

Доклады Российской академии наук. Химия, науки о материалах , 2021, T. 498, № 1, стр. 3-29
Реакция Дильса–Альдера как индикатор внутренних и внешних воздействий на скорость и равновесие. кинетика, термохимия, катализ, высокое давление. Обзор
Академик РАН А. И. Коновалов 1, Д. А. Корнилов 1, В. Д. Киселев 1, *
1 Химический институт им. А.М. Бутлерова
Казанского федерального университета
420008 Казань, Россия
* E-mail: vkiselev.ksu@gmail.com
Поступила в редакцию 02.04.2021
После доработки 12.05.2021
Принята к публикации 27.05.2021
Аннотация
Реакция Дильса–Альдера (РДА), кроме огромного синтетического потенциала, интересна тем, что является наиболее надежным полигоном для количественного исследования влияния различных внутренних и внешних факторов на скорость и равновесие. В обзоре рассмотрено влияние энергии межмолекулярного орбитального взаимодействия, затем влияние энтальпии реакции. Показано, что скорость РДА можно описать при совместном учете этих факторов. Обсуждены кинетические и термохимические данные для РДА в присутствии кислот Льюиса. Новые данные о скорости РДА при перетирании реагентов в твердой фазе генерируют много задач для дальнейшего изучения. И, наконец, анализ обширных данных о влиянии высокого давления на скорость и равновесие РДА привел к заключению, что различия в объемах активации и объемах реакции не связаны с механизмом протекания РДА.
ВВЕДЕНИЕ
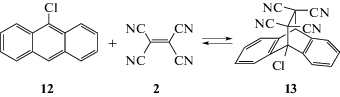
Первые данные о реакции циклоприсоединения между замещенными 1,3-диенами и активированными алкенами О. Дильс (O. Diels) и К. Альдер (K. Alder) описали 95 лет назад [1–4]. Интенсивное изучение реакции образования шестичленных карбо- и гетероциклов проводилось и проводится почти во всех крупных химических центрах мира [5–10]. Б. Арбузов, А. Вассерманн (A. Wassermann), Р. Вудвард (R. Woodward), М. Дьюар (M. Dewar), А. Петров, Дж. Робертс (J. Roberts), В. Тартаковский, К. Фукуи (K. Fukui), Х. Фуджимото (H. Fujimoto), Р. Хоффман (R. Hoffman), Р. Хьюзген (R. Huisgen) и многие другие ученые внесли значительный вклад в развитие теории и практики реакции Дильса–Альдера (РДА). В последние десятилетия глубокие и детальные работы Ю. Зауера (J. Sauer) [11–14], Ф-Г. Клярнера (F.-G. Klärner) [15, 16], К. Хука (K. Houk) [17, 18], Р. Зустмана (R. Sustmann) [13, 19, 20] и Г. Дженера (G. Jenner) [21] расширили наши знания об этом процессе. Первые исследования связаны с выяснением возможностей и условий для вовлечения разнообразных субстратов в качестве диенов и диенофилов для синтеза новых продуктов [1–14]. Значительный объем работ связан с количественным изучением реакционной способности разнообразных систем, скорость реакции для которых различается почти на 20 порядков [11–14, 22–31]. В обзоре детально рассмотрено влияние растворителя, образования межмолекулярных комплексов, влияние каталитических и солевых добавок на скорость реакции и проведены успешные попытки обобщенного учета энергии орбитальных взаимодействий реагентов, баланса энергии разрыва и образования связей и структурных особенностей диен-диенофильных пар при формировании переходного состояния. Критически рассмотрены значения объемных параметров РДА и возможности использования повышенного гидростатического давления для получения трудносинтезируемых соединений и для уточнения механизма реакции [21, 27, 32–39]. Убедительная однозначность протекания РДА и надежность количественного описания ее параметров позволили применить эту реакцию в качестве базового индикатора для выяснения влияния различных факторов на скорость и равновесие, а также для понимания “аномалий” их воздействия.
К настоящему времени имеется более полумиллиона работ в области РДА, что делает невозможным их полный анализ. Имеется ряд книг [5–10, 34] и обзоров [11, 21, 24–27, 40], посвященных этой реакции. Поэтому здесь будут изложены основные результаты, полученные при изучении РДА в нашей лаборатории.
I. ЭНЕРГИЯ МЕЖМОЛЕКУЛЯРНОГО ОРБИТАЛЬНОГО ВЗАИМОДЕЙСТВИЯ
При спектрофотометрическом изучении скорости РДА было обнаружено образование молекулярных комплексов π–π-типа между диенами и диенофилами [41–43]. Оказалось, что изменение значений потенциалов ионизации (IP, ionization potential) в ряду диенов-доноров и энергии сродства к электрону (EA, electron affinity energy) в ряду диенофилов-акцепторов пропорциональны изменению прочности межмолекулярных комплексов и изменению свободной энергии активации в РДА [43–47]. Изменение энергии стабилизации в этих процессах при смене растворителя тоже оказалось пропорциональным [24–26, 47, 48]. Дополнительно кинетические и термохимические измерения показали пропорциональное изменение энтальпии образования молекулярных комплексов и энтальпии активации в РДА внутри структурных рядов реагентов [47–52]. Однако эта пропорциональность не позволяет однозначно решить вопрос о месте молекулярных комплексов (МС, molecular complex) в схеме реакции (схема 1).
Схема 1.
Возможные направления образования продукта (Р) реакции Дильса–Альдера между диеном (Д) и диенофилом (А) с участием молекулярного комплекса (МС).
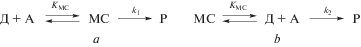
При проведении реакции в условиях большого избытка одного из реагентов (например, сА$ \gg $ сД) наблюдаемая константа скорости реакции (kэксп.) для этих направлений описывается соотношениями (1) и (2) соответственно:
(1)
${{k}_{{{\text{эксп}}{\text{.}}}}} = {{k}_{1}}{{K}_{{{\text{MC}}}}}{\text{/}}(1 + {{K}_{{{\text{MC}}}}} \cdot {{с}_{{\text{А}}}})$(2)
${{k}_{{{\text{эксп}}{\text{.}}}}} = {{k}_{2}}{\text{/}}(1 + {{K}_{{{\text{MC}}}}} \cdot {{с}_{{\text{А}}}}),$Рассчитанные значения констант равновесия из зависимости (1/kэксп.) от (сА) (уравнения 1 и 2) близко соответствуют данным прямых измерений KMC [44–47, 53]. Понятно, что и эти результаты не позволяют сделать выбор между направлениями реакции (a) и (b) (схема 1). Если создать условия, при которых произведение (KMC ∙ сА) значительно меньше единицы, то экспериментальные значения энтальпии активации для двух возможных направлений (a) и (b) определяются соотношениями (3) и (4) соответственно:
(3)
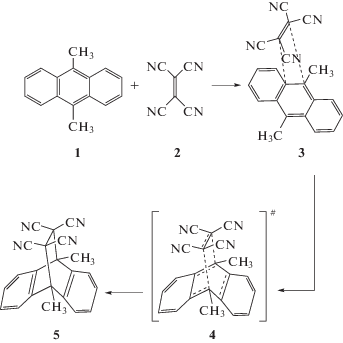
$\Delta H_{{{\text{эксп}}{\text{.}}}}^{ \ne } = \Delta H_{{\text{1}}}^{ \ne } + \Delta H_{{{\text{MC}}}}^{0}$Для реагентов, способных к образованию прочного МС, значение $\Delta H_{{{\text{эксп}}{\text{.}}}}^{ \ne }$ может быть отрицательным только при реализации направления (a), когда |ΔHMC| > ΔH$_{{\text{1}}}^{ \ne }$ (уравнение 3). Большое различие значений энтальпии растворения твердого тетрацианоэтилена в хлорбензоле (23.1 кДж моль–1, IP = 9.10 эВ) и в мезитилене (–2.7 кДж моль–1, IP = 8.14 эВ) можно отнести к различию энтальпий образования этих комплексов [54–56]. На примере реакции Дильса–Альдера между более сильным π-донором 9,10-диметилантраценом 1 (IP = 7.04 эВ) и тетрацианоэтиленом 2 (EА = 2.88 эВ) (схема 2) в серии растворителей было обнаружено уменьшение константы скорости реакции с ростом температуры [49], что соответствует отрицательным значениям наблюдаемой энтальпии активации в ацетонитриле (–5.0 ± 0.8 кДж моль–1), дихлорметане (–7.5 ± 1.0 кДж моль–1), 1,2-дихлорэтане (–7.9 ± ± 1.0 кДж моль–1) и хлороформе (–11.5 ± ± 1.5 кДж моль–1). Полученные результаты соответствуют тому, что энергия межмолекулярного взаимодействия этих реагентов при образовании МС превышает по величине затраты энергии в процессе перераспределения связей при достижении активированного комплекса. Дополнительно следует, что для данной реакции не требуется переориентация структуры молекулярного комплекса 3 при дальнейшем его переходе в активированный комплекс 4 и продукт реакции 5 [49].
Схема 2.
Образование молекулярного комплекса 3 между 9,10-диметилантраценом 1 и тетрацианоэтиленом 2 на пути к аддукту 5 [27].
Это означает, что основной вклад энергии стабилизации структурных рядов диенов-доноров и диенофилов-акцепторов можно описать в виде взаимодействия граничных орбиталей [57]:
где β – коэффициент перекрывания взаимодействующих орбиталей.Все три варианта орбитального взаимодействия (диен-донор, диенофил-акцептор; диен-акцептор, диенофил-донор и промежуточный) можно объяснить на качественном уровне в рамках изменения энергии орбитального взаимодействия. Промежуточный тип орбитального взаимодействия экспериментально был обнаружен и надежно доказан в работах [58–72].
Понятно, что энергия стабилизации характеризует лишь снижение активационного барьера за счет орбитального взаимодействия реагирующих систем, но не формирует величину самого барьера активации. При анализе большого объема накопленных кинетических данных [58–97] было замечено, что в структурных рядах диенов или диенофилов часто наблюдается достаточно четкая корреляция ln k vs. 1/(IP – EА), хотя и не всегда. Причем подобные зависимости в этих структурных рядах реагентов не располагаются на общей прямой, а смещены друг относительно друга [24–27, 48, 71, 97]. До выявления этой закономерности ранее указывалось на возможность нарушения рядов активности реагентов, причем причина этих нарушений была неизвестной [11].
Таким образом, можно заключить, что энергия межмолекулярной стабилизации при взаимодействии граничных орбиталей часто контролирует изменение прочности МС и изменение скорости РДА в одном структурном ряду, причем МС могут быть промежуточными на пути реакции.
II. ВЛИЯНИЕ ЭНЕРГИИ РАЗРЫВА И ОБРАЗОВАНИЯ СВЯЗЕЙ
В соответствии с принципом термохимической кинетики баланс энергии разрыва и образования связей всегда формирует величину энергии активационного барьера [98–100]. Доля вклада энергии разрыва связей лежит в интервале от 0 до 1 в зависимости от вклада других факторов. Для более сопряженных систем с большей энергией разрыва π-связей при постоянном вкладе других факторов следует ожидать повышенного барьера активации. Поскольку энергия σ-связей в ряду замещенных реагентов изменяется значительно меньше, по сравнению с энергией π-связей, то различие в энтальпии реакции в основном обусловлено различием в энергии π-связей. Благодаря большому объему калориметрических данных об энтальпии РДА с участием разнообразных партнеров становятся понятными многие “аномалии”, сопровождающие эти реакции [24–27, 73–75, 77, 78, 87, 88, 97]. Понятно, что все доступные РДА обязаны сопровождаться выделением тепла, причем экзотермичность процесса должна превышать 40 кДж моль–1, для того чтобы зафиксировать хотя бы следовые концентрации аддукта. Это обусловлено отрицательным и довольно постоянным значением энтропии процесса (–150 ± ± 20 Дж моль–1 K–1). Накопленный банк кинетических и термохимических данных позволяет выяснить, почему пропорциональность между изменением кинетической активности реагентов и термодинамической стабильности образующихся продуктов наблюдается далеко не всегда [24–31, 97]. Наглядным примером служит сопоставление активности циклопентадиена (IP = 8.58 эВ) и 9,10-диметилантрацена (IP = 7.04 эВ) в реакции с цианоэтиленами [11, 24–27]. Скорость реакции между π-акцепторными тетрацианоэтиленом и трицианоэтиленом с сильным π-донором, 9,10-диметилантраценом, выше, чем с циклопентадиеном (контроль энергией орбитального взаимодействия). С менее акцепторными диенофилами – акрилонитрилом, фумародинитрилом и малеодинитрилом – скорость реакции уже выше с циклопентадиеном как менее сопряженным диеном (табл. 1). Было установлено, что при значительном изменении энергии сопряжения в ряду замещенных бутадиенов и замещенных антраценов корреляции скорости реакции от энергии стабилизации могут нарушаться даже внутри каждой из этих серий [24–27, 74, 97]. Например, из-за повышенной энергии сопряжения в 9-метокси- и 9,10-диметоксиантрацене экзотермичность РДА с их участием значительно меньше, чем в реакции с соизмеримыми по π-донорным свойствам 9-метил- и 9,10-диметилантраценом (табл. 1). Это объясняет не только малое значение константы равновесия, но и пониженное значение константы скорости реакции [74, 97]. Из-за значительно меньшей энергии сопряжения в ряду замещенных бутадиенов экзотермичность реакции с их участием значительно выше, чем в реакции с другими диенами (табл. 1). По сравнению с 2,3-диметилбутадиеном (8.40 эВ) для более π-донорного диена – антрацена (IP = = 7.33 эВ) – наблюдается одинаковая активность в реакции с сильным π-акцептором, тетрацианоэтиленом (ЕА = 2.88 эВ), благодаря значительному различию в энтальпии этих процессов (–77 и –176 кДж моль–1 соответственно). Уменьшение энергии стабилизации при переходе к менее π-акцепторному диенофилу, малеиновому ангидриду (ЕА = 0.97 эВ), приводит к тому, что активность антрацена уже в 56 раз меньше, чем 2,3-диметилбутадиена (табл. 1).
Таблица 1.
Константы скорости реакции Дильса–Альдера k2a (л моль–1 с–1), потенциалы ионизации диенов IP, энергии сродства к электрону диенофилов EА, и энтальпии этих реакций в растворе ΔHr – n при 25°С [27]
| № | Реакция | 8 + lg k2 | IP, эВ | EА, эВ | 100/(IP – EА), эВ–1 | –ΔHr – n, кДж моль–1 |
|---|---|---|---|---|---|---|
| Тетрацианоэтилен + | ||||||
| 1. | 2,5-Дифенилизобензофуран | ∼16.0 | 7.62 | 2.88 | 21.09 | 69 |
| 2. | 9,10-Диметилантрацен | 12.97 | 7.04 | 2.88 | 24.04 | 88 |
| 3. | 9-Метоксиантрацен | 11.71 | 7.17 | 2.88 | 23.31 | 61 |
| 4. | 9-Метилантрацен | 11.35 | 7.17 | 2.88 | 23.31 | 85 |
| 5. | 9,10-Диметоксиантрацен | 10.94 | 7.09 | 2.88 | 23.75 | 53 |
| 6. | Антрацен | 8.48 | 7.33 | 2.88 | 22.47 | 77 |
| 7. | 9-Хлорантрацен | 7.74 | 7.39 | 2.88 | 22.17 | 66 |
| 8. | 2-Метоксибутадиен | 8.47 | 8.62 | 2.88 | 17.42 | 159 |
| 9. | 2,3-Диметилбутадиен | 8.40 | 8.61 | 2.88 | 17.45 | 176 |
| 10. | транс-1-Фенилбутадиен | 7.81 | 8.16 | 2.88 | 18.94 | 142 |
| 11. | транс-1-Метилбутадиен | 7.23 | 8.61 | 2.88 | 17.45 | 163 |
| 12. | транс,транс-1,4-Дифенилбутадиен | 6.87 | 8.09 | 2.88 | 19.19 | 97 |
| 13. | 2-Метилбутадиен | 6.87 | 8.89 | 2.88 | 16.64 | 166 |
| 14. | Бутадиен | 5.23 | 9.03 | 2.88 | 16.26 | 154 |
| 15. | Циклопентадиен | 10.63 | 8.58 | 2.88 | 17.54 | 113 |
| 16. | Циклогексадиен-1,3 | 8.14 | 8.25 | 2.88 | 18.62 | 130 |
| Трицианоэтилен + | ||||||
| 17. | 2,5-Дифенилизобензофуран | 12.0 | 7.62 | 2.10 | 18.12 | 71 |
| 18. | 9,10-Диметилантрацен | 8.77 | 7.04 | 2.10 | 20.24 | 90 |
| 19. | Циклопентадиен | 8.68 | 8.58 | 2.10 | 15.43 | 115 |
| 1,1-Дицианоэтилен + | ||||||
| 20. | 2,5-Дифенилизобензофуран | 11.0 | 7.62 | 1.53 | 16.42 | 77 |
| 21. | 9,10-Диметилантрацен | 7.10 | 7.04 | 1.53 | 18.15 | 96 |
| 22. | 9-Метилантрацен | 6.18 | 7.17 | 1.53 | 17.73 | 93 |
| 23. | Антрацен | 4.52 | 7.33 | 1.53 | 17.24 | 85 |
| 24. | Циклопентадиен | 7.66 | 8.58 | 1.53 | 14.18 | 121 |
| Фумародинитрил + | ||||||
| 25. | 2,5-Дифенилизобензофуран | 7.49 | 7.62 | 0.78 | 14.62 | 82 |
| 26. | Циклопентадиен | 4.91 | 8.58 | 0.78 | 12.82 | 126 |
| 27. | 9,10-Диметилантрацен | 4.14 | 7.04 | 0.78 | 15.97 | 101 |
| 28. | 9-Метилантрацен | 3.08 | 7.17 | 0.78 | 15.65 | 98 |
| 29. | 9-Метоксиантрацен | 2.28 | 7.17 | 0.78 | 15.65 | 74 |
| 30. | Антрацен | 1.62 | 7.33 | 0.78 | 15.27 | 90 |
| 31. | 9,10-Диметоксиантрацен | 1.08 | 7.09 | 0.78 | 15.85 | 66 |
| Акрилонитрил + | ||||||
| 32. | 2,5-Дифенилизобензофуран | 5.34 | 7.62 | 0.02 | 13.16 | 87 |
| 33. | Циклопентадиен | 3.02 | 8.58 | 0.02 | 11.68 | 131 |
| 34. | 9,10-Диметилантрацен | 1.95 | 7.04 | 0.02 | 14.24 | 106 |
| 35. | 9-Метилантрацен | 1.04 | 7.17 | 0.02 | 13.99 | 103 |
| 36. | 9-Метоксиантрацен | 0.70 | 7.17 | 0.02 | 13.99 | 79 |
| 37. | Антрацен | –0.07 | 7.33 | 0.02 | 13.68 | 95 |
| Малеиновый ангидрид + | ||||||
| 38. | 2,5-Дифенилизобензофуран | 8.53 | 7.62 | 0.97 | 15.04 | 85 |
| 39. | Циклопентадиен | 6.96 | 8.58 | 0.97 | 13.14 | 129 |
| 40. | 9,10-Диметилантрацен | 6.09 | 7.04 | 0.97 | 16.47 | 104 |
| 41. | 9-Метилантрацен | 4.54 | 7.17 | 0.97 | 16.13 | 101 |
| 42. | 9-Метоксиантрацен | 3.45 | 7.17 | 0.97 | 16.13 | 77 |
| 43. | Антрацен | 2.78 | 7.33 | 0.97 | 15.72 | 93 |
| 44. | 9,10-Диметоксиантрацен | 2.49 | 7.09 | 0.97 | 16.33 | 69 |
| 45. | 9-Хлорантрацен | 2.16 | 7.39 | 0.97 | 15.57 | 82 |
| 46. | 2,3-Диметилбутадиен | 4.53 | 8.61 | 0.97 | 13.09 | 194 |
| 47. | транс-1-Метилбутадиен | 4.36 | 8.61 | 0.97 | 13.09 | 179 |
| 48. | 2-Метилбутадиен | 4.19 | 8.89 | 0.97 | 12.63 | 182 |
| 49. | транс-1-Фенилбутадиен | 4.05 | 8.16 | 0.97 | 13.91 | 158 |
| 50. | Бутадиен | 3.83 | 9.03 | 0.97 | 12.41 | 170 |
| 51. | транс,транс-1,4-Дифенилбутадиен | 2.47 | 8.09 | 0.97 | 14.04 | 113 |
| 52. | Пентацен | 6.13 | 6.64 | 0.97 | 17.64 | 132 |
| 53. | Тетрацен | 4.88 | 7.01 | 0.97 | 16.55 | 103 |
| 54. | Гексахлорциклопентадиен | 0.06 | 8.96 | 0.97 | 12.56 | 58 |
| Хлормалеиновый ангидрид + | ||||||
| 55. | 2,5-Дифенилизобензофуран | 8.58 | 7.62 | 1.08 | 15.29 | 82 |
| 56. | 9,10-Диметилантрацен | 5.81 | 7.04 | 1.08 | 16.78 | 101 |
| 57. | 9-Метилантрацен | 4.35 | 7.17 | 1.08 | 16.42 | 98 |
| 58. | Антрацен | 2.49 | 7.33 | 1.08 | 16.00 | 90 |
| 59. | 9-Хлорантрацен | 1.83 | 7.39 | 1.08 | 15.85 | 79 |
| Метилмалеиновый ангидрид + | ||||||
| 60. | 9,10-Диметилантрацен | 3.38 | 7.04 | 0.83 | 16.10 | 103 |
| 61. | 9-Метилантрацен | 1.93 | 7.17 | 0.83 | 15.77 | 100 |
| N-п-Нитрофенилмалеинимид + | ||||||
| 62. | 2,5-Дифенилизобензофуран | 9.16 | 7.62 | 1.01 | 15.13 | 96 |
| 63. | Пентацен | 7.75 | 6.64 | 1.01 | 17.76 | 143 |
| 64. | Тетрацен | 5.96 | 7.01 | 1.01 | 16.67 | 124 |
| 65. | Циклопентадиен | 7.31 | 8.58 | 1.01 | 13.21 | 140 |
| 66. | 9-Метилантрацен | 5.23 | 7.17 | 1.01 | 16.23 | 112 |
| 67. | Антрацен | 3.93 | 7.33 | 1.01 | 15.82 | 104 |
| 68. | 9-Хлорантрацен | 2.92 | 7.39 | 1.01 | 15.67 | 93 |
| N-Фенилмалеинимид + | ||||||
| 69. | 2,5-Дифенилизобензофуран | 8.57 | 7.62 | 0.89 | 14.86 | 98 |
| 70. | Циклопентадиен | 6.94 | 8.58 | 0.89 | 13.00 | 142 |
| 71. | 9,10-Диметилантрацен | 6.48 | 7.04 | 0.89 | 16.26 | 117 |
| 72. | 9-Метилантрацен | 4.71 | 7.17 | 0.89 | 15.92 | 114 |
| 73. | 9-Метоксиантрацен | 3.45 | 7.17 | 0.89 | 15.92 | 90 |
| 74. | Антрацен | 2.86 | 7.33 | 0.89 | 15.53 | 106 |
| 75. | 9-Хлорантрацен | 1.45 | 7.39 | 0.89 | 15.38 | 95 |
| 76. | Пентацен | 6.98 | 6.64 | 0.89 | 17.39 | 145 |
| 77. | Тетрацен | 5.34 | 7.01 | 0.89 | 16.34 | 126 |
| 78. | 5,11-Дихлортетрацен | 4.43 | 6.81 | 0.89 | 16.89 | 107 |
| Акролеин + | ||||||
| 79. | 2,5-Дифенилизобензофуран | 5.92 | 7.62 | 0.15 | 13.39 | 78 |
| 80. | Циклопентадиен | 3.30 | 8.58 | 0.15 | 11.87 | 122 |
| 81. | 2-Метилбутадиен | 0.7 | 8.89 | 0.15 | 11.44 | 175 |
| 82. | Бутадиен | 0.0 | 9.03 | 0.15 | 11.26 | 163 |
| Некоторые другие реакции | ||||||
| 83. | N-п-Нитрофенилмалеинимид + + 2,5-дифенилизобензофуран | 9.22 | 7.62 | 0.99 | 15.08 | 95 |
| 84. | N-п-Бромфенилмалеин имид + + 2,5-дифенилизобензофуран | 8.87 | 7.62 | 0.95 | 14.99 | 98 |
| 85. | N-п-Толилмалеинимид + 2,5-дифенилизобензофуран | 8.56 | 7.62 | 0.83 | 14.73 | 98 |
| 86. | N-п-Метоксифенилмалеинимид + + 2,5-дифенилизобензофуран | 8.50 | 7.62 | 0.81 | 14.68 | 99 |
| 87. | N-п-Диметиламинофенилмалеин-имид + 2,5-дифенилизобензофуран | 8.36 | 7.62 | 0.73 | 14.51 | 100 |
| 88. | Малеодинитрил + 2,5-дифенилизобензофуран | 8.12 | 7.62 | 0.78 | 14.62 | 84 |
| 89. | Акриловая кислота + 2,5-дифенилизобензофуран | 6.18 | 7.62 | 0.09 | 13.28 | 84 |
| 90. | Метилакрилат + 2,5-дифенилизобензофуран | 5.28 | 7.62 | 0.19 | 13.46 | 85 |
| 91. | Акриловая кислота + + циклопентадиен | 4.77 | 8.58 | 0.09 | 11.78 | 128 |
| 92. | Метилакрилат+ циклопентадиен | 3.51 | 8.58 | 0.19 | 11.91 | 129 |
| 93. | Бутадиен + этилен | –2.7 | 9.03 | –1.8 | 9.22 | 166 |
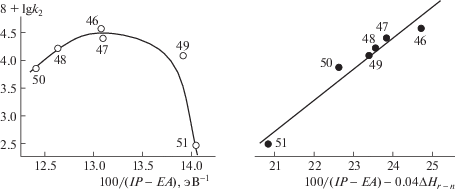
Необходимость совместного учета двух факторов (орбитального и баланса энергии связи) наиболее ярко проявилась в ряду замещенных 1,3-бутадиенов [27, 97]. Изменение энтальпии реакции малеинового ангидрида с 2,3-диметилбутадиеном (–194 кДж моль–1) при переходе к транс-1-фенилбутадиену (–158 кДж моль–1) и, особенно, к транс,транс-1,4-дифенилбутадиену (–113 кДж моль–1) оказалось столь значительным, что здесь учет только энергии стабилизации не может предсказать даже направление изменения скорости реакции в ряду бутадиенов без привлечения вклада энергии разрыва и образования связей (рис. 1).
Рис. 1.
Зависимость логарифма констант скорости k2 (л моль–1 с–1) реакции Дильса–Альдера замещенных бутадиенов с малеиновым ангидридом от донорно-акцепторных свойств реагентов 100/(IP – ЕА) ($\bigcirc $) и при дополнительном учете энтальпии реакции ΔНr – n (кДж моль–1) ($ \bullet $) [27]. Номера точек соответствуют номерам реакций в табл. 1.
Из анализа массива данных, представленных в табл. 1, видно, что подобных “нарушений” довольно много. Таким образом, обычное и “аномальное” изменение активности для различных классов реагентов в реакции Дильса–Альдера становится понятным только при совместном учете влияния энергии орбитального взаимодействия и энтальпии реакции, причем не только при сравнении различных реакционных серий, но даже иногда внутри одной реакционной серии. С целью количественного описания скорости реакции Дильса–Альдера с участием различных классов реагентов со связями С=С были рассмотрены три основных вклада: 1) различие энергий граничных орбиталей диена и диенофила [57]; 2) баланс энергий разрыва и образования связей [98–100] и 3) изменение коэффициентов перекрывания орбиталей [12, 19]. Проведение такого расширенного анализа стало возможным после накопления большого объема кинетических, термохимических и орбитальных характеристик (табл. 1).
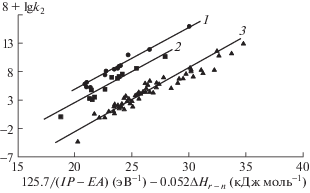
Интересно отметить, что при описании скорости реакции с учетом энергии граничных орбиталей 1/(IP – ЕА) и баланса энергии разрыва и образования связей (ΔНr – n) весь массив кинетических данных (табл. 1) распределяется в виде трех отдельных линейных зависимостей (рис. 2). Уменьшение активности в этих рядах диенов (замещенные фураны > циклопентадиены > антрацены и 1,3-бутадиены) соответствует уменьшению коэффициента перекрывания орбиталей из-за увеличения межатомного расстояния: R1–4 = 2.19 Å в фуране, 2.36 Å в циклопентадиене, 2.81 Å в антрацене и 2.90 Å в s-цис-бутадиене-1,3. Было обнаружено, что между коэффициентом перекрывания орбиталей (β2, уравнение 5) и межатомным расстоянием в диене R1–4 существует линейная зависимость [12, 19]. При совместном учете этих трех факторов прослеживается [23, 25–27, 97] (рис. 3) общая корреляция для всего массива реакций при r = 0.972, n = 93, s0 = 0.9:
(6)
$\begin{gathered} \lg {{k}_{2}} = --28.81 + 316.3{\text{/}}(IP--ЕА)-- \\ - \;69.9{{R}_{{1--4}}}{\text{/}}(IP--ЕА)--0.054\Delta {{H}_{r}}_{{\,--\,n}} \\ \end{gathered} $Рис. 2.
Зависимость логарифма констант скорости k2 (л моль–1 с–1) реакций Дильса–Альдера диенофилов с производными фурана (прямая 1), циклопентадиена (прямая 2), антрацена и бутадиена (прямая 3) от донорно-акцепторных свойств реагентов 1/(IP – ЕА), и энтальпии реакций ΔHr – n, по данным табл. 1 [27].
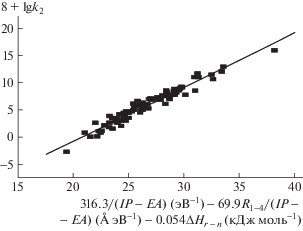
Здесь значения IP и EА выражены в эВ, R1–4 – в Å, ΔHr – n – в кДж моль–1, k2 – в л моль–1 с–1 при 25°С. Влияние изменения энтальпии процесса на скорость изученного массива РДА (рис. 3, уравнение 6) оказалось соизмеримым с вкладом изменения коэффициентов перекрывания орбиталей реагирующих реагентов, и каждый из этих вкладов вдвое меньше вклада различий в величине энергии донорно-акцепторного взаимодействия.
Интересно отметить, что, в отличие от классических соотношений активности и селективности для реакций замещения в ароматическом ряду (более активный реагент – менее селективный), в реакции Дильса–Альдера обнаружено “аномальное” соотношение: более активный реагент – более селективный и наоборот [24, 25, 81, 82]. Показано, что “аномальная” селективность полностью оправдана, если изменение скорости контролируется энергией стабилизации (уравнение 5). Тем не менее немало примеров [24–31, 48], где “аномальная” селективность инвертируется в “нормальную”, если скорость контролируется энергией разрыва π-связей. Эти противоречия становятся понятными при совместном учете основных факторов, определяющих скорость реакции (уравнение (6).
Рассмотренные в этом разделе данные позволяют заключить, что скорость РДА с участием связей С=С определяется влиянием энергии стабилизации при взаимодействии граничных орбиталей 1/(IP – EA) с учетом их перекрывания R1, 4 и балансом энергий разрыва старых π-связей и образования новых σ-связей ΔНr – n. Отметим, что изменение энтальпии РДА с участием С=С-связей в широком пределе (50–200 кДж моль–1) обусловлено большим различием энергии π-связей при слабом изменении энергии σ-связей. Поэтому скорость РДА с участием гетероатомных связей (С=С, N=N, C=N, C=S) нельзя описать общим соотношением, подобным уравнению (6).
III. КАТАЛИЗ КИСЛОТАМИ ЛЬЮИСА
Из соотношения (6) следует, что при постоянных температуре и давлении можно изменить скорость реакции, если создать условия, в которых изменяются орбитальные свойства реагентов, либо энергии реагирующих связей, либо структура диенов. На основании многочисленных исследований молекулярных комплексов можно сделать заключение [52, 101–111], что перевод диенофила в n,ν-комплекс с кислотой Льюиса резко повышает его π-акцепторные свойства. Это проявляется в изменении максимума полосы поглощения комплекса с π-донором, например, с гексаметилбензолом (табл. 2). Обнаруженное увеличение π-акцепторных свойств диенофилов в n,ν-комплексах с кислотами Льюиса [107, 111] послужило мощным стимулом для широкого исследования каталитического эффекта в РДА, а также данных об энтальпии образования n,ν-комплексов, энтальпии катализируемых реакций (табл. 3) и о влиянии среды на сольватацию реагентов, n,ν-комплексов и переходного состояния [25–27, 102–113].
Таблица 2.
Длины волн λmax, энергии Еch.tr. максимумов полос π → π* переходов комплексов гексаметилбензола с обычными и активированными диенофилами и увеличение энергии сродства к электрону этих диенофилов ΔЕА [107, 111]
| Диенофил | Кислота Льюиса | λ, нм | Еch.tr., эВ | ΔЕА, эВ |
|---|---|---|---|---|
| N-п-Толилмалеинимид | Нет | 330 | 3.76 | – |
| GaCl3 | 475 | 2.61 | 1.15 | |
| AlCl3 | 488 | 2.54 | 1.22 | |
| N-Фенилмалеинимид | Нет | 337 | 3.68 | – |
| GaCl3 | 470 | 2.64 | 1.05 | |
| AlCl3 | 481 | 2.58 | 1.10 | |
| N-п-Бромфенилмале-инимид | Нет | 345 | 3.58 | – |
| GaCl3 | 470 | 2.64 | 0.95 | |
| AlCl3 | 480 | 2.59 | 1.00 | |
| N-п-Нитрофенилмалеинимид | Нет | 365 | 3.40 | – |
| GaCl3 | 464 | 2.67 | 0.73 | |
| Малеиновый ангидрид | Нет | 353 | 3.51 | – |
| GaCl3 | 440 | 2.82 | 0.69 | |
| AlCl3 | 470 | 2.64 | 0.87 | |
| Цитраконовый ангидрид | Нет | 340 | 3.65 | – |
| GaCl3 | 440 | 2.82 | 0.83 | |
| AlCl3 | 455 | 2.73 | 0.92 | |
| Хлормалеиновый ангидрид | Нет | 365 | 3.40 | – |
| GaCl3 | 480 | 2.58 | 0.82 | |
| AlCl3 | 505 | 2.46 | 0.94 | |
| Дихлормалеиновый ангидрид | Нет | 388 | 3.20 | – |
| GaCl3 | 500 | 2.48 | 0.72 |
Таблица 3.
Энтальпии образования n,${v}$-комплексов (ΔНМС, кДж моль–1), константы скорости (k2, л моль–1 с–1), энтальпии (ΔН≠, кДж моль–1) и энтропии (ΔS≠, Дж моль–1 K–1) активации и наблюдаемый эффект ускорения (kcat/k) в катализируемых реакциях Дильса–Альдера (бензол, 25°С) [26–28, 103–113]
| №№ | Реакция | MX3 | –ΔНМС | k2 | ΔН≠ | –ΔS≠ | kcat/k |
|---|---|---|---|---|---|---|---|
| 1. | 9,10-Диметилантрацен + + Метилакрилат | Нет | – | 7.8 × 10–7 | 63 | 150 | – |
| 2. | AlCl3 | – | 0.15 | 31 | 155 | 1.9 × 105 | |
| 3. | GaCl3 | 52 | 3.5 × 10–2 | – | – | 4.5 × 104 | |
| 4. | 9,10-Диметилантрацен + + Метилметакрилат | Нет | – | 1.3 × 10–7 | 63 | 163 | – |
| 5. | AlCl3 | – | 3.8 × 10–2 | 35 | 155 | 2.8 × 105 | |
| 6. | 9,10-Диметилантрацен + + Диметилфумарат | Нет | – | 6.7 × 10-6 | 52 | 167 | – |
| 7. | AlCl3 | – | 0.43 | 23 | 171 | 6.4 × 104 | |
| 8. | Антрацен + N-p-нитрофенилмалеинимид | Нет | – | 8.5 × 10–5 | 57 | 129 | – |
| 9. | AlCl3 | – | 8.2 | 33 | 113 | 9.6 × 104 | |
| 10. | GaCl3 | 48 | 1.0 | – | – | 1.2 × 104 | |
| 11. | Антрацен + N-p-бромфенилмалеинимид | Нет | – | 2.5 × 10–5 | 60 | 129 | – |
| 12. | AlCl3 | – | 2.9 | 34 | 121 | 1.2 × 105 | |
| 13. | GaCl3 | 53 | 0.65 | 34 | 135 | 2.6 × 104 | |
| 14. | Антрацен + N-p-метоксифенилмалеинимид | Нет | – | 8.1 × 10–6 | 68 | 112 | – |
| 15. | AlCl3 | – | 1.2 | 38 | 113 | 1.5 × 105 | |
| 16. | Антрацен + N-фенилмалеинимид | Нет | – | 7.3 × 10–6 | 68 | 112 | – |
| 17. | AlCl3 | – | 1.2 | 48 | 105 | 1.7 × 105 | |
| 18. | AlBr3 | 78 | 5.7 | – | – | 7.9 × 105 | |
| 19. | GaCl3 | 55 | 0.35 | – | – | 4.9 × 104 | |
| 20. | Антрацен + N-p-толилмалеинимид | Нет | – | 6.8 × 10–6 | 68 | 113 | – |
| 21. | AlCl3 | – | 1.2 | 41 | 105 | 1.8 × 105 | |
| 22. | GaCl3 | 57 | 0.35 | – | – | 5.2 × 104 | |
| 23. | 9,10-Диметилантрацен + + N-фенилмалеинимид | Нет | – | 3.0 × 10–2 | – | – | – |
| 24. | SnCl4 | 8 | 3.6 × 10–2 | – | – | 1.2 | |
| 25. | Et2O · BF3 | 5 | 3.0 × 10–2 | – | – | 1.0 | |
| 26. | Антрацен + малеиновый ангидрид | Нет | – | 6.0 × 10–6 | 65 | 126 | – |
| 27. | BCl3 | – | 4.1 × 10–2 | – | – | 6.9 × 103 | |
| 28. | GaCl3 | 41 | 0.40 | – | – | 6.7 × 104 | |
| 29. | AlCl3 | – | 1.6 | – | – | 2.6 × 105 | |
| 30. | AlBr3 | – | 7.2 | – | – | 1.2 × 106 | |
| 31. | Антрацен + 1,4-нафтохинон | Нет | – | 1.8 × 10–7 | 66 | 140 | – |
| 32. | GaCl3 | 54 | 2.3 | 34 | 120 | 1.3 × 107 | |
| 33. | 2GaCl3 | – | 6.0 × 102 | – | – | 3.4 × 108 | |
| 34. | Антрацен + 1,4-бензохинон | Нет | – | 2.4 × 10–6 | 64 | 123 | – |
| 35. | GaCl3 | 53 | 1.9 | – | – | 7.8 × 105 | |
| 36. | 2,5-Дифенилизобензофуран + О,О-диэтил-β-карбометоксивинилфосфонат | Нет | – | 7.0 × 10–4 | – | – | – |
| 37. | GaCl3 | 90; Р=О | 0.37 | – | – | 5.3 × 102 | |
| 38. | 2,5-Дифенилизобензофуран + О,О-дифенил-β-карбо-метоксивинилфосфонат | Нет | – | 3.7 × 10–5 | 58 | 121 | – |
| 39. | GaCl3 | 76; Р=О | 3.4 × 10–2 | 42 | 121 | 9.2 × 102 | |
| 40. | 9,10-Диметилантрацен + + О,О-диэтил-β-карбо-метоксивинилфосфонат | Нет | – | 8.9 × 10-10 | 90 | 155 | – |
| 41. | 2GaCl3 | 148; | 1.7 × 10–2 | 40 | 140 | 1.9 × 107 | |
| Р=О, | |||||||
| С=О | |||||||
| 42. | Тетрафенилциклопента-диенон + транс-стильбен | Нет | – | 2.8 × 10–8 | 70 | 144 | – |
| 43. | GaCl3 | 39 | 5.6 × 10–5 | 39 | 185 | 2.0 × 103 | |
| 44. | Тетрафенилциклопента-диенон + норборнадиен | Нет | – | 6.0 × 10–7 | 64 | 137 | – |
| 45. | GaCl3 | 39 | 5.0 × 10–4 | 34 | 181 | 8.4 × 102 | |
| 46. | Тетрафенилциклопента-диенон + N-фенилмале-инимид | Нет | – | 3.9 × 10–5 | 59 | 120 | – |
| 47. | GaCl3 | 55 имид | 1.4 × 10–4 | – | – | 3.6 | |
| 48. | AlCl3 | имид | 1.1 × 10–3 | 45 | 137 | 28 | |
| 49. | 2 AlCl3 | оба | 4.2 × 10–5 | – | – | 1.2 | |
| 50. | Тетрафенилциклопента-диенон + N-п-толилмале-инимид | Нет | – | 6.3 × 10–5 | 57 | 121 | – |
| 51. | GaCl3 | 57 имид | 1.5 × 10–4 | – | – | 2.4 | |
| 52. | AlCl3 | имид | 7.1 × 10–4 | – | – | 11 | |
| 53. | 2 AlCl3 | оба | 6.5 × 10–5 | – | – | 1.0 | |
| 54. | С-Фенил, N-метилнитрон + + N-фенилмалеинимид | Нет | – | 2.0 × 10–4 | – | – | – |
| 55. | GaCl3 | 55 имид | 50 | – | – | 2.5 × 105 | |
| 56. | 9,10-Диметилантрацен + + акрилонитрил | Нет | – | 2.2 × 10–6 | 63 | 146 | – |
| 57. | GaCl3 | 51 | 0.13 | 36 | 130 | 5.9 × 104 | |
| 58. | 9-Метилантрацен + + акрилонитрил | Нет | – | 1.1 × 10–7 | 74 | 122 | – |
| 59. | GaCl3 | 51 | 1.7 × 10–2 | 42 | 126 | 1.5 × 105 | |
| 60. | Антрацен + акрилонитрил | Нет | – | 8.6 × 10–9 | 76 | 138 | – |
| 61. | GaCl3 | 51 | 5.3 × 10–4 | 47 | 141 | 6.2 × 104 | |
| 62. | 9-Хлорантрацен + акрилонитрил | Нет | – | 1.9 × 10–9 | 79 | 140 | – |
| 63. | GaCl3 | 51 | 1.6 × 10–4 | 49 | 145 | 8.4 × 104 | |
| 64. | 9-Метилантрацен + метилакрилат | Нет | – | 5.8 × 10–8 | – | – | – |
| 65. | GaCl3 | 52 | 1.3 × 10–2 | – | – | 2.2 × 105 | |
| 66. | 9,10-Диметилантрацен + + цианоацетилен | Нет | – | 4.0 × 10–6 | – | – | – |
| 67. | GaCl3 | – | 1.1 | – | – | 2.8 × 105 | |
| 68. | 9,10-Диметилантрацен + + метилпропиолат | Нет | – | 2.0 × 10–8 | – | – | – |
| 69. | GaCl3 | – | 6.0 × 10–3 | – | – | 3.0 × 105 | |
| 70. | 9,10-Диметилантрацен + + малеиновый ангидрид | Нет | – | 1.2 × 10–2 | 43 | 138 | – |
| 71. | GaCl3 | 41 | 2.8 × 102 | – | – | 2.3 × 104 | |
| 72. | AlCl3 | – | 2.0 × 103 | – | – | 1.7 × 105 | |
| 73. | 9-Метилантрацен + мале-иновый ангидрид | Нет | – | 3.4 × 10–4 | 53 | 134 | – |
| 74. | GaCl3 | 41 | 12.8 | 27 | 140 | 3.8 × 104 | |
| 75. | AlCl3 | – | 95.2 | – | – | 2.8 × 105 | |
| 76. | 9-Хлорантрацен + мале-иновый ангидрид | Нет | – | 1.4 × 10–6 | 67 | 130 | – |
| 77. | GaCl3 | 41 | 5.2 × 10–2 | 36 | 153 | 3.7 × 104 | |
| 78. | AlCl3 | – | 0.38 | – | – | 2.8 × 105 | |
| 79. | Антрацен + цитраконовый ангидрид | Нет | – | 1.5 × 10–8 | 73 | 135 | – |
| 80. | GaCl3 | 49 | 1.3 × 10–4 | 9.0 × 103 | |||
| 81. | AlCl3 | – | 8.3 × 10–3 | 32 | 164 | 5.7 × 105 | |
| 82. | 9-Хлораантрацен + цитраконовый ангидрид | Нет | – | 1.5 × 10–8 | 73 | 135 | – |
| 83. | GaCl3 | 49 | 1.3 × 10–4 | – | – | 9.0 × 103 | |
| 84. | AlCl3 | – | 8.3 × 10–3 | 32 | 164 | 5.7 × 105 | |
| 85. | 9-Метилантрацен + цитраконовый ангидрид | Нет | – | 8.5 × 10–7 | 64 | 135 | – |
| 86. | GaCl3 | 49 | 2.0 × 10–2 | 41 | 129 | 2.3 × 104 | |
| 87. | AlCl3 | – | 0.14 | 28 | 154 | 1.7 × 105 | |
| 88. | 9,10-Диметилантрацен + + цитраконовый ангидрид | Нет | – | 2.4 × 10–5 | 45 | 169 | – |
| 89. | GaCl3 | 49 | 0.44 | 31 | 137 | 1.9 × 104 | |
| 90. | AlCl3 | – | 2.2 | 29 | 128 | 9.1 × 104 | |
| 91. | Антрацен + хлормалеиновый ангидрид | Нет | – | 3.1 × 10–6 | 62 | 128 | – |
| 92. | GaCl3 | 40 | 0.11 | – | – | 3.6 × 104 | |
| 93. | AlCl3 | – | 2.3 | – | – | 7.5 × 105 | |
| 94. | 9-Хлорантрацен + хлормалеиновый ангидрид | Нет | – | 6.8 × 10–7 | – | – | – |
| 95. | GaCl3 | 40 | 2.3 × 10–2 | – | – | 3.4 × 104 | |
| 96. | AlCl3 | – | 0.14 | – | – | 2.1 × 105 | |
| 97. | 9-Метилантрацен + хлормалеиновый ангидрид | Нет | – | 2.3 × 10–4 | 49 | 139 | – |
| 98. | GaCl3 | 40 | 11.9 | 28 | 118 | 5.3 × 104 | |
| 99. | AlCl3 | – | 39.7 | – | – | 1.8 × 105 | |
| 100. | 9,10-Диметилантрацен + + хлормалеиновый ангидрид | Нет | – | 6.4 × 10–3 | 46 | 118 | – |
| 101. | GaCl3 | 40 | 2.4 × 102 | – | – | 3.8 × 104 | |
| 102. | AlCl3 | – | 1.4 × 103 | – | – | 2.2 × 105 | |
| 103. | Тетрациклон + N-п-нитрофенилмалеинимид | Нет | – | 4.0 × 10–5 | 64 | 100 | – |
| 104. | GaCl3 | 48 имид | 2.8 × 10–4 | – | – | 7.1 | |
| 105. | AlCl3 | имид | 1.2 × 10–3 | 57 | 96 | 30 | |
| 106. | Тетрациклон + N-п-бромфенилмалеинимид | Нет | – | 3.6 × 10–5 | 64 | 100 | – |
| 107. | GaCl3 | 53 имид | 2.2 × 10–4 | – | – | 6.2 | |
| 108. | AlCl3 | имид | 1.1 × 10–3 | 53 | 111 | 29.1 | |
| 109. | Фенциклон + N-п-нитрофенилмалеинимид | Нет | – | 2.12 | 31 | 133 | – |
| 110. | GaCl3 | 48 имид | 36.3 | 20 | 135 | 17.1 | |
| 111. | AlCl3 | 83.3 | – | – | 30.3 | ||
| 112. | Фенциклон + N-п-бромфенилмалеинимид | Нет | – | 1.42 | 33 | 137 | – |
| 113. | GaCl3 | 53 имид | 21.0 | 20 | 140 | 14.8 | |
| 114. | AlCl3 | имид | 51.6 | – | – | 36.3 | |
| 115. | Фенциклон + N-фенилмалеинимид | Нет | – | 0.76 | 30 | 146 | – |
| 116. | GaCl3 | 55 имид | 9.4 | 28 | 121 | 12.4 | |
| 117. | AlCl3 | имид | 46.5 | 14 | 155 | 61.2 | |
| 118. | Фенциклон + N-п-толилмалеинимид | Нет | – | 0.75 | 34 | 133 | – |
| 119. | GaCl3 | 57 имид | 10.4 | 24 | 134 | 13.9 | |
| 120. | AlCl3 | имид | 40.1 | 22 | 127 | 53.4 | |
| 121. | Фенциклон + метилакрилат | Нет | – | 1.2 × 10–3 | 46 | 133 | – |
| 122. | AlCl3 | – | 0.63 | 32 | 129 | 5.5 × 102 | |
| 123. | Фенциклон + метилметакрилат | Нет | – | 7.5 × 10–4 | 46 | 139 | – |
| 124. | AlCl3 | – | 0.22 | 30 | 144 | 2.9 × 102 | |
| 125. | Фенциклон + диметилфумарат | Нет | – | 4.7 × 10–5 | 54 | 136 | – |
| 126. | AlCl3 | – | 2.3 × 10–2 | 41 | 126 | 4.9 × 102 | |
| 127. | Транс,транс-1,4-дифенилбутадиен + N-p-толилмалеинимид | Нет | – | 9.3 × 10–7 | 54 | 168 | – |
| 128. | AlCl3 | – | 5.9 × 10–3 | 38 | 148 | 6.3 × 102 | |
| 129. | Транс, транс-1,4-дифенилбутадиен + N-фенилмале-инимид | Нет | – | 1.4 × 10–6 | 50 | 177 | – |
| 130. | AlCl3 | – | 5.6 × 10–3 | 39 | 143 | 3.9 × 102 | |
| 131. | Транс, транс-1,4-дифенилбутадиен + N-p-бромфенилмалеинимид | Нет | – | 2.1 × 10–6 | 48 | 180 | – |
| 132. | AlCl3 | – | 7.1 × 10–3 | – | – | 3.4 × 102 |
Широко распространенный термин – “катализ” кислотами Льюиса РДА – не является здесь строгим, поскольку реально образуется новая, более активная супрамолекула реагента в виде n,ν-комплекса между кислотой Льюиса и (обычно) диенофилом. Как видно из накопленных данных (табл. 3), образование прочных n,ν-комплексов приводит к резкому (до 1 × 108 раз) увеличению скорости процесса, позволяя вовлекать в реакцию чрезвычайно инертные в обычных условиях реагенты.
Было обнаружено [103–113], что увеличение скорости катализируемых РДА типа “диен-донор, диенофил-акцептор” в сравнительно инертных по отношению к кислотам Льюиса средах (бензол, толуол, дихлорметан, 1,2-дихлорэтан) довольно постоянно и при мольном соотношении диенофил : кислота Льюиса = 1 : 1 составляет обычно 1, 3, 4, 5 и 6 порядков в присутствии SnCl4, BBr3, GaCl3, AlCl3 и AlBr3 соответственно. Особенно “удобен” здесь хлорид галлия, который легко очищается перегонкой и хорошо растворим даже в инертных растворителях. В реакциях с обращенным электронным характером реагентов – “диен-акцептор, диенофил-донор” – координация этих кислот Льюиса с диеном тоже приводит к увеличению скорости реакции (табл. 3), хотя здесь эффект ускорения заметно меньше [109, 110].
Из табл. 2 видно, что величины снижения энергии переноса заряда в π,π-комплексах, следовательно, увеличения энергии сродства к электрону, довольно велики (0.75–1.20 эВ) [107, 111]. Сопоставление скорости реакции Дильса–Альдера и энергии переноса заряда в π,π-комплексах гексаметилбензола с рядом диенофилов показало общую зависимость для обычных и активированных малеинимидов, подобную широко известной зависимости для обычных реагентов [24–27].
Нами обнаружено лишь незначительное различие в энтальпии образования n,ν-комплексов хлорида галлия с диенофилами или с аддуктами в растворе [103]. Это соответствует малому различию в энтальпии обычной и активированной кислотами Льюиса РДА. С учетом изменения энергии сродства к электрону (ΔЕА) диенофилов при координации с кислотами Льюиса уравнение (6) довольно точно предсказывает скорость и для катализируемого процесса. Это позволяет направленно выбирать условия для проведения таких реакций. Таким образом, резкое увеличение скорости реакции Дильса–Альдера в присутствии кислот Льюиса обусловлено сильным увеличением орбитального взаимодействия между диеном и активированным диенофилом. Интересно отметить, что довольно умеренные по активности малеинимиды в n,ν-комплексе с хлоридом галлия и, особенно, в n,ν-комплексе с хлоридом алюминия по скорости реакции уже близки к самому активному диенофилу – тетрацианоэтилену (табл. 3).
Энтальпии образования n,ν-комплексов были определены калориметрическим методом [103–105, 108]. Обнаружено [101, 102, 107], что каталитический эффект ускорения РДА, ln(kcat./k), пропорционален (r = 0.985) энтальпии образования комплекса между диенофилом и кислотой Льюиса:
(7)
$\ln ({{k}_{{{\text{cat}}{\text{.}}}}}{\text{/}}k) = 0.19\Delta {{H}_{{{\text{МС}}}}},\;{\text{кДж}}\;{\text{мол}}{{{\text{ь}}}^{{--1}}}$Выбор растворителя чрезвычайно важен для проведения катализируемой реакции. В присутствии n-донорных добавок происходит перераспределение кислоты Льюиса из-за ее связывания не только с диенофилом, но и с конкурентным n-донором [112–114].
Эти данные позволяют понять, что огромные эффекты ускорения РДА обусловлены значительными изменениями значений энергии сродства к электрону диенофилов при образовании n,ν-комплексов. Эффект ускорения можно плавно изменять от максимального значения до полного подавления как подбором кислоты Льюиса, так и выбором конкурентного n-донорного растворителя.
IV. УСКОРЕНИЕ РЕАКЦИИ ДИЛЬСА–АЛЬДЕРА В ПРИСУТСТВИИ ПЕРХЛОРАТА ЛИТИЯ
Огромный объем экспериментальных данных свидетельствует о слабом влиянии полярности растворителей на скорость РДА [11–14, 24–27, 52, 87, 88]. Было обнаружено, что в 5 М растворе перхлората лития в диэтиловом эфире многие реакции Дильса–Альдера протекают значительно быстрее [115–118]. Причины ускорения реакций в концентрированных солевых растворах до конца еще не выяснены. Здесь обсуждаются процессы сольватации, координация катиона лития с неподеленной электронной парой в активирующей группе диенофилов, стабилизация зарядов. Возможный механизм активации реакций циклоприсоединения в солевых растворах рассмотрен в работах [117, 118]. Интересно отметить, что для равновесных реакций с участием фурана в присутствии перхлората лития в эфире увеличиваются константы и скорости, и равновесия, а для реакций с C,N-дифенилнитроном наблюдается снижение этих характеристик (табл. 4). Это существенно отличает поведение перхлората лития в эфире от реакций в присутствии обычных кислот Льюиса или при проведении при повышенном гидростатическом давлении [117, 118].
Таблица 4.
Константы скорости k2 и равновесия Keq для реакции фурана с N-фенилмалеимидом (реакция I), C,N-дифенилнитрона с N-п-бромфенилмалеимидом (реакция II) и C,N-дифенилнитрона с N-фенилмале-имидом (реакция III) в растворах перхлората лития ${{C}_{{{\text{LiCl}}{{{\text{O}}}_{{\text{4}}}}}}}$ при 25°C [115, 118]
| Реакция | Растворитель | ${{c}_{{{\text{LiCl}}{{{\text{O}}}_{{\text{4}}}}}}}$а, моль л–1 | k2, л моль–1 с–1 | Keq, л моль–1 |
|---|---|---|---|---|
| I | Диэтиловый эфир | 0 | 6.6 × 10–6 | 3.6 |
| I | Диэтиловый эфир | 0.05 | 5.5 × 10–5 | 25 |
| I | Диэтиловый эфир | 4.0 | 5.3 × 10–3 | 1180 |
| II | Диэтиловый эфир | 0 | 6.1 × 10–3 | 4500 |
| II | Диэтиловый эфир | 3.0 | – | 100 |
| II | Ацетон | 0 | 2.4 × 10–3 | 2200 |
| II | Ацетон | 1.0 | 1.5 × 10–3 | 530 |
| II | Ацетон | 3.0 | 5.8 × 10–4 | 310 |
| III | Диэтиловый эфир | 0 | 5.3 × 10–3 | 913 |
| III | Диэтиловый эфир | 4.0 | 5.9 × 10–4 | 303 |
Хотя причины влияния растворов перхлората лития на скорость и равновесие реакций до конца еще не выяснены, эти растворы нашли широкое применение в органическом синтезе. Например, ряд циклобутановых продуктов был впервые получен в среде перхлората лития в диэтиловом эфире вследствие благоприятного для реакции изменения скорости и равновесия в этих условиях [116–118]. Калориметрическое определение энтальпий растворения перхлоратов лития и магния показало, что во всех n-донорных растворителях процесс растворения экзотермичен, причем в диэтиловом эфире он минимален. Обнаружены огромные изменения парциальных мольных объемов перхлоратов лития и магния вплоть до их отрицательных значений в растворе [119–124]. Поведение 5 М раствора перхлората лития в эфире уже подобно ионной жидкости [125].
Анализ накопленных данных об эффектах ускорения реакции Дильса–Альдера в присутствии галогенидов алюминия, галлия и бора показал, что такие соли более благоприятны для проведения реакций циклоприсоединения с орбитальным контролем. В концентрированных растворах перхлората лития в органических растворителях результирующее влияние на скорость и равновесие различных типов реакций определяется предпочтительной координацией соли и различием эффектов сольватации реагентов, продуктов и переходного состояния.
V. СОЛЬВАТАЦИОННЫЕ ЭФФЕКТЫ В РЕАКЦИИ ДИЛЬСА–АЛЬДЕРА
Комбинация калориметрических и кинетических измерений оказалась весьма результативной для описания влияния среды на изменение энтальпии сольватации переходного состояния в реакциях и энтальпии сольватации молекулярных комплексов [52, 126–132]. Из термодинамического цикла следует, что различие в энергии сольватации активированного комплекса δHsolv,TS в растворителе S относительно растворителя сравнения S0 определяется соотношением (8):
(8)
$\delta {{H}_{{{\text{solv,TS}}}}}\left( {{\text{S/}}{{{\text{S}}}_{0}}} \right) = \delta {{H}^{ \ne }}({\text{S/}}{{{\text{S}}}_{0}}) + \delta {{H}_{{{\text{solv,L}}}}}({\text{S/}}{{{\text{S}}}_{0}}),$Нами было показано, что энергетический уровень переходного состояния в некатализируемых процессах изменяется в ряду растворителей слабо [52, 87, 88, 127]. Различие в энергии активации в изученных средах в большей степени обусловлено изменением энтальпийного уровня реагентов [53, 126–128]. Исключение составляют слабо ассоциированные растворители, способные к образованию Н-связи с диенофилом. Скорость большинства реакций Дильса–Альдера в таких средах, как хлороформ и пентахлорэтан, выше из-за повышенной стабилизации энтальпийного уровня активированного комплекса [87, 88]. Интересно отметить, что изменение энтальпии сольватации переходного состояния в ряду растворителей (уравнение (8)), по данным для прямого процесса, совпадает с результатами измерений для процесса распада аддукта на реагенты [127]. При переходе к реакциям в присутствии кислот Льюиса большое снижение энтальпийного уровня реагентов сопровождается еще большей стабилизацией активированного комплекса [103, 112, 114, 126]. Изменения энтальпии сольватации N-фенилмалеинимида, хлорида галлия и n,ν-комплекса между ними определены на основании данных термохимических измерений [126]. Влияние среды на энтальпийный уровень самого n,${v}$-комплекса оказалось очень слабым. Следует отметить, что надежность данных об энтальпии реакции в серии растворителей ΔHr–n,S, энтальпии растворения реагентов ΔHsol,L и продуктов ΔHsol,P проверяется постоянством значения энтальпии реакции в стандартном состоянии ΔHr–n, st.st.:
VI. ТВЕРДОФАЗНЫЕ РЕАКЦИИ ДИЛЬСА–АЛЬДЕРА
Особый интерес в рамках “зеленой химии” представляет изучение скорости РДА без растворителя при перетирании эквимольных долей реагентов в твердой фазе с образованием целевых продуктов реакции (схема 3).
Схема 3.
Твердофазные реакции 9,10-диметилантрацена 1 и антрацена 6 с тетрацианоэтиленом 2, N-фенилмалеин- имидом 7 и 4-фенил-1,2,4-триазолин-3,5-дионом 8 с образованием аддуктов 5, 9, 10 и 11 [133].
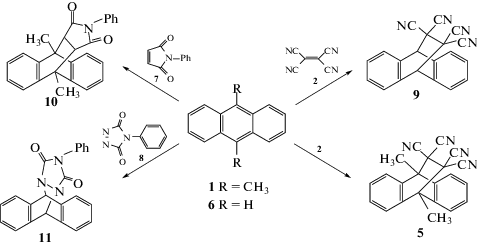
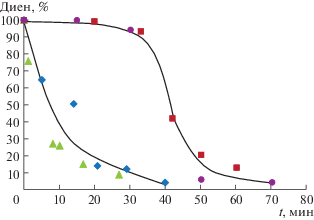
Благодаря высокому коэффициенту поглощения антрацена 6 и 9,10-диметилантрацена 1 можно отслеживать (350–400 нм) их очень малые концентрации в растворе (менее 1 × 10–4 М), при которых скорость реакции в растворе низкая. Отбор проб при равномерном перетирании эквимольных смесей кристаллов диена 6 с тетрацианоэтиленом 2 и диена 1 с N-фенилмалеимидом 7 позволил определить скорость этих реакций по уменьшению концентраций диенов (рис. 4) [133].




Поскольку реакция 1 + 2 → 5 (схема 3) в растворе протекает очень быстро [49], ее скорость в твердой фазе определяли по изменению интенсивности полос ИК-спектров в отобранных пробах порошков (рис. 5) [133].
Рис. 5.
Изменение доли 9,10-диметилантрацена 1 (– – серия измерений 1, –
– серия измерений 1, – – серия измерений 2) при перетирании эквимолярной смеси кристаллов 1 и 2 [133].
– серия измерений 2) при перетирании эквимолярной смеси кристаллов 1 и 2 [133].
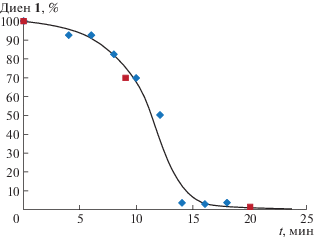
Интересно отметить, что в растворе константа скорости реакции 1 + 2 → 5 (табл. 1, № 2) на 4 порядка больше, чем для реакции 6 + 2 → 9 (табл. 1, № 6) [49], тогда как при перетирании порошков полупериоды этих реакций (рис. 4 и 5) почти одинаковы (∼10 мин) [133]. Причины таких различий пока не известны. Протекание реакции эквимолярной смеси белых кристаллов антрацена 6 и красных кристаллов 4-фенил-1,2,4-триазолин-3,5-диона 8 в вибрационной мельнице показано на рис. 6.

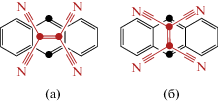
Перетирание в агатовой ступке кристаллов нафталина (IP = 8.15 эВ) или гексаметилбензола (IP = 7.85 эВ) с белыми кристаллами тетрацианоэтилена сразу приводит к образованию ярко окрашенных красных или зеленых твердых молекулярных комплексов. Значения максимумов их полос поглощения в спектрах отражения близки значениям максимумов в растворе [133]. РДА этих π-доноров с тетрацианоэтиленом невозможны из-за полного смещения равновесия в сторону реагентов. Эти же π-доноры образуют окрашенные комплексы при перетирании с тетрахлор-п-бензохиноном (хлоранилом). Перетирание антрацена и 9,10-диметилантрацена с хлоранилом также приводит к образованию цветных комплексов. Здесь аддукты не образуются из-за высокой энергии сопряжения в реагентах. Неожиданным оказалось отсутствие окраски при перетирании порошков активных диенов 6 (IP = 7.33 эВ) и 1 (IP = 7.04 эВ) с тетрацианоэтиленом. Следует ожидать, что, если ориентация структур молекулярного комплекса и активированного комплекса в реакциях 6 + 2 → 9 и 1 + 2 → 5 (схема 3) не совпадает (рис. 7a), окраска комплексов должна появляться при перетирании реагентов. При близком же соответствии этих структур (рис. 7б) можно ожидать лишь небольшой энергии активации и быстрого перехода молекулярных комплексов в неокрашенный аддукт [133].
Рис. 7.
Неблагоприятная (a) и благоприятная (б) геометрия молекулярного комплекса для реакций между антраценами 6 или 1 и тетрацианоэтиленом 2 [133].
Протекание реакции 1 + 2 → 5 (схема 3) в растворе через промежуточное образование молекулярного комплекса между реагентами было доказано ранее [49].
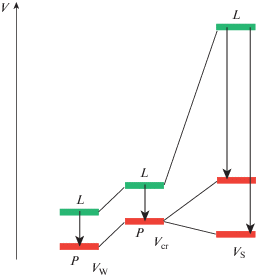
VII. РЕАКЦИИ ДИЛЬСА–АЛЬДЕРА ПРИ ПОВЫШЕННОМ ДАВЛЕНИИ
Изучение реакций в растворе при высоком гидростатическом давлении преследует две главные цели: 1) практическое применение для увеличения констант скорости и равновесия, особенно при синтезе труднодоступных соединений, и 2) получение дополнительной информации для уточнения механизма реакций [32, 134, 135]. Применение высокого давления позволило получить недоступные в обычных условиях аддукты РДА с нафталином и тиофеном и РСА данные для этих аддуктов [136]. Объемы активации (ΔV≠ = VAC – VL) и реакции (ΔVr – n = VР – VL) можно определить из зависимости констант скорости и равновесия реакции от давления при постоянной температуре Т, диэлектрической проницаемости растворителя ε, его плотности d и вязкости η:
(10)
$\begin{gathered} {{(\partial \ln k{\text{/}}\partial p)}_{{{\text{T}},\varepsilon ,d,\eta }}} = --{{(\partial \Delta {{G}^{ \ne }}{\text{/}}\partial p)}_{{{\text{T}},\varepsilon ,d,\eta }}}{\text{/}}RT = \\ = --\Delta {{Z}^{ \ne }}{\text{/}}RT \approx --\Delta {{V}^{ \ne }}{\text{/}}RT \\ \end{gathered} $(11)
$\begin{gathered} {{(\partial \ln K{\text{/}}\partial p)}_{{{\text{T}},\varepsilon ,d,\eta }}} = --{{(\partial \Delta {{G}_{{r\, - \,n}}}{\text{/}}\partial p)}_{{{\text{T}},\varepsilon ,d,\eta }}}{\text{/}}RT = \\ = --\Delta {{Z}_{{r\, - \,n}}}{\text{/}}RT \approx --\Delta {{V}_{{r\, - \,n}}}{\text{/}}RT \\ \end{gathered} $Для подбора практических условий проведения реакций под давлением достаточно знать значения ΔZ и их изменения в зависимости от давления [137]. Однако уточнение природы активированного комплекса по данным объемных параметров сопряжено с рядом проблем. Уравнения (10) и (11) позволяют рассчитать значения объемов ΔV≠ и ΔVr – n, если изменение свободных энергий (ΔG≠) или (ΔGr – n) обусловлены только вкладами PΔV≠ или PΔVr – n. Однако известно, что повышение давления вызывает изменение всех физических свойств растворителя, таких как, например, объем (и концентрация) (∂V/∂P), диэлектрическая проницаемость (dε/dP), вязкость (∂η/∂P) [36, 138]. То есть, при повышенном давлении изменение этих свойств растворителя может привести к дополнительному изменению скорости, не связанному с величиной PΔV≠, что приводит к “фантомным” вкладам (ΔV$_{2}^{ \ne }$, ΔV$_{3}^{ \ne }$, ΔV$_{4}^{ \ne }$) в значение ΔV≠:
(12)
$\begin{gathered} {{(\partial \ln \Sigma k{\text{/}}\partial p)}_{{\text{Т}}}} = {{(\partial \ln {{k}_{1}}{\text{/}}\partial p)}_{{{\text{Т}},(P\Delta {{V}^{ \ne }})}}} + \\ + \;{{(\partial \ln {{k}_{2}}{\text{/}}\partial p)}_{{{\text{Т}},(\partial C/\partial P)}}} + {{(\partial \ln {{k}_{3}}{\text{/}}\partial p)}_{{{\text{Т}},(\partial \varepsilon /\partial P)}}} + \\ + \;{{(\partial \ln {{k}_{4}}{\text{/}}\partial p)}_{{{\text{Т}},(\partial \eta /\partial P)}}} \\ \end{gathered} $(13)
$\begin{gathered} \Delta {{Z}^{ \ne }} = \Delta V_{{1(P \cdot \Delta {{V}^{ \ne }})}}^{ \ne } + \Delta V_{{2(\partial C/\partial P)}}^{ \ne } + \\ + \;\Delta V_{{3(\partial \varepsilon /\partial P)}}^{ \ne } + \Delta V_{{4{\text{ }}(\partial \eta /\partial P)}}^{ \ne } \\ \end{gathered} $Нами было показано [138], что вклад $\Delta V_{{2,(dC/dP)}}^{ \ne }$ (∼2–3 см3 моль–1) можно легко определить из соотношения (n – 1)βTRT, где n − порядок реакции, а βT − коэффициент сжимаемости растворителя [139]. Поправка $\Delta V_{{3,(d\varepsilon /dP)}}^{ \ne }$ обычно важна для полярных и ионных процессов [140, 141]. Влияние увеличения вязкости с ростом давления $\Delta V_{{4,(d\eta /dP)}}^{ \ne }$ проявляется только для очень быстрых реакций. Для молекулярных реакций диффузионный контроль наблюдали лишь в очень вязких средах [142, 143]. Следует подчеркнуть еще раз, что наиболее надежные количественные данные о влиянии высокого гидростатического давления на скорость и равновесие можно получить именно для реакции Дильса–Альдера, поскольку вкладами $\Delta V_{{3(d\varepsilon /dP)}}^{ \ne }$ и $\Delta V_{{4(d\eta /dP)}}^{ \ne }$ можно пренебречь, а вклад $\Delta V_{{2,(dC/dP)}}^{ \ne }$ можно легко определить [27, 139, 144]. Накоплен большой объем данных о том, что для неполярной РДА с участием реагентов разной структуры наблюдаются большие различия в значении объема активации и объема реакции. Эти результаты были непонятны и не имели объяснения. Для решения этой проблемы было предложено проводить анализ не объемных параметров, а их отношения ΔV≠/ΔVr – n [33, 34, 37, 38]. Поскольку в переходном состоянии РДА следует ожидать лишь частичное завязывание новых связей по сравнению с аддуктом, то должно выполняться соотношение ΔV≠/ΔVr – n < 1. Однако во многих работах для неполярных РДА обнаружено необъяснимое соотношение объемов активации и реакции в растворе ΔV≠/ΔVr – n > 1 [27–31, 33–39]. Это означает, что мольный объем активированного комплекса по какой-то причине меньше мольного объема аддукта. Важно отметить, что и в процессе распада аддукта на исходные компоненты отмечены как положительные, так и отрицательные значения объемов активации в растворе [15, 144–146]. Такое аномальное соотношение объемных параметров ΔV≠/ΔVr – n > 1 можно было бы объяснить большой электрострикцией растворителя при сольватации зарядов в переходном состоянии РДА. Однако оснований для предположения электрострикции растворителя при сольватации переходного состояния неполярной или изополярной РДА не найдено [144–146].
Значение объема активации можно определить только лишь из зависимости константы скорости реакции от внешнего давления (уравнение (10)). Объем реакции можно определить разными путями. Это дает возможность проверить наличие или отсутствие осложнений при определении объемных параметров из зависимости констант скорости и равновесия реакции от внешнего давления. Установка [147, 148] позволила изучить влияние давления на скорость реакции и на равновесие РДА 9-хлорантрацена 12 с тетрацианоэтиленом 2 в 1,2-дихлорэтане (схема 4). Объем этой реакции был определен тремя независимыми способами [149–152]. По данным о влиянии давления на константу равновесия рассчитано значение объема этой реакции ΔVr–n, S, равное –20.6 ± 1.5 см3 моль–1 [150, 152]. Согласно второму способу, разность объемов активации для прямого (–28.5 см3 моль–1) и обратного процесса (–6.5 см3 моль–1) также приводит к довольно близкому значению объема реакции (–22.0 ± ± 1.5 см3 моль–1) [149, 151]. Наконец, объем реакции был определен при обычном давлении по разности парциальных мольных объемов аддукта 13 (255.5 см3 моль–1) и реагентов 12 (170.7 см3 моль–1) и 2 (107.8 см3 моль–1), что дает близкое значение для ΔVr – n, S, равное –23.2 ± ± 2.0 см3 моль–1 [149].
Отсюда можно заключить, что объемные параметры активации и реакции Дильса–Альдера по данным о влиянии давления на скорость и равновесие надежно определяются соотношениями (10) и (11).
Даже в отсутствие сильных специфических взаимодействий реагентов с растворителем наблюдается заметное влияние среды на величины объемных параметров РДА [39, 54–56]. Для серии реакций с участием тетрацианоэтилена нами показано, что большой вклад энергии специфического взаимодействия тетрацианоэтилена с растворителем резко уменьшается при переходе к активированному комплексу и полностью исчезает при переходе к аддукту [54–56]. В среде алкилбензолов наблюдаются пропорциональные изменения энтальпии сольватации тетрацианоэтилена, его мольного объема, энтальпии и свободной энергии образования его π,π-комплексов с растворителем и скорости РДА (табл. 5) [54–56].
Таблица 5.
Потенциалы ионизации растворителей IP, парциальные мольные объемы тетрацианоэтилена V, энтальпии растворения ΔHsol и сольватации ΔHsolv, свободные энергии ΔGMC, образования комплексов тетрацианоэтилена с алкилбензолами и константы скорости k2 в реакции Дильса–Альдера тетрацианоэтилена с антраценом при 25°C [54–56]
| Растворитель | IP, эВ | V, см3 моль–1 | ΔHsol, кДж моль–1 | –ΔHsolv, кДж моль–1 | –ΔGMC, кДж моль–1 | k2, л моль–1 с–1 |
|---|---|---|---|---|---|---|
| Хлорбензол | 9.10 | 109.23 | 23.1 | 58.1 | –0.65 | 1.82 |
| Бензол | 9.25 | 108.40 | 14.9 | 66.3 | 1.72 | 0.38 |
| Толуол | 8.82 | 104.56 | 9.7 | 71.5 | 3.24 | 0.13 |
| o-Ксилол | 8.58 | 102.06 | 1.4 | 79.8 | 4.81 | 0.061 |
| p-Ксилол | 8.48 | 101.46 | 0 | 81.2 | 5.04 | – |
| Mезитилен | 8.14 | 98.07 | –2.7 | 83.9 | 7.07 | 0.010 |
| Ацетонитрил | 12.12 | 109.97 | 15.2 | 66.0 | – | 2.18 |
| Этилацетат | 9.54 | 112.09 | 9.2 | 72.0 | – | 0.24 |
| Циклогексанон | 9.14 | 110.42 | 7.6 | 73.6 | – | 0.20 |
| 1,4-Диоксан | 9.13 | 105.72 | 4.3 | 76.9 | – | 0.34 |
| 1,2-Дихлорэтан | 11.12 | 107.81 | 21.3 | 59.9 | – | 3.82 |
| Дихлорметан | 11.35 | 107.50 | 23.4 | 57.8 | – | 4.28 |
Для реакций с участием тетрацианоэтилена (схема 5) было показано, что с ростом экзотермичности процесса в ряду растворителей растет отрицательное значение объема активации и объема реакции [54–56, 153]. Поэтому для максимального и благоприятного изменения скорости и равновесия таких реакций в условиях повышенного гидростатического давления следует применять наиболее инертный растворитель.
Схема 5.
Реакция Дильса–Альдера циклопентадиена 14, 1,3-бутадиена 15, транс,транс-1,4-дифенилбутадиена 16 и 9-хлорантрацена 12 с тетрацианоэтиленом 2.

Нормальное соотношение ΔV$_{{\text{W}}}^{ \ne }$/ΔVr – n,W < 1 выполняется всегда при сравнении собственных, т.е. ван-дер-ваальсовых объемных параметров. Мы определили, что значения объемов реакций в твердой фазе ΔVr – n, cr тоже малы и близки значениям ΔVr – n,W [154, 155]. Но сами значения ΔV$_{{\text{W}}}^{ \ne }$, ΔVr – n, W и ΔVr – n, cr значительно меньше по величине, чем эти параметры для РДА в растворе [34, 154–156]. Собственные значения ΔV$_{{\text{W}}}^{ \ne }$, ΔVr – n, W и ΔVr – n, cr малы и не зависят от природы реагентов в РДА.
Из полученных данных [154, 155] установлено, что мольные объемы реагентов и продуктов при переходе из твердой фазы в раствор изменяются непропорционально. Были получены количественные данные об изменении мольных объемов [154–156]. Мольный объем в твердой фазе можно определить, используя данные РСА об их плотности, а в растворе – из зависимости объема раствора от его концентрации ∂V/∂С. Поскольку объем реакции определяется проще и надежнее (уравнения (14) и (15)), чем объем активации, были рассмотрены изменения мольных объемов при переходе продуктов (Р) и реагентов (L) из твердой фазы в раствор.
Было изучено влияние растворителя на изменение мольного объема 2D-структур реагентов и 3D-структур аддуктов РДА. Можно ожидать, что 2D-структуры молекул реагентов L будут полностью доступны для межмолекулярного взаимодействия в собственном окружении в кристалле L‒L и в растворе L‒S. С другой стороны, 3D-структуры молекул аддуктов Р в собственном окружении в кристалле могут иметь пониженный коэффициент упаковки η = VW/Vcr из-за стерических затруднений и менее эффективного вовлечения своей поверхности в межмолекулярное взаимодействие Р‒Р. В растворе возможно проникновение Р‒S молекул растворителя или их фрагментов в “карманы” или “пустоты” таких 3D-молекул. Это может привести к тому, что молекулы растворителя будут полнее перекрывать поверхность 3D-молекул, чем 3D-молекулы в собственном окружении. Если это так, то этот эффект должен проявляться в непропорциональном изменении мольных объемов и, возможно, в заметном различии энтальпии растворения таких 2D- и 3D-молекул в ряду растворителей. Такой анализ был проведен [154, 155] для твердых 2D-молекул реагентов 2, 6, 7, 16, 20 и 3D-молекул их аддуктов 9, 19, 21, 22 (рис. 8). В табл. 6 для соединений 2, 6, 7, 9, 16, 19–22 приведены данные их собственных мольных объемов VW, объемов в твердой фазе Vcr, и в растворе VS, [154, 155]. Надо отметить (табл. 6), что мольные объемы твердых 2D-реагентов 2, 6, 7, 16, 20 увеличиваются на 10–15% при переходе в раствор. Подобное увеличение объема наблюдается при их плавлении [156, 157]. Напротив, изменение мольного объема 3D-аддуктов 9, 19, 21, 22 при переходе в раствор оказалось небольшим (последняя колонка в табл. 6).
Рис. 8.
2D-реагенты: тетрацианоэтилен 2, малеиновый ангидрид 20, N-фенилмалеинимид 7, антрацен 6, транс,транс-1,4-дифенилбутадиен-1,3 16 и их 3D-аддукты (9, 19, 21, 22).
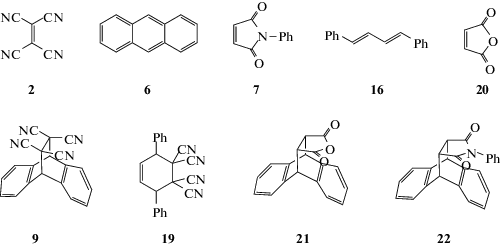
Таблица 6.
Ван-дер-ваальсовы мольные объемы VW соединений 2, 6, 7, 9, 16, 19–22, их мольные объемы в твердой фазе Vcr и в растворе 1,4-диоксана VS1, ацетонитрила VS2, 1,2-дихлорэтана VS3, этилацетата VS4, хлорбензола VS5, циклогексанона VS6, коэффициенты упаковки η, собственные объемы реакций ΔVr‒n,W, объемы реакций в твердой фазе ΔVr‒n, cr и в растворе ΔVr ‒ n,S, энтальпии растворения ΔHsol при 25°Cа [154, 155]
| Соединение | VW (ηW) | Vcr (ηcr) | VS1 (ηS1) ΔHsol,S1 | VS2 (ηS2) ΔHsol,S2 | VS3 (ηS3) ΔHsol,S3 | VS4 (ηS4) ΔHsol,S4 | VS5 (ηS5) ΔHsol,S5 | VS6 (ηS6) ΔHsol,S6 | (VS – Vcr)/Vcr , % S1; S2; S3; S4 ; S5; S6 |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 62.6 (1) | 97.5 (0.642) | 105.7 (0.592); 4.3 | 108.7 (0.576); 15.2 | 107.8 (0.581); 21.3 | 112.1 (0.558); 9.2 | 109.2 (0.573); 23.1 | 110.4 (0.567); 7.6 | 8.4; 11.5; 10.6; 15.0; 12.0; 13.2 |
| 20 | 43.9 (1) | 65.5 (0.670) | 71.3 (0.615); 10.4 | 70.4 (0.623); 13.1 | 71.1 (0.617); 15.9 | 69.2 (0.634); 11.4 | 73.9 (0.594); 18.3 | 74.5 (0.589); ‒ | 8.8; 7.5; 8.5; 5.6; 12.8; 13.7 |
| 7 | 88.6 (1) | 123.2 (0.719) | 137.1 (0.646) | 133.3 (0.665) | 138.1 (0.642) | ‒ | ‒ | ‒ | 11.3; 8.2; 12.1; ‒; ‒; ‒ |
| 6 | 101.3 (1) | 143.0 (0.708) | 159.5 (0.635); 22.7 | 158.2 (0.640); 28.0 | 161.9 (0.626); 24.9 | 153.7 (0.659); 25.1 | 160.1 (0.633); 24.6 | 159.8 (0.634); 20.6 | 11.5; 10.6; 13.2; 7.5; 12.0; 11.7 |
| 16 | 123.8 (1) | 179.4 (0.690) | 200.4 (0.618); 22.3 | 200.0 (0.619); 30.1 | 202.7 (0.611); 24.3 | 195.2 (0.634); 23.1 | ‒ | 201.7 (0.614); ‒ | 11.7; 11.5; 13.0; 8.8; ‒; 12.4 |
| 9 (2 + 6) | 160.5 (1) | 235.5 (0.682) | 234.6 (0.684); –7.5 | 237.9 (0.675); 4.4 | 242.3 (0.662); 8.6 | 234.3 (0.685); | 237.9 (0.675); 13.8 | 244.8 (0.656); –4.4 | –0.4; 0.9; 2.8; –0.5; 1.0; 3.9 |
| 21 (20 + 6) | 141.2 (1) | 198.9 (0.710) | 205.2 (0.688) | 202.1 (0.699) | 207.8 (0.679) | 198.9 (0.710) | 210.7 (0.670) | 207.1 (0.682) | 3.2; 1.6; 4.5; 0.0; 5.9; 4.1 |
| 22 (7 + 6) | 185.6 (1) | 256.0 (0.725) | 276.9 (0.670) | 272.6 (0.681) | 279.8 (0.663) | ‒ | ‒ | ‒ | 8.1; 6.4; 9.2; ‒; ‒; ‒ |
| 19 (2 + 16) | 183.2 (1) | 267.5 (0.684) | 279.8 (0.655) | 275.5 (0.665) | 278.8 (0.657) | 272.8 (0.672) | ‒ | 278.1 (0.659) | 4.6; 3.0; 4.2; 2.0; –; 4.0 |
| Объем реакции (±2 см3 моль‒1) | |||||||||
| Реакция | ΔVr‒n,W | ΔVr‒n,cr | ΔVr‒n,S1 | ΔVr‒n,S2 | ΔVr‒n,S3 | ΔVr‒n,S4 | ΔVr‒n,S5 | ΔVr‒n,S6 | ΔVr‒n,cr ‒ ΔVr‒n,W |
| 2 + 6 → 9 | –3.3 | –5.0 | –30.6 | –29.0 | –27.4 | –31.5 | –31.4 | –25.4 | –1.7 |
| 20 + 6 → 21 | –4.0 | –9.6 | –25.6 | –26.5 | –25.2 | ‒24.0 | –23.3 | –27.2 | –5.6 |
| 7 + 6 → 22 | –4.3 | –10.2 | –19.7 | –18.9 | –20.2 | ‒ | ‒ | ‒ | –5.9 |
| 2 + 16 → 19 | –3.2 | –9.4 | –26.3 | –33.2 | –31.7 | –34.5 | ‒ | –34.0 | –6.2 |
Это означает, что ожидаемое увеличение в межмолекулярном расстоянии и в мольном объ- еме при переходе твердых 3D-аддуктов 9, 19, 21, 22 в раствор почти полностью компенсируется частичным уменьшением общего объема раствора из-за более эффективного проникновения молекул растворителя ко всей поверхности этих 3D-молекул. Коэффициенты упаковки VW/Vcr кристаллических 3D-аддуктов 9, 19, 21, 22 мало отличаются от значений VW/VS в растворе. Вследствие этого растворение аддукта 9 в 1,4-диоксане и циклогексаноне сопровождалось даже экзо-эффектом (табл. 6). Несмотря на отсутствие специфических взаимодействий с растворителем наблюдали неожиданно огромное различие в изменении мольных объемов 2D-реагентов и 3D-аддуктов при их переходе из твердой фазы в раствор. Вновь значения ван-дер-ваальсовых объемов реакции ΔVr ‒ n, W и объемов реакции в твердой фазе (ΔVr ‒ n, cr) оказались невысокими для всех рассмотренных реакций (табл. 6). Изменения значений объема реакций в этом ряду растворителей не коррелируют с любой шкалой полярности растворителя.
Становится понятным, что большое различие между значениями объемов реакции в растворе ΔVr ‒ n, S и в твердой фазе ΔVr ‒ n, cr обусловлено большими и непропорциональными изменениями в упаковке 2D-молекул реагентов и 3D-молекул аддуктов при их переходе из твердой фазы в раствор (табл. 7).
Таблица 7.
Изменения мольных объемов при переходе твердых 2D-реагентов 2, 6, 7, 16, 20 и 3D-аддуктов 9, 19, 21, 22 в раствор (VS – Vcr) при 25°C
| Растворитель | VS – Vcr, см3 моль–1 | |||||||
|---|---|---|---|---|---|---|---|---|
| 2 + 6 | 20 + 6 | 7 + 6 | 2 + 16 | 9 | 21 | 22 | 19 | |
| 1,4-Диоксан | 24.7 | 22.3 | 30.4 | 29.2 | –0.9 | 6.3 | 20.9 | 12.3 |
| Ацетонитрил | 26.4 | 20.1 | 25.3 | 31.8 | 2.4 | 3.2 | 16.6 | 8.0 |
| 1,2-Дихлорэтан | 29.2 | 24.5 | 33.8 | 33.6 | 6.8 | 8.9 | 23.8 | 11.3 |
| Этилацетат | 25.3 | 14.4 | ‒ | 30.4 | –1.2 | 0 | ‒ | 5.3 |
| Хлорбензол | 28.8 | 25.5 | ‒ | ‒ | 2.4 | 11.8 | ‒ | ‒ |
| Циклогексанон | 29.7 | 25.8 | ‒ | 35.2 | 9.3 | 8.2 | ‒ | 10.6 |
Рис. 9.
Изменения объемов 2D-реагентов L и 3D-продуктов P при переходе от ван-дер-ваальсовых объемов (VW) в твердую фазу (Vcr) и в раствор (VS).
Огромные различия в изменении мольных объемов и энтальпий растворения твердых 2D- и 3D-соединений в РДА (рис. 8) позволяют ожидать подобного поведения и для других 3D-молекул, таких как 1,3,5-трифенилбензол 23, гексафенилбензол 24 и 9,10-дифенилантрацен 25 (рис. 10, табл. 8).

Таблица 8.
Ван-дер-ваальсовы мольные объемы VW соединений 23–25, их мольные объемы в твердой фазе Vcr и в растворе VS и энтальпии растворения (ΔHsol,) при 25°C [155]. Мольные объемы даны в см3 моль‒1
| Растворитель | VS | VW/VS | ΔHsol, кДж моль‒1 |
|---|---|---|---|
| Антрацен 6: VW = 101.3; Vcr = 143.0; VW/Vcr = 0.708 | |||
| 1,4-Диоксан | 159.5 ± 0.5 | 0.635 | 22.7 ± 1.0 |
| Бензол | 157.2 ± 0.5 | 0.644 | 24.7 ± 1.0 |
| 23: VW = 176.8; Vcr = 254.3; VW/Vcr = 0.695 | |||
| 1,4-Диоксан | 254.2 ± 1.0 | 0.697 | 17.8 ± 1.0 |
| Бензол | 270.1 ± 1.0 | 0.655 | 20.1 |
| 24: VW = 304.5; Vcr = 456.2; VW/Vcr = 0.667 | |||
| 1,4-Диоксан | 417 ± 2 | 0.730 | –16 ± 2 |
| Бензол | 452 ± 2 | 0.674 | 4.0 ± 1.0 |
| Этилацетат | 462 ± 4 | 0.659 | ‒ |
| Толуол | 469 ± 3 | 0.649 | 5.0 ± 1.0 |
| Циклогексанон | 470 ± 3 | 0.648 | 0 ± 1.0 |
| 25: VW = 186.6; Vcr = 267.1; VW/Vcr = 0.699 | |||
| 1,4-Диоксан | 251 ± 3 | 0.743 | 1.0 ± 1.0 |
| Бензол | 282 ± 1 | 0.662 | 17.5 ± 1.0 |
Плоские 2D-молекулы, такие как антрацен 6, плотно упакованы в кристалле и их переход в раствор сопровождается увеличением мольного объема и высоким значением эндо-эффекта растворения, близким энтальпии плавления. По данным РСА фенильные группы соединений 23‒25 выведены из плоскости центрального кольца. Здесь увеличение объема при разрушении кристаллов 23‒25 сопровождается уменьшением объема раствора за счет доступного проникновения молекул растворителя к поверхности этих 3D-молекул. Особенно резко это проявляется при растворении гексафенилбензола 24, для которого наблюдается резкое уменьшение мольного объема и даже экзо-эффект растворения в 1,4-диоксане. 3D-структура 9,10-дифенилантрацена 25 препятствует приближению любого диенофила к активному 9,10-реакционному центру. Даже очень активный 4-фенил-1,2,4-триазолин-3,5-дион 8 реагирует не по 9,10-положению диена 25, как предполагалось ранее [158]. Данные РСА соответствуют протеканию реакции по менее активному, но доступному 1,4-реакционному центру диена 25 [159]. Для этого диена окружение молекулами 1,4-диоксана энергетически более выгодно, чем в кристалле (V12, cr/V12, диоксан = = 1.064). Вновь энтальпия растворения 25 в 1,4-диоксане и бензоле резко различается (табл. 8).
Для твердого конгрессана 26 (рис. 11) Vcr равен 172, в растворе н-гексана VS равен 160 и в бензоле ‒ 177 см3 моль‒1 [157]. Это означает, что н-гексан легче проникает к поверхности молекулы 26, по сравнению с бензолом. Для шарообразной молекулы фуллерена С-60 27 объемные параметры равны: VW = 317, Vcr = 429 см3 моль‒1 и VW/Vcr = 0.74 [157]. В большинстве растворов его мольные объемы VC-60, S значительно меньше, чем в кристалле: 351 в сероуглероде, 365 в толуоле и 400 см3 моль‒1 в декалине [157]. Важно отметить экзо-эффект растворения ΔHsol фуллерена 27 в этом ряду растворителей: –20, –8.6 и –5.0 кДж моль‒1 соответственно [160].
Изучение скорости и равновесия РДА при высоком гидростатическом давлении позволяет сделать некоторые обобщения. Во-первых, можно определить значения ΔZ≠ и ΔZr ‒ n (уравнения (10) и (11)) в широком интервале давлений [137], которые необходимы для планирования условий синтеза труднодоступных соединений. Во-вторых, эти данные позволили понять причины больших различий значений ΔV≠ и ΔVr ‒ n РДА в растворе с участием различных структур реагентов и “аномальное” соотношение ΔV≠/ΔVr ‒ n > 1. Значительные увеличения мольных объемов для 2D-молекул реагентов и малые нерегулярные увеличения или даже уменьшения мольных объемов для 3D-молекул аддуктов при переходе из кристаллов в раствор (рис. 8) формируют реальные значения объемов реакции в растворе. Подобные непропорциональные изменения мольных объемов следует ожидать в растворе для 2D-реагентов и 3D-активированного комплекса.
Полученные результаты позволяют заключить, что значения объемов активации и объемов реакции Дильса‒Альдера в растворе и их соотношения ΔV≠/ΔVr ‒ n формируются, главным образом, различной возможностью упаковки молекул растворителя в сольватной оболочке реагентов, активированного комплекса и аддуктов. Поэтому значения объемов активации и реакции не пригодны для уточнения природы активированного комплекса и его места на координате реакции.
“Дьявол прячется в деталях”. Теплота растворения 2D-кристаллов обычно ожидаемо близка энтальпии их плавления. Большая доля свободной, не вовлеченной в межмолекулярное взаимодействие, поверхности 3D-молекул в твердой фазе становится более доступной в растворе для молекул растворителя. Это объясняет, почему растворение кристаллов больших 3D-молекул может сопровождаться небольшими эндо- или даже экзо-эффектами в отсутствие специфических взаимодействий. В результате можно отметить, что шкала полярности растворителя ЕТ и многопараметровые корреляции, пригодные для описания сольватации 2D-молекул, могут оказаться неспособными описать сольватацию 3D-молекул.
ЗАКЛЮЧЕНИЕ
Знание основных факторов, влияющих на реакционную способность реакции Дильса‒Альдера, объясняет многие особенности и кажущиеся “аномалии” ее поведения. Обнаруженные закономерности протекания обычной и катализируемой РДА позволяют планировать оптимальные условия ее проведения. Надежные данные об изменении объемов активации и реакции позволили понять, что эти параметры не связаны с механизмом РДА.
Описанные особенности протекания РДА в отсутствие и в присутствии катализаторов, при обычном и повышенном гидростатическом давлении могут быть полезными для понимания поведения и других органических реакций.
Список литературы
Diels O., Alder K. // Justus Liebigs Ann. Chem. 1926. V. 450. P. 237−254. https://doi.org/10.1002/jlac.19264500119
Diels O., Alder K. // Justus Liebigs Ann. Chem. 1929. V. 470. P. 62−103. https://doi.org/10.1002/jlac.19294700106
Diels O., Alder K. // Justus Liebigs Ann. Chem. 1931. V. 490. P. 236−242. https://doi.org/10.1002/jlac.19314900109
Diels O., Alder K. // Justus Liebigs Ann. Chem. 1928. V. 460. P. 98−122. https://doi.org/10.1002/jlac.19284600106
Онищенко А.С. Диеновый синтез. Москва: АН СССР. 1963. 650 с.
Carruthers W. Cycloaddition Reactions in Organic Synthesis. Oxford: Pergamon Press, 1990. 373 p.
Desimoni G., Tacconi G., Barco A., Polloni G. Natural Product Synthesis Through Pericyclic Reactions. Washington, D.C.: American Chemical Society. 1983. 443 p.
Fringuelli F., Tatichi A. Dienes in the Diels‒Alder Reactions. Chichester: Wiley, 1990. 348 p.
Kobayashi S., Jorgensen K.A. Cycloaddition reaction in organic synthesis. Weinheim, Wiley. 2001. 332 p. https://doi.org/10.1002/3527600256.ch8
Fringuelli F., Tatichi A. The Diels-Alder reaction: Selected practical methods. Chichester: Wiley, 2002. 340 p.
Sauer J. // Angew. Chem. 1966. V. 78. P. 233−252. https://doi.org/10.1002/ange.19660780403
Sauer J., Sustmann R. // Angew. Chem. 1980. V. 92. P. 773−801. https://doi.org/10.1002/ange.19800921004
Sauer J., Wiest H., Mielert A. // Chem. Ber. 1964. V. 97. P. 3183−3207. https://doi.org/10.1002/cber.19640971129
Rohr U., Schatz J., Sauer J. // Eur. J. Org. Chem. 1998. V. 1998. P. 2875−2883. https://doi.org/10.1002/(SICI)1099-0690(199812)1998:12<2875::AID-EJOC2875>3.0.CO;2-N
Klärner F.-G., Breitkopf V. // Eur. J. Org. Chem. 1999. V. 1999. P. 2757−2762. https://doi.org/10.1002/(SICI)1099-0690(199911)1999:11<2757::AID-EJOC2757>3.0.CO;2-J
Deiters U., Klärner F.-G., Krawczyk B., Ruster V. // J. Am. Chem. Soc. 1994. V. 116. P. 7646−7657. https://doi.org/10.1021/ja00096a023
Houk K.N. Perycyclic Reactions. New York: Academic Press, 1977. 181 p.
Houk K.N., Li Y., Evanseck J.D. // Angew. Chem., Int. Ed. 1992. V. 31. P. 682−708. https://doi.org/10.1002/anie.199206821
Sustmann R., Bohm M., Sauer J. // Chem. Ber. 1979. V. 112. P. 883−889. https://doi.org/10.1002/cber.19791120313
Sustmann R., Schubert R. // Angew. Chem. 1972. V. 84. P. 888−889. https://doi.org/10.1002/ange.19720841807
Jenner G. // J. Phys. Org. Chem. 2002. V. 15. P. 1−13. https://doi.org/10.1002/poc.458
Коновалов А.И. Дис. докт. хим. наук. Казань: Казанский университет, 1973. 306 с.
Киселев В.Д. Дис. докт. хим. наук. Казань: Казанский университет, 1986. 373 с.
Коновалов А.И. // Усп. Хим. 1983. Т. 52. С. 1852−1878.
Киселев В.Д., Коновалов А.И. // Усп. Хим. 1989. Т. 58. С. 383−416.
Коновалов А.И., Киселев В.Д. // Изв. АН, Сер. хим. 2003. С. 279−294.
Kiselev V.D., Konovalov A.I. // J. Phys. Org. Chem. 2009. V. 22. P. 466−483. https://doi.org/10.1002/poc.1503
Kiselev V.D., Kornilov D.A., Anikin O.V., Sedov I.A., Konovalov A.I. // J. Phys. Org. Chem. 2018. V. 31. Article no. e3737. https://doi.org/10.1002/poc.3737
Киселёв В.Д., Корнилов Д.А., Аникин О.В., Латыпо-ва Л.И., Бермешев М.В., Чапала П.П., Конова- лов А.И. // ЖОрХ. 2016. Т. 52. С. 793–795.
Аникин О.В., Корнилов Д.А., Никитина Т.В., Кисе-лев В.Д. // Хим. Физ. 2018. Т. 37. С. 3–6. https://doi.org/10.1134/S0207401X18080022
Kiselev V.D., Kornilov D.A., Anikin O.V., Shulyatiev A.A., Konovalov A.I. // J. Sol. Chem. 2019. V. 48. P. 31–44. https://doi.org/10.1007/s10953-019-00846-6
Гоникберг М.Г. Химическое равновесие и скорость реакции при высоких давлениях. Москва: АН СССР, 1969. 426 с.
Drljaca A., Hubbard C.D., Van Eldik R., Asano T., Basilevsky M.A., le Noble W.J. // Chem. Rev. 1998. V. 98. P. 2167–2290. https://doi.org/10.1021/cr970461b
Van Eldik R., Klärner F.-G. High pressure chemistry. Weinheim: Wiley-VCH, 2002. 458 p.
Le Noble W.J. Organic High Pressure Chemistry. Amsterdam–Oxford–New York–Tokyo: Elsevier, 1988. 489 p.
Isaacs N.S. Liquid Phase High Pressure Chemistry. Chichester–New York–Brisbane–Toronto: Wiley, 1981. 414 p.
Asano T., le Noble W.J. // Chem. Rev. 1978. V. 78. P. 407–489. https://doi.org/10.1021/cr60314a004
Van Eldick R., Asano T., le Noble W.J. // Chem. Rev. 1989. V. 89. P. 549–688. https://doi.org/10.1021/cr00093a005
Grieger R.A., Eckert C.A. // Trans. Faraday Soc. 1970. V. 66. P. 2579–2584. https://doi.org/10.1039/TF9706602579
Киселев В.Д., Шакирова И.И., Коновалов А.И. // Изв. АН, Серия хим. 2013. P. 290–307.
Арбузов Б.А., Коновалов А.И. // Изв. АН СССР, Сер. хим. 1959. С. 2130–2134.
Арбузов Б.А., Коновалов А.И. // Изв. АН СССР, Сер. хим. 1960. С. 68–72.
Арбузов Б.А., Коновалов А.И. // Изв. АН СССР, Сер. хим. 1965. С. 1290–1291.
Коновалов А.И., Киселев В.Д. // ЖОрХ. 1966. Т. 2. С. 142–144.
Коновалов А.И., Киселев В.Д., Вигдорович О.А. // ЖОрХ. 1967. Т. 3. С. 2085–2088.
Коновалов А.И., Киселев В.Д., Метелина М.Б. // ЖОрХ. 1970. Т. 6. С. 1548–1552.
Коновалов А.И., Киселев В.Д. // ЖОрХ. 1974. Т. 10. С. 6–10.
Киселев В.Д., Коновалов А.И. // Рос. Хим. Журн. 1999. Т. 43. С. 94–104.
Kiselev V.D., Miller J.G. // J. Am. Chem. Soc. 1975. V. 97. P. 4036–4039. https://doi.org/10.1021/ja00847a028
Коновалов А.И., Киселев В.Д., Самуилов Я.Д. // Докл. АН СССР. 1968. Т. 179. С. 866–869.
Коновалов А.И., Киселев В.Д., Самуилов Я.Д. // Докл. АН СССР. 1969. Т. 186. С. 347–349.
Киселев В.Д., Маврин Г.В., Коновалов А.И. // ЖОрХ. 1980. Т. 16. С. 1435–1441.
Киселев В.Д., Коновалов А.И. // ЖОХ. 1982. Т. 52. С. 1474–1477.
Киселев В.Д., Кашаева Е.А., Шихаб М.С., Медведе-ва М.Д., Коновалов А.И. // Изв. АН. Сер. хим. 2000. С. 1046–1050.
Kiselev V.D., Kashaeva E.A., Shihab M.S., Konova-lov A.I. // Mendeleev Commun. 2000. V. 10. P. 49–51. https://doi.org/10.1070/MC2000v010n02ABEH001176
Kiselev V.D., Konovalov A.I., Asano T., Iskhakova G.G., Kashaeva E.A., Shihaab M.S., Medvedeva M.D. // J. Phys. Org. Chem. 2001. V. 14. P. 636–643. https://doi.org/10.1002/poc.398
Fujimoto H., Inagaki S., Fukui K. // J. Am. Chem. Soc. 1976. V. 98. P. 2670–2671. https://doi.org/10.1021/ja00425a048
Коновалов А.И., Соломонов Б.Н. // Докл. АН CCCР. 1973. Т. 211. С. 1115–1117.
Коновалов А.И., Самуилов Я.Д., Слепова Л.Ф., Бреус В.А. // ЖОрХ. 1973. Т. 9. С. 2087–2089.
Коновалов А.И., Самуилов Я.Д., Слепова Л.Ф., Бреус В.А. // ЖОрХ. 1973. Т. 9. С. 2519–2521.
Коновалов А.И., Соломонов Б.Н., Чертов О.Ю. // ЖОрХ. 1975. Т. 11. С. 106–109.
Коновалов А.И., Соломонов Б.Н., Чертов О.Ю. // ЖОрХ. 1975. Т. 11. С. 110–112.
Коновалов А.И., Самуилов Я.Д., Урядова Л.Ф., Бердников Е.А. // ЖОрХ. 1976. Т. 12. С. 645–648.
Самуилов Я.Д., Урядова Л.Ф., Коновалов А.И. // ЖОрХ. 1979. Т. 15. С. 977–983.
Коновалов А.И., Урядова Л.Ф., Самуилов Я.Д. // ЖОрХ. 1976. Т. 12. С. 2610–2615.
Самуилов Я.Д., Коновалов А.И., Урядова Л.Ф. // ЖОрХ. 1974. Т. 10. С. 1934–1936.
Коновалов А.И., Соломонов Б.Н. // ЖОрХ. 1975. Т. 11. С. 2144–2149.
Самуилов Я.Д., Урядова Л.Ф., Коновалов А.И. // ЖОрХ. 1976. Т. 12. С. 810–812.
Коновалов А.И., Самуилов Я.Д. // Докл. АН СССР. 1972. Т. 204. С. 359–361.
Коновалов А.И., Самуилов Я.Д., Бердников Е.А., Племенков В.В. // Докл. АН СССР. 1973. Т. 208. С. 862–863.
Коновалов А.И. // ЖОрХ. 1969. Т. 5. С. 1713–1718.
Коновалов А.И. // Докл. АН СССР. 1975. Т. 223. С. 613–616.
Киселев В.Д., Устюгов А.Н., Бреус И.П., Конова- лов А.И. // Докл. АН СССР. 1977. Т. 234. С. 1089–1092.
Киселев В.Д., Коновалов А.И., Вейсман Е.А., Устюгов А.Н. // ЖОрХ. 1978. Т. 14. С. 128–134.
Коновалов А.И., Соломонов Б.Н. // Докл. АН СССР. 1972. Т. 202. С. 1331–1333.
Киселев В.Д., Адигезалов Н.Р., Коновалов А.И. // ЖОрХ. 1989. Т. 25. С. 539–542.
Киселев В.Д., Малков В.Б., Коновалов А.И. // ЖОрХ. 1990. Т. 26. С. 229–240.
Киселев В.Д., Кашаева Е.А., Галиакберова М.Г., Коновалов А.И., Литвинов И.А., Катаева О.Н., Зверев В.В., Наумов В.А. // ЖОрХ. 1993. Т. 29. С. 1719–1726.
Киселев В.Д., Пацановский И.И., Кашаева Е.А., Попова Е.В., Мюллер Хр., Шмутцлер Р., Ишмаева Э.А., Коновалов А.И. // ЖОрХ. 1996. Т. 32. С. 1853–1856.
Коновалов А.И., Соломонов Б.Н., Устюгов А.Н. // Докл. АН СССР. 1973. Т. 211. С. 102–105.
Коновалов А.И., Соломонов Б.Н., Устюгов А.Н. // Докл. АН СССР. 1973. Т. 213. С. 349–352.
Самуилов Я.Д., Урядова Л.Ф., Соломонов Б.Н., Коновалов А.И. // ЖОрХ. 1975. Т. 11. С. 1917–1921.
Коновалов А.И., Самуилов Я.Д., Уба В.М. // ЖОрХ. 1973. Т. 9. С. 2084–2086.
Самуилов Я.Д., Урядова Л.Ф., Коновалов А.И. // ЖОрХ. 1974. Т. 10. С. 1931–1933.
Самуилов Я.Д., Нуруллина Р.Л., Урядова Л.Ф., Коновалов А.И. // ЖОрХ. 1980. Т. 16. С. 1858–1863.
Коновалов А.И., Самуилов Я.Д., Хайруллин З.А. // ЖОрХ. 1972. Т. 8. С. 229–232.
Коновалов А.И., Киселев В.Д., Устюгов А.Н., Гесс Н.Г. // ЖОрХ. 1976. Т. 12. С. 2541–2546.
Коновалов А.И., Бреус И.П., Шарагин И.А., Кисе-лев В.Д. // ЖОрХ. 1979. Т. 15. С. 361–367.
Коновалов А.И. // Докл. АН СССР. 1963. Т. 149. С. 1334–1338.
Коновалов А.И., Верещагин А.Н., Камашева Г.И. // Докл. АН СССР. 1969. Т. 185. С. 597–599.
Коновалов А.И., Камашева Г.И., Лоскутов М.П. // Докл. АН СССР. 1971. Т. 201. С. 363–365.
Коновалов А.И., Камашева Г.И., Лоскутов М.П. // Докл. АН СССР. 1972. Т. 204. С. 103–106.
Коновалов А.И., Камашева Г.И. // ЖОрХ. 1972. Т. 8. С. 1446–1448.
Коновалов А.И., Камашева Г.И. // ЖОрХ. 1972. Т. 8. С. 1831–1833.
Коновалов А.И., Камашева Г.И., Лоскутов М.П. // ЖОрХ. 1972. Т. 8. С. 2088–2090.
Коновалов А.И., Камашева Г.И., Лоскутов М.П. // ЖОрХ. 1973. Т. 9. С. 2048–2056.
Киселев В.Д., Коновалов А.И. // ЖОрХ. 1986. Т. 22. С. 1133–1145.
Bell R.P. Acid-base catalysis. London: Oxford University Press, 1941. 211 p.
Evans M.G., Polanyi M. // Trans. Faraday Soc. 1936. V. 32. P. 1333–1360. https://doi.org/10.1039/TF9363201333
Семенов Н.Н. О некоторых проблемах химической кинетики и реакционной способности. Москва: АН СССР, 1958, 686 с.
Киселев В.Д., Шакиров И.М., Коновалов А.И. // ЖОрХ. 1986. Т. 22. С. 1034–1045.
Киселев В.Д., Шакиров И.М., Коновалов А.И. // ЖОрХ. 1984. Т. 20. С. 1454–1459.
Киселев В.Д., Маврин Г.В., Коновалов А.И. // ЖОрХ. 1982. Т. 18. С. 2505–2509.
Киселев В.Д., Хузяшева Д.Г., Коновалов А.И. // ЖОрХ. 1983. Т. 19. С. 1268–1273.
Киселев В.Д., Шакиров И.М., Хузяшева Д.Г., Коновалов А.И. // ЖОрХ. 1983. Т. 19. С. 2064–2068.
Киселев В.Д., Хузяшева Д.Г., Коновалов А.И. // ЖОрХ. 1983. Т. 19. С. 884–885.
Киселев В.Д., Шакиров И.М., Коновалов А.И. // ЖОрХ. 1984. Т. 20. С. 1454–1459.
Киселев В.Д., Шакиров И.М., Коновалов А.И. // ЖОрХ. 1985. Т. 21. С. 1215–1221.
Киселев В.Д., Шакиров И.М., Коновалов А.И. // ЖОрХ. 1985. Т. 21. С. 665–666.
Киселев В.Д., Шакиров И.М., Коновалов А.И. // ЖОрХ. 1986. Т. 22. С. 311–315.
Киселев В.Д., Шакиров И.М., Коновалов А.И. // ЖОрХ. 1986. Т. 22. С. 1034–1045.
Киселев В.Д., Вейсман Е.А., Коновалов А.И. // ЖОХ. 1986. Т. 56. С. 2378–2386.
Киселев В.Д., Кашаева Е.А., Коновалов А.И. // ЖОХ. 2002. Т. 72. С. 1898–1901.
Киселев В.Д., Вейсман Е.А., Коновалов А.И. // ЖОрХ. 1985. Т. 21. С. 1145–1150.
Штырлин Ю.Г., Киселев В.Д., Мурзин Д.Г., Садюкова О.Н., Коновалов А.И. // ЖОрХ. 1993. Т. 29. С. 1713–1718.
Мурзин Д.Г., Штырлин Ю.Г., Садюкова О.Н., Киселев В.Д., Коновалов А.И. // ЖОрХ. 1994. Т. 30. С. 150–151.
Киселев В.Д., Штырлин Ю.Г., Мурзин Д.Г., Коновалов А.И. // Докл. РАН. 1995. Т. 345. С. 64–67.
Shtyrlin Yu.G., Murzin D.G., Iskhakova G.G., Luzano-va N.A., Kiselev V.D., Konovalov A.I. // Tetrahedron. 1998. V. 54. P. 2631–2646. https://doi.org/10.1016/S0040-4020(98)00025-8
Киселев В.Д., Кашаева Е.А., Лузанова Н.А., Коновалов А.И. // ЖОХ. 1997. Т. 67. С. 1449–1454.
Kiselev V.D., Kashaeva E.A., Luzanova N.A., Konova-lov A.I. // Thermochim. Acta. 1997. V. 303. P. 225–228. https://doi.org/10.1016/S0040-6031(97)00272-4
Kiselev V.D., Kashaeva E.A., Iskhakova G.G., Pota-pova L.N., Konovalov A.I. // J. Phys. Org. Chem. 2006. V. 19. P. 179–186. https://doi.org/10.1002/poc.1012
Kiselev V.D. // Mendeleev Commun. 2008. V. 18. P. 59–61. https://doi.org/10.1016/j.mencom.2008.01.022
Kiselev V.D. Lithium and magnesium perchlorate solutions and their influence on the rate and equilibrium of some cycloaddition reactions. In: Matthews L.V. (ed.) Perchlorates: Production, Uses and Health Effects / New York, Nova Science Publishers, 2011. P. 51–82.
Kiselev V.D., Bolotov A.V., Kashaeva H.A., Potapova L.N., Shakirova I.I., Konovalov A.I. // Fluid Phase Equilib. 2011. V. 312. P. 1–6. https://doi.org/10.1016/j.fluid.2011.09.004
Kiselev V.D., Kashaeva H.A., Shakirova I.I., Potapo-va L.N., Konovalov A.I. // J. Sol. Chem. 2012. V. 41. P.1375–1387. https://doi.org/10.1007/s10953-012-9881-9
Киселев В.Д., Заботина О.А., Вейсман Е.А., Коновалов А.И. // ЖОрХ. 1983. Т. 19. С. 1617–1621.
Киселев В.Д., Малков В.Б., Коновалов А.И. // ЖОрХ. 1990. Т. 26. С. 229–240.
Киселев В.Д., Вейсман Е.А., Заботина О.А. // ЖОХ. 1982. Т. 52. С. 333–342.
Киселев В.Д., Коновалов А.И., Вейсман Е.А., Соломонов Б.Н. // ЖОХ. 1985. Т. 55. С. 1965–1969.
Киселев В.Д., Малков В.Б., Коновалов А.И. // ЖОрХ. 1988. Т. 24. С. 1926–1931.
Киселев В.Д., Кашаева Е.А., Коновалов А.И. // ЖОХ. 1998. Т. 68. С. 1309–1311.
Киселев В.Д., Малков В.Б., Коновалов А.И. // ЖОрХ. 1991. Т. 27. С. 922–928.
Kiselev V.D., Kolesnikova A.O., Dinikaev I.F., Shulya-tiev A.A., Klimovitskii A.E., Kornilov D.A. // Int. J. Chem. Kinet. 2021. V. 53. P. 207–212. https://doi.org/10.1002/kin.21434
El’yanov B.S., Gonikberg E.M. // J. Chem. Soc., Faraday Trans. 1. 1979. V. 75. P. 172–191. https://doi.org/10.1039/F19797500172
El’yanov B.S., Vasylvitskaya E.M. // Rev. Phys. Chem. Jpn. 1980. V. 50. P. 169–184.
Iskhakova G.G., Kiselev V.D., Kashaeva E.A., Potapova L.N., Berdnikov E.A., Krivolapov D.B., Litvi-nov I.A. // Arkivoc. 2004. P. 70–79. https://doi.org/10.3998/ark.5550190.0005.c07
Корнилов Д.А., Киселев В.Д., Коновалов А.И. // Изв. АН, Сер. хим. 2017. С. 564–566.
Kiselev V.D. // Int. J. Chem. Kinet. 2013. V. 45. P. 613–622. https://doi.org/10.1002/kin.20800
Kiselev V.D., Bolotov A.V., Satonin A.P., Shakirova I.I., Kashaeva H.A., Konovalov A.I. // J. Phys. Chem. B. 2008. V. 112. P. 6674–6682. https://doi.org/10.1021/jp800513d
Киселев В.Д., Коновалов А.И. // Докл. РАН. 1998. Т. 363. С. 780–782.
Kiselev V.D., Kornilov D.A., Konovalov A.I. // J. Phys. Chem. B. 2014. V. 118. P. 3702–3709. https://doi.org/10.1021/jp501344t
Asano T. High pressure kinetics and highly viscous media. In: van Eldik R., Klärner F.G. (eds.) High Pressure Chemistry: Synthetic, Mechanistic, and Supercritical Applications. Weinheim: Wiley-VCH, 2002, P. 97–128.
Киселев В.Д., Кашаева Е.А., Шихаб М.С., Потапо-ва Л.Н., Исхакова Г.Г. // Изв. АН, Сер. хим. 2004. С. 45–50.
Kiselev V.D. // Int. J. Chem. Kinet. 2010. V. 42. P. 117–125. https://doi.org/10.1002/kin.20462
Jenner G., Papadopoulos M., Rimmelin J. // J. Org. Chem. 1983. V. 48. P. 748–749. https://doi.org/10.1021/jo00153a029
George A.V., Isaacs N.S. // J. Chem. Soc., Perkin Trans. 2. 1985. V. 48. P. 1845–1847. https://doi.org/10.1039/ P29850001845
Киселев В.Д., Коновалов А.И. // Патент РФ № 2161790. 2001.
Киселев В.Д., Коновалов А.И. // Патент РФ № 2188408. 2002.
Киселев В.Д., Кашаева Е.А., Дмитриев В.П., Коновалов А.И. // ЖОХ. 1998. Т. 68. С. 1690–1694.
Kiselev V.D., Kashaeva E.A., Konovalov A.I. // Mendeleev Commun. 1998. V. 8. P. 192–193. https://doi.org/10.1070/MC1998v008n05ABEH000985
Kiselev V.D., Kashaeva E.A., Konovalov A.I. // Tetrahedron. 1999. V. 55. P. 1153–1162. https://doi.org/10.1016/S0040-4020(98)01093-X
Киселев В.Д., Исхакова Г.Г., Шихаб М.С., Конова-лов А.И. // ЖОХ. 2001. Т. 71. С. 1652–1657.
Kornilov D.A., Dinikaev I.F., Kiselev V.D. // High Press. Res. 2019. V. 39. P. 640–654. https://doi.org/10.1080/08957959.2019.1672678
Киселев В.Д., Исхакова Г.Г., Кашаева Е.А., Потапова Л.Н., Коновалов А.И. // Изв. АН, Сер. хим. 2004. С. 2490–2495.
Kiselev V.D., Kolesnikova A.O., Kornilov D.A. // J. Mol. Liq. 2020. V. 311. Article Number 113356. https://doi.org/10.1016/j.molliq.2020.113356
Lide D.R., Frederikse H.P.R. Handbook of Chemistry and Physics, 75th ed. Boca Raton–Ann Arbor–London–Tokyo: CRC Press, 1994–1995.
Ruelle P., Farina-Cuendet A., Kesselring U.W. // J. Am. Chem. Soc. 1996. V. 118. P. 1777–1784. https://doi.org/10.1021/ja953467w
Roy N., Lehn J.-M. // Chem. Asian J. 2011. V. 6. P. 2419–2425. https://doi.org/10.1002/asia.201100244
Kiselev V.D., Shakirova I.I., Kashaeva E.A., Potapova L.N., Kornilov D.A., Krivolapov D.B., Konovalov A.I. // Mendeleev Commun. 2013. V. 23. P. 235–236. https://doi.org/10.1016/j.mencom.2013.07.021
Smith A.L., Walter E., Korobov M.V., Gurvich O.L. // J. Phys. Chem. 1996. V. 100. P. 6775–6780. https://doi.org/10.1021/jp952873z
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Химия, науки о материалах