацией [11–13].
Для проверки предположения о формировании межцепных агрегатов в процессе полимеризации ПФС мы исследовали поведение разбавленного раствора H180F195 (табл. 1) при [H180F195] = = 1 мг/мл в ДМФА при 60°С методом дина-мического светорассеяния (ДСР) на гониометре ALVDLS/SLS-SP 5022F (Германия) с помощью ALV 5000/E коррелятора. Измерения проводили при 60°С при λ = 632.8 нм (He-Ne лазер). Для обработки спектров применяли метод CONTIN и рассчитывали гидродинамические радиусы каждой моды по формуле Стокса–Эйнштейна.
Таблица 1.
Условия получения и молекулярно-массовые характеристики ПГЭМА и ДС
| Образец | Шифр1) | [ОПЦ]/[ДАК] | Время, ч | q, %2) | Mn × 10–3 | Mw/Mn4) | Состав ДС, мол. %5) | ||
|---|---|---|---|---|---|---|---|---|---|
| Теор3) | ГПХ4) | ГЭМА | ПФС | ||||||
| ПГЭМА | H180 | 2.5/1 | 18 | 91 | 12.0 | 23.5 | 1.24 | 100 | 0 |
| ДС | H180F195 | 4/1 | 24 | 29 | 51.2 | 36.6 | 1.30 | 48 | 52 |
1) Шифр “Н180”используется для обозначения ПГЭМА со степенью полимеризации Pn = Mn/130.143, где Mn – среднечисловая молекулярная масса ПГЭМА, определенная ГПХ, 130.143 – молекулярная масса ГЭМА(Н). Шифр “H180F195” относится к ДС, состоящему из 180 звеньев ГЭМА и 195 звеньев ПФС (F). Степень полимеризации ПФС рассчитывали, принимая во внимание состав ДС. 2) Конверсию q определяли гравиметрически. 3) Теоретические значения Mn рассчитывали, полагая, что одна молекула ОПЦ агента (ЦПТБ или ПГЭМА ОПЦ-агент) приводит к контролируемому росту одной макромолекулы (со)полимера, по уравнению: ${{M}_{n}} = ~{{M}_{{{\text{ОПЦ}}}}} + \frac{{q{{{[{\text{ПФС}}]}}_{0}}{{M}_{{{\text{ПФС}}}}}}}{{{{{[{\text{ОПЦ}}]}}_{0}}}}$, где МОПЦ и МПФС – молекулярные массы ЦПТБ или ПГЭМА ОПЦ-агента, и ПФС, [ОПЦ]0 и [ПФС]0 – их начальные концентрации, q – конверсия ПФС. 4) Условия эксперимента: прибор Agilent 1200 с рефрактометрическим детектором, колонка G-gel [14, 15], элюент – смесь ТГФ : 0.03 M LiBr в ДМФА (50 : 50 об. %), температура 30°С и скорость 0.5 мл/мин. Калибровка выполнена по полистирольным стандартам. 5) Состав ДС определяли ЭА.
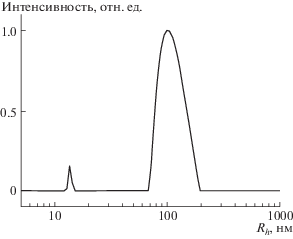
Исследованный образец в разбавленном растворе находится в агрегированном состоянии. На кривой распределения интенсивности рассеяния света по размеру частиц (рис. 3) наблюдается два пика: минорный пик при Rh = 13.5 и основной пик при 100 нм, которые можно отнести к макромолекулам и агрегатам соответственно. В условиях проведения полимеризации ПФС концентрация полимеров существенно выше, например, исходная концентрация ПГЭМА ОПЦ-агента в реакционной смеси при синтезе H180F195 составляет примерно 80 мг/мл, что в 80 раз превышает концентрацию H180F195 в образце, который исследовали методом ДСР. На растворимость образующегося полимера в среде синтеза оказывает положительное влияние присутствие мономерного ПФС. Проведенные модельные эксперименты продемонстрировали полное молекулярное растворение (контроль методом ДРС) ППФС при концентрации 1% в среде ПФС/ДМФА 40/60 (по объему) при температуре 25°С, которое сменяется на частичную растворимость (осадок/коллоидный раствор с Rh = 1 мкм) при уменьшении содержания мономера до 30/70. Все эти данные согласуются с предположением о том, что по мере формирования ППФС-блока и уменьшения количества ПФС мономера начинается микрофазное расслоение системы с образованием мицеллоподобных агрегатов с ППФС ядром.
ИСТОЧНИКИ ФИНАНСИРОВАНИЯ
Рис. 3.
Зависимость интенсивности рассеяния света под углом 150°от радиуса частиц (“невзвешенное” распределение) для раствора H180F195 в ДМФА при 60°С. [H180F195] = 1 мг/мл.
Работа выполнена при поддержке Российского научного фонда (проект № 17–13–01359). Регистрация спектров ЯМР, элементный анализ проведены при поддержке Министерства науки и высшего образования Российской Федерации с использованием научного оборудования Центра исследования строения молекул ИНЭОС РАН.
Список литературы
Chekurov K.E., Barabanova A.I., Blagodatskikh I.V., Lokshin B.V., Peregudov A.S., Abramchuk S.S., Khokhlov A.R. // Dokl. Chem. 2019. V. 484. № 2. P. 33–36.
Chekurov K.E., Barabanova A.I., Blagodatskhikh I.V., Peregudov A.S., Khokhlov A.R. // Fluorine Notes. 2019. V. 123. № 2. P. 1–2.
Chekurov K.E., Barabanova A.I., Blagodatskhikh I.V., Lokshin B.V., Kondratenko M.S., Gallyamov M.O., Abramchuk S.S., Peregudov A.S., Khokhlov A.R. // Polymer. 2019 (in editors).
Chang H., Tu K., Wang X., Liu J. // RSC Advances. 2015. V. 39. № 5. P. 30647–30653.
Chu Z., Seeger S. // Chem. Soc. Rev. 2014. V. 43. № 8. P. 2784–2798.
Tuteja A., Choi W., Mabry J.M., McKinley G.H., Co-hen R.E. // Proceedings of the National Academy of Sciences of the United States of America. 2008. V. 105. №. 47. P. 18200–18205.
Chekurov K.E., Barabanova A.I., Blagodatskikh I.V., Peregudov A.S., Khokhlov A.R. // Fluorine Notes. 2019. V. 124. № 3. P. 1–2.
Barabanova A.I., Gromov V.F., Bune E.V., Bogachev Y.S., Kozlova N.V., Teleshov E.N. // Vysokomol. Soedin. 1994. V. 36. № 6. P. 901–907.
Bune Y.V., Barabanova A.I., Bogachev Y.S., Gromov V.F. // Eur. Polym. J. 1997. V. 33. № 8. P. 1313–1323.
Moad G. // Macromol. Chem. Phys. 2014. V. 215. P. 9–26.
Derry M.J., Fielding L.A., Armes S.P. // Progress in Polymer science. 2016. V. 52. P. 1–18.
Zhou D., Dong. S., Kuchel R.P., Perrier S., Zetter-lund P.B. // Polym. Chem. 2017. V. 8. P. 3082–3089.
Zhou D., Kuchel R.P., Zetterlund P.B. // Polym. Chem. 2017. V. 8. № 29. P. 4177–4181.
Tennikova T. B., Horak D., Svec F., Kolar J., Coupek J., Trushin A., Maltzev V.G., Belenki B.G. // J. Chromatigr. A. 1988. V. 435. P. 357–362.
Фоменков А.И., Благодатских И.В., Пономарев Ив.И., Волкова Ю.А., Пономарев И.И., Хохлов А.Р. // Высокомол. Соед. Б. 2009. Т. 51. № 5. С. 874–882.
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Химия, науки о материалах