

Доклады Российской академии наук. Химия, науки о материалах , 2020, T. 495, № 1, стр. 66-70
Специфика зависимости скорости реакций горения газов от температуры
Член-корреспондент РАН В. В. Азатян *
Объединенный институт высоких температур Российской академии наук
Москва, Россия
* E-mail: vylenazatyan@yandex.ru
Поступила в редакцию 26.05.2020
После доработки 22.10.2020
Принята к публикации 29.10.2020
Аннотация
Выяснена основная причина важнейших особенностей газофазного горения, ранее не находивших объяснения. Показано, что резкое ускорение горения при нагревании и большие скорости вызваны прежде всего не увеличением константы скорости лимитирующей стадии, как это считалось ранее, а ростом концентраций активных промежуточных частиц с температурой по закону экспоненты, содержащей фактор Больцмана в положительном показателе степени. Такой характер температурной зависимости определяет переход горения во взрыв и в детонацию.
Зависимость скорости реакций горения от температуры определяет их важнейшие закономерности. Осуществляя положительную обратную связь между температурой горючей газовой смеси и скоростью процесса, температурная зависимость обеспечивает возгорание при нагревании, ускорение реакций под воздействием выделяющегося тепла, переход горения во взрыв и в детонацию. При давлениях выше десятой доли атмосферного давления горение завершается за доли миллисекунды, несмотря на большую прочность химических связей в исходных молекулах. С этой особенностью горения неразрывно связана проблема чрезвычайно сильной температурной зависимости скорости реакции, легко переходящей во взрыв и в детонацию. Актуальность этих проблем определяется значимостью не только для теории, но также для практики, в том числе, в связи с работой силовых установок, с проблемами взрывобезопасности. До недавнего времени химический процесс горения при давлениях выше сотых долей атмосферного давления гипотетически представляли одностадийной реакцией, которой приписывали константу скорости реакции второго или первого кинетического порядка. Самоускорение считали результатом только саморазогрева (например, [1–7]). Позже, однако, было показано [8–10], что эти представления находятся в коренном противоречии со многими наблюдаемыми закономерностями и, прежде всего, с очень большими энергиями активации межмолекулярных реакций, исключающими большие скорости процессов, необходимые для горения. Известно также, что численное решение уравнений с записью реакций атомов и радикалов не объясняет, а лишь описывает частные закономерности в меру достоверности принятой реакционной схемы и входящих параметров.
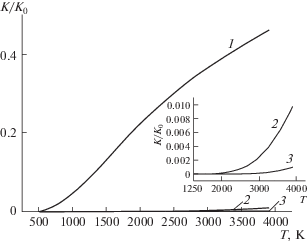
Ответ на фундаментальный вопрос о факторах, определяющих высокие скорости реакций горения, взрыва и детонации газов был получен на базе экспериментальных и теоретических исследований, выявивших цепной характер этих процессов [8–10], который и определяет их особенности. Было показано также, что, вопреки сложившимся представлениям, чем ниже энергия активации, тем сильнее зависят константа скорости и скорость реакции от температуры (рис. 1). Однако выяснилось, что сильная температурная зависимость константы не обеспечивает наблюдаемое резкое самоускорение горения.
Рис. 1.
Графики функции k/k0= e–E/RT при значениях энергии активации, кДж моль–1: 25 (кривая 1), 150 (кривая 1), 225 (кривая 1).
Настоящая работа посвящена выяснению факторов, определяющих специфику температурной зависимости скорости реакций горения газов на примере модельной реакции водорода с кислородом. Горение протекает с участием следующих основных реакций [1, 11]:
В реакциях атомов и радикалов – носителей цепей (НЦ) образуются новые ветви, и цепь разветвляется. Лимитирующей стадией процесса является реакция (I) размножения свободных валентностей, поскольку энергия активации этой стадии выше энергий активации стадий (II) и (III) [8, 11]. Атомы Н вступают также в реакции образования малоактивных продуктов, не участвующих в цепном горении. В таких реакциях цепь обрывается. Это, например, гетерогенная рекомбинация активных частиц, а также реакция
(IV)
${\text{Н}} + {{{\text{О}}}_{{\text{2}}}} + {\text{М}} = {\text{Н}}{{{\text{О}}}_{2}} + {\text{М}},$В силу очень высоких величин констант скорости стадий (II) и (III) роль реакций обрыва цепей с участием активных частиц ОН и О незначительна, и их можно не учитывать. Известным методом частичных квазистационарных концентраций [11] система кинетических уравнений активных частиц приводится к одному уравнению, относящемуся к той частице, реакция которой осуществляет разветвление и этим лимитируют размножение активных промежуточных частиц. В данном процессе это атомы Н.
Из приведенной схемы реакции следует, что скорость расходования кислорода равна:
(1)
$\begin{gathered} W = - d[B]{\text{/}}dt = \\ = {{\omega }_{0}} + \left\{ {{{k}_{1}} + {{k}_{5}}[M]} \right\}[B]n = {{\omega }_{0}} + {{k}_{{\text{c}}}}[B]n, \\ \end{gathered} $Согласно схеме реакции, изменению концентрации атомов Н соответствует уравнение (2):
(2)
$\frac{{dn}}{{dt}} = {{\omega }_{{\text{o}}}} + \left( {f - g} \right)n = {{\omega }_{{\text{o}}}} + \varphi n,$(3)
$\begin{gathered} f = 2{{k}_{1}}\left[ B \right] = 2k_{1}^{0}{{e}^{{--~\frac{{{{E}_{1}}}}{{RT}}}}}\left[ B \right], \\ g = {{k}_{{{\text{эфф}}}}}, \\ \end{gathered} $Из уравнения (2) следует, что в условиях g > f $~$устанавливается низкая стационарная концентрация активных частиц, определяемая соотношением скоростей их образования в очень медленной реакции (0) и гибели. Если же f > g, т.е. если размножение НЦ быстрее их гибели, то происходит лавинное размножение активных частиц. Соответственно, лавинообразно ускоряется и расходование исходных реагентов, реагирующих с этими НЦ. Таким образом, условием цепного воспламенения является неравенство:
При давлениях выше сотых долей атмосферного давления горение сопровождается интенсивным саморазогревом и в определенных условиях переходит во взрыв. При реальных предэкспоненциальных множителях, относящихся к бимолекулярным реакциям, функция ${{e}^{{--~\frac{E}{{RT}}}}}^{~}$ не описывает наблюдаемый резкий рост скорости независимо от величины Е. Например, при нагревании стехиометрической смеси Н2 с О2 при 1 атм. от 823 до 853 К константа скорости межмолекулярной реакции (0), характеризующейся энергией активации 225 кДж моль–1 [12], возрастает приблизительно лишь на 5%. На столько же ускоряется реакция (0). Между тем, при таком нагреве смесь воспламеняется и выгорает за доли миллисекунды. Таким образом, скорость реакции горения от температуры зависит несравненно сильнее, чем скорость межмолекулярной реакции, приписываемой ранее горению.
Проведенный нами анализ показал, что температурная зависимость скорости реакций горения определяется, прежде всего, следующей особенностью кинетики изменения концентраций НЦ: в каждый момент времени и при каждых данных величинах f и g скорость изменения концентрации НЦ находится в обратной связи с концентрацией n, как это видно также из уравнения (5). При этом, если f > g, то обратная связь положительная, и, значит, в развивающемся процессе величина n возрастает во времени экспоненциально даже при постоянной температуре. При саморазогреве рост концентрации активных частиц становится еще сильнее. Вызвано это тем, что в показатель степени в качестве множителя времени t входит разность (f – g). В величину же f входит константа скорости k1 со своим фактором Больцмана в соответствии с выражением (3). Таким образом, при f > g величины n и W зависят от температуры по закону экспоненты, находящейся в положительном показателе степени. Поскольку такая функциональная зависимость осуществляется при каждой данной температуре, то она выполняется в ходе горения.
Описанное резкое самоускорение процесса при повышении температуры вызвано, прежде всего, сильным увеличением скоростей реакций (II) и (III). Благодаря малым энергиям активаций этих реакций и, соответственно, сильному росту их скоростей с температурой (рис. 1) происходит экспоненциально ускоряющееся размножение активных частиц и, значит, так же сильно ускоряющееся расходование исходных реагентов, т.е. скорость горения.
Приведенный выше цепной механизм позволяет выразить описанную специфику зависимости скорости процесса и концентрации НЦ от времени, от концентраций реагентов и от температуры и выразить закон в аналитическом виде. При t > to ≅ 2.5/φ, когда в уравнениях (1) и (2) можно пренебречь величиной ω0, интегрирование уравнения (2) с учетом формулы (3) и температурной зависимости k1 приводит к следующей зависимости n от Т и от t:
(5)
$n = {{n}_{0}}\exp \mathop \smallint \limits_{{{t}_{0}}}^t \left[ {{{f}_{0}}\exp \left( {--\frac{{{{E}_{{\text{p}}}}}}{{RT}}} \right) - g} \right]dt.$Здесь Eр – энергия активации разветвления, n0 – концентрация НЦ при t0, величина f0 равна 2$k_{1}^{0}$[В], где $k_{1}^{0}$ – предэкспоненциальный множитель константы скорости k1.
При подстановке n из выражения (5) в уравнение скорости (1) получается [8–10]:
(6)
$\frac{W}{{\left[ B \right]}} = {{k}_{1}}{{n}_{{0~}}}\exp \mathop \smallint \limits_{{{t}_{0}}}^t \,[{{f}_{0}}\exp ( - E{\text{/}}RT)--g]\,dt.$Из уравнения (6) следует, что в режиме развивающегося горения скорость возрастает по закону экспоненты, находящейся в положительном показателе степени. По такому же закону возрастает величина $\frac{{\partial W}}{{\partial T}},$ т.е. температурная зависимость скорости. Согласно выражению (6), скорость и $\frac{{\partial W}}{{\partial T}}$ возрастают также вследствие увеличения времени реакции. Такая сильная температурная зависимость скорости обусловливает распространение пламени, легкий переход горения в цепно-тепловой взрыв и определяет резкое самоускорение реакции в режимах цепно-теплового взрыва и детонации.
В общем виде обратная связь между ni и $~\frac{{\partial {{n}_{i}}}}{{\partial t}}$ относится ко всем активным частицам, это следует из самого цепного механизма. Математически обратная связь в общем виде следует также из анализа системы уравнений, учитывающих разного типа реакции активных частиц [13]. В эти уравнения входят произведения концентраций констант скорости реакций элементарных реакций, относящихся ко всем компонентам цепей. Учитывается также диффузия частиц. Наличие диффузионного члена не устраняет указанную обратную связь. Система уравнений, учитывающая реакции всех компонентов, вообще говоря, не линейная. Но в каждой точке пространства и в каждый момент времени ее решение определяется свойствами линеаризованной системы, куда входит и вклад диффузионных членов. Концентрации НЦ изменяются по закону exp(Φt), где Φ играет роль величины φ в формуле (4) и равна разности величин f1 и g1, являющихся аналогами f и g. Произведения констант скорости на концентрации, относящиеся к разветвлению и к обрыву цепей, входят в величину Φ соответственно с положительным и c отрицательным знаком, также как и в рассмотренной выше одноцентровой схеме. Константы скорости, входящие в величину Φ, и, значит, сама величина Φ с изменением температуры, конечно, изменяются. Однако экспоненциальный характер зависимости концентраций НЦ от произведения Φt сохраняется. Скорость разветвления и, значит, величина f1 со своими факторами Больцмана зависят от температуры сильнее, чем скорость обрыва. Поэтому при f1> g1 с повышением температуры скорость цепной реакции возрастает фактически по “двойной экспоненте”, аналогично показанной уравнением (19).
Сделанные выше выводы о роли активных частиц в температурной зависимости скорости далее сравниваются с результатами экспериментов. Реакция проводилась при двух разных, практически постоянных температурах. Ставилась цель выяснить, определяется ли рост скорости реакции цепного горения только увеличением константы скорости лимитирующей стадии, как это считалось ранее, или сказывается также рост концентраций активных частиц. На рис. 2 приведены осциллограммы давления, хемилюминесценции и температуры, зарегистрированные в ходе горения стехиометрической смеси Н2 с О2 в термостатированном кварцевом реакторе при постоянных температурах 773 и 768 К и начальном давлении 2.25 Торр. Давление регистрировалось с помощью чувствительного манометра. Микротермопара размещалась в кварцевом капилляре, промытом плавиковой кислотой для уменьшения эффективности рекомбинации атомов и рекомбинационного разогрева. На рис. 2 видно, что различие разогревов при начальных температурах 668 и 773 К не превышает 0.15°, что составляет лишь 3% от разности начальных температур. Регистрированный же рост температуры примерно на 4° отражает в основном рекомбинационный разогрев капилляра с термопарой.
Рис. 2.
Осциллограммы давления (кривые 1, 1 '), хемилюминесценции (кривые 2, 2 ') и температуры (кривые 3, 3 ') при 768 К (кривые 1, 2, 3) и 773 К (кривые 1 ', 2 ', 3 ').
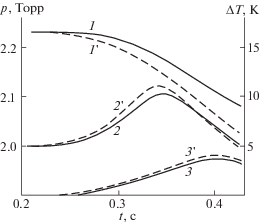
Рассмотрим сначала роль изменения концентраций активных частиц в температурной зависимости скорости реакции горения. Как это видно из стехиометрии брутто-реакции:
уменьшение числа молей смеси в ходе горения равно уменьшению числа молей О2, и, соответственно, снижение давления в замкнутом реакторе равно снижению парциального давления О2. Поэтому отношение максимальных величин угловых коэффициентов кинетических кривых 1 и 1' на рис. 2 равно отношению максимальных скоростей расходования О2 при рассматриваемых 773 и 768 К. Согласно закону Аррениуса, это отношение и заданные температуры связаны известным выражением:(7)
$\ln \frac{{W{\kern 1pt} '}}{W} = \frac{{E\left( {{{T}_{2}}--{{T}_{1}}} \right)}}{{R{{T}_{1}}{{T}_{2}}}}.$Подставив отношение скоростей, равное 1.29, и приведенные значения температуры в выражение (7), получаем эффективную энергию активации, равную 60.0 ккал моль–1. Между тем энергия активации константы скорости лимитирующей стадии (I) равна лишь 16.7 ккал моль–1 [9, 12]. Поэтому при традиционном игнорировании роста концентрации атомов Н расчетная скорость возрастает на 7% против 29% роста скорости процесса в целом. Это значит, что в согласии с уравнением (6), при повышении температуры всего лишь на 5° влияние роста концентрации НЦ на скорость процесса значительно больше по сравнению с влиянием увеличения k1. Росту концентрации атомов Н в этом интервале температур соответствует эффективная энергия активации, равная 43.3 ккал моль–1.
Скорость ω0 несравненно меньше скоростей остальных реакций. В силу низких давлений незначительна также роль реакции (VI). Поэтому, согласно выражению (1), отношение скорости реакции W к величине k1[O2] равно величине n, т.е. концентрации атомов Н. Величинам W, k1 и [O2], относящимся к максимальному наклону осциллограммы (рис. 1, кривая 1'), соответствует концентрация атомов Н, равная 2 × 1011 ат. см–3. В силу цепного механизма эта величина на много порядков больше равновесной концентрации атомов Н.
Температурная зависимость скорости в области атмосферного давления была изучена с использованием приведенных в работе [1] кинетических данных по реакции Н2 с О2 в замкнутом кварцевом реакторе при 830 К вблизи третьего предела воспламенения, а также скорости этой реакции при температуре 1320 К, определенной нами по измеренной видимой скорости дефлаграционного распространения пламени в кварцевой трубке диаметром 1.5 см. Согласно данным работы [1], скорость W3 реакции горения Н2 с О2 при 830 К в самых начальных стадиях горения равна 4.7 × 1014 молек. см–3 с–1. Скорость реакции W4 при 1320 К определяли как отношение падения концентрации О2 к продолжительности нахождения смеси в пламени. Это время рассчитывалось как отношение длины зоны реакции δ к величине измеренной видимой скорости пламени, равной 1000 см с–1. Величина δ, рассчитанная по методу, описанному в работе [14], составляла приблизительно 1 × 10–2 см. Определенная таким образом из экспериментальных данных скорость W4 оказалась равной 2.4 × 1022 молек. см–3 с–1, что в 5 × 105 раз больше, чем при 830 К. Между тем при данном повышении температуры скорость, рассчитанная традиционно без учета роста концентрации атомов Н, оказывается равной лишь 2 × 1016 молек. см–3 с–1, что в 1.2 × 106 раз меньше реальной скорости реакции горения. Таким образом, при игнорировании роста концентрации активных частиц расчетная скорость процесса возрастает лишь в 100 раз, что в 5000 раз меньше реального роста.
При допущении одностадийности реакции, подчиняющейся закону Аррениуса, из величин скорости при температурах 830 и 1320 К по уравнению (7) получается эффективная “энергия активации”, равная 78.4 ккал моль–1. При значении константы скорости k1 = 16.7 ккал моль–1 разность этой величины и энергии активации составит 61.47 ккал моль–1, что можно рассматривать как эффективную “энергию активации” роста концентрации атомов Н.
Концентрация атомарного водорода, определенная как отношение скорости реакции при 1320 К к произведению константы скорости k1 и средней концентрации О2, равна 4.7 × 1016 ат. см–3, что составляет 0.8% от общей смеси и согласуется с результатами масс-спектрометрических измерений концентраций атомов Н в пламени смеси водорода с воздухом, также бедной водородом [15].
Таким образом, установлены причины важнейших особенностей газофазного горения, ранее не находивших объяснения. Показано, что резкое ускорение горения при нагревании и большие скорости вызваны прежде всего не увеличением константы скорости лимитирующей стадии, как это считалось ранее, а ростом концентраций активных промежуточных частиц с температурой по закону экспоненты, содержащей фактор Больцмана в положительном показателе степени.
Список литературы
Lewis B., Von Elbe G. Combustion, explosions and flame in gases / N.Y.–L.: Acad. Press, 1987. 592 p.
Химическая энциклопедия / М.; Сов. энциклопедия. 1988. Т. 1. Горение. С. 1164.
Физическая энциклопедия / М.: Сов. энциклопедия, 1988. Т. 1. Горение. С. 515.
Франк–Каменецкий Д.А. Основы макрокинетики, диффузия, теплопередача в химической кинетике. Учебник-монография / Долгопрудный: “Интеллект”, 2008. 407 с.
Tang S., Chernovsky M.K., Im H.G., Atreya A. // Combust. Flame. 2010. V. 157. P. 118. https://doi.org/10.1016/j.combustflame.2009.09.010
Кукин П.П., Юшин В.В., Емельянов С.Г. Теория горения и взрыва. Учеб. пособие / М.: Юрайт-Издат, 2012. 435 с.
Девисилов В.А., Дроздова Т.И., Тимофеева С.С. Теория горения и взрыва / М.: Форум, 2012. 351 с.
Азатян В.В. // Журн. физ. химии. 2011. Т. 85. № 8. С.1405–1418.
Азатян В.В. Цепные реакции в процессах горения, взрыва и детонации газов / Черноголовка: Редакционно-издательский отдел ИПХФ РАН, 2017. 448 с.
Азатян В.В. // Кинетика и катализ. 2020. Т. 61. № 3. С. 291–301. https://doi.org/10.31857/SO453881120030041
Семенов Н.Н. Избранные труды / М.: Наука, 2005. Т. 3. 499 с.
Baulch D.L., Bowman T., Cobos C.J. et al. // J. Phys. Chem. Ref. Data. 2005. V. 34. № 3. P. 757. https://doi.org/10.1063/1.1748524
Иванова А.Н., Тернопольский Б.Л., Карнаух А.А. // Кинетика и катализ. 1997. Т. 38. № 4. С. 485–492.
Зельдович Я.Б., Компанеец А.С. Теория детонации / М.: Гостехиздат, 1955. 268 с.
Korobeinichev O.P., Shmakov A.G., Rybitskaya I.V., Bol’- shova T.A., Chernov A.A., Knyaz’kov D.A., Konnov A.A. // Kinet. Catal. 2009. V. 50. P. 156–161. https://doi.org/10.1134/S0023158409020025
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Химия, науки о материалах