

Доклады Российской академии наук. Химия, науки о материалах , 2022, T. 507, № 1, стр. 46-58
Адамантил-замещенный триплетный бирадикал: синтез, структура, окислительно-восстановительные и магнитные свойства
М. В. Михайлова 1, Е. М. Дудко 1, 2, Д. И. Насырова 1, 2, А. Я. Акыева 1, М. А. Сыроешкин 1, А. С. Богомяков 3, Н. А. Артюхова 3, М. В. Федин 3, Д. Е. Горбунов 4, Н. П. Грицан 4, Е. В. Третьяков 1, *, академик РАН В. И. Овчаренко 1, 3, академик РАН М. П. Егоров 1
1 Институт органической химии им. Н.Д. Зелинского Российской академии наук
119991 Москва, Россия
2 Национальный исследовательский университет “Высшая школа экономики”
109028 Москва, Россия
3 Институт “Международный томографический центр” Сибирского отделения Российской академии наук
630090 Новосибирск, Россия
4 Институт химической кинетики и горения
им. В.В. Воеводского Сибирского отделения
Российской академии наук
630090 Новосибирск, Россия
* E-mail: tretyakov@ioc.ac.ru
Поступила в редакцию 20.07.2022
После доработки 11.10.2022
Принята к публикации 14.10.2022
- EDN: UYPTZX
- DOI: 10.31857/S2686953522600532
Аннотация
Взаимодействием литиированного 4,4,5,5-тетраметил-4,5-дигидро-1H-имидазол-3-оксид-1-оксила с 1-нитрозоадамантаном с последующим окислением полученного гидроксиламина синтезирован стабильный бирадикал – 2-{N-[(3s,5s,7s)-адамантил-1]-N-оксиламино}-4,4,5,5-тетраметил-4,5-дигидро-1H-имидазол-3-оксид-1-оксил, являющийся гетероатомным аналогом триметиленметана. По данным РСА в парамагнетике значение двугранного угла между плоскостями парамагнитных фрагментов близко к 60°. Магнетохимические измерения показали, что в бирадикале энергетический зазор между триплетным и синглетными состояниями (2J/kB; H = –2JS1/2·S1/2) составляет 880 K. Спектр ЭПР бирадикала моделируется с использованием параметров: S = 1, gxx = 2.007, gyy = 2.005, gzz = 2.006, D = 825 МГц, Е = 60 МГц. По данным циклической вольтамперометрии в анодной области бирадикал подвергается электрохимическому окислению при потенциалах 0.79, 1.63, 1.89 и 2.25 В, причем первый процесс химически и электрохимически обратим. В катодной области бирадикал восстанавливается квазиобратимо при –1.18 и необратимо при –2.59 В.
ВВЕДЕНИЕ
Стабильные органические парамагнетики различного структурного типа находят применение в каталитических процессах, химии полимерных материалов, биологических исследованиях и медицине. Органические радикалы активно используются в дизайне магнетиков на молекулярной основе, устройств спинтроники, магнитноактивных сенсоров, контрастных агентов для магнитнорезонансной томографии, перезаряжаемых электрических батарей [1–6]. При этом в последнее время заметно вырос интерес к изучению и практическому использованию стабильных би- и полирадикалов, в том числе с носителями спина разной природы [7–11]. Последнее стало возможным вследствие развития химии стабильных органических радикалов, в частности соединений нитронилнитроксильного ряда. Так, разработка синтеза разнообразных металлоорганических производных нитронилнитроксилов, изучение их реакционной способности открыли новые пути целенаправленной функционализации атома углерода парамагнитного фрагмента в нитронилнитроксилах [12–15]. К числу наиболее важных методов можно отнести реакции разнообразных электрофилов с литиированным нитронилнитроксилом 2-имидазолинового ряда [16–27], давшие возможность получения и последующего исследования целого ряда высокоспиновых систем, в том числе топологически сходных с каноническими углеводородными бирадикалами (схема 1) [28–34]. В ряду последних можно выделить уникальные двух-спиновые системы TD1 и TD2, обладающие высокоспиновым основным состоянием и большими значениями энергий внутримолекулярного обменного взаимодействия (2J/kB > 700 K).
Схема 1.
Примеры высокоспиновых систем, полученных с использованием литиированного нитронилнитроксила 2-имидазолинового ряда.
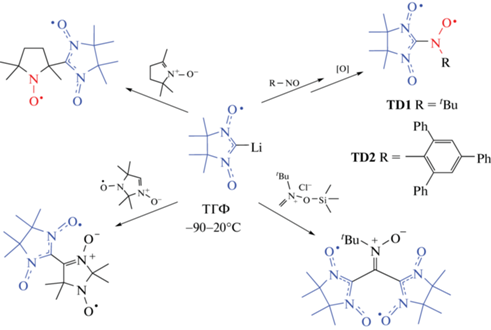
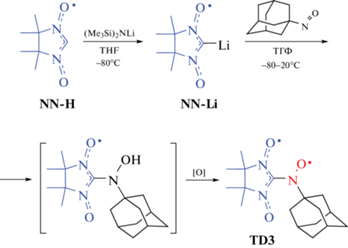
С целью расширения ряда триплетных бирадикалов типа TD с последующей перспективой построения магнитно-структурных корреляций в ходе настоящего исследования синтезирован и полностью охарактеризован стабильный парамагнетик TD3, обладающий топологией триметиленметана и являющийся редким примером парамагнитного производного адамантана.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Синтез TD3 осуществляли путем взаимодействия литиевого производного NN-Li [12] с 1-нитрозоадамантаном с образованием соответствующего продукта присоединения, который в процессе выделения подвергался гидролизу и окислению в целевой парамагнетик TD3 (схема 2).
Бирадикал TD3 менее устойчив, чем трет-бутильное производное TD1, и в растворе, а также при хранении в твердом виде (кристаллы) при –15°C он постепенно разлагается. Тем не менее, используя свежеприготовленные образцы, бирадикал TD3 был полностью охарактеризован как в растворе, так и в кристаллическом состоянии. На рис. 1 приведены экспериментальный и расчетный спектры ЭПР соединения TD3 в стекле толуола при 80 К. Спектр в области g = 2 моделировали с использованием следующего набора параметров: S = 1, gxx = 2.007, gyy = 2.005, gzz = 2.006, D = 825 МГц, Е = 60 МГц; где gii (i = x, y, z) – главные компоненты g-тензора, D и Е – скалярные параметры тензора расщепления в нулевом поле. Дополнительный малоинтенсивный сигнал в центре спектра (отмечен знаком *), не описываемый расчетом, соответствует примеси монорадикала, интегральная интенсивность которого не превышает 10% относительно бирадикала TD3.
Рис. 1.
Спектр стационарного ЭПР Х-диапазона (9.73 ГГц) бирадикала TD3 в стекле толуола при 80 К. 1 – эксперимент, 2 – расчет (см. параметры в тексте; * – примесь монорадикала).

Электрохимические параметры процессов окисления и восстановления TD3 определяли методом циклической вольтамперометрии на стеклоуглеродном дисковом электроде в растворе фонового электролита на основе ацетонитрила. В области окисления бирадикала зарегистрирован ряд пиков при 0.79 (I), 1.63 (II), 1.89 (III) и 2.25 В (IV) (рис. 2). На рис. 2 видно, что пики II, III и IV на кривых 2, 3 и 4 соответствуют химически необратимым процессам. Напротив, пик I (кривая 1 на рис. 2), отвечающий отрыву одного электрона от бирадикала, имеет обратный пик восстановления. Более детальный анализ обратимости данного процесса проведен путем обработки ЦВА кривых, полученных при различных скоростях развертки потенциала: 0.05, 0.1, 0.2, 0.5 и 1 В с–1 (рис. 3). Показано, что отношение токов в обратном и прямом пиках не зависит от скорости развертки потенциала и близко к 1 ($I_{{\text{p}}}^{{{\text{red}}}}{\text{/}}I_{{\text{p}}}^{{{\text{ox}}}}$ = 0.90), а величина интервала между прямым и обратным пиками (ΔЕ = 0.063 В) сопоставима с теоретическим значением для электрохимически обратимых процессов (ΔЕ = 0.059 В [35]). Это свидетельствует о химической и электрохимической обратимости процесса и стабильности образующегося парамагнитного катиона во временном диапазоне эксперимента. Для данного процесса также были определены потенциалы пиков окисления и ответного восстановления без влияния нескомпенсированного сопротивления ($E_{{\text{p}}}^{{{\text{ox}}}}$ = 0.780 В и $E_{{\text{p}}}^{{{\text{red}}}}$ = = 0.717 В) и потенциал полуволны (Е1/2 = 0.749 В).
Рис. 2.
ЦВА-кривые окисления и восстановления TD3 (С = 2 × 10–3 М) в 0.1 M растворе Bu4NBF4 / MeCN на стеклоуглеродном дисковом электроде при скорости развертки потенциала 0.1 В с–1. Кривые 1, 2, 3 и 5 показывают обратимое (пик I), квазиобратимое (пик V) и необратимое (пики II, III) электрохимическое поведение TD3.
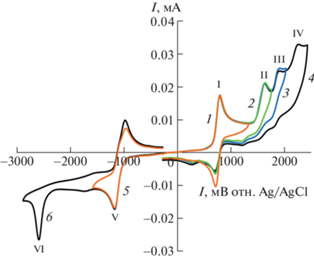
Рис. 3.
(а) ЦВА кривые процесса окисления (пик I) TD3 (С = 2 × 10–3 М) в 0.1 M растворе Bu4NBF4/MeCN на стеклоуглеродном дисковом электроде при скоростях развертки потенциала 0.05, 0.1, 0.2, 0.5 и 1.0 В с–1. (б) Зависимости потенциалов пиков окисления и ответного восстановления от тока в пике для соответствующего процесса. Аппроксимация зависимостей до нулевого тока дает потенциалы пиков окисления и ответного восстановления без влияния нескомпенсированного сопротивления и потенциал полуволны.

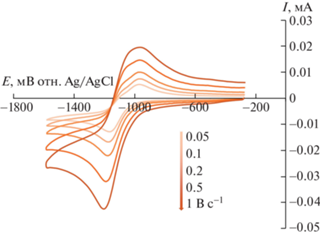
На кривой восстановления TD3 наблюдаются два пика при –1.18 (V) и –2.59 В (VI). Пик VI химически необратим (кривая 6 на рис. 2), пик V имеет ответную волну окисления (кривая 5 на рис. 2). Обратный пик для последнего процесса характеризуется сложной формой, что связано с протеканием двух электрохимических реакций при близких потенциалах. Эти реакции могут соответствовать окислению образовавшегося в прямом процессе парамагнитного аниона и продукта его химического превращения. На ЦВА кривых для пика V, полученных при различных скоростях развертки потенциала (рис. 4), прослеживается рост тока первой реакции по отношению ко второй с увеличением скорости развертки. Это говорит о снижении количества анион-радикальных частиц, подвергшихся химическому превращению, с уменьшением временного промежутка между прямым и обратным пиками.
Рис. 4.
ЦВА-кривые процесса восстановления (пик V) TD3 (С = 2 × 10–3 М) в 0.1 M раствора Bu4NBF4 / MeCN на стеклоуглеродном дисковом электроде при скоростях развертки потенциала 0.05, 0.1, 0.2, 0.5 и 1.0 В с–1.
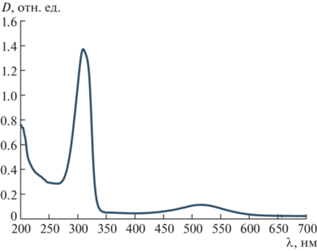
Раствор бирадикала TD3 обладает винным цветом (бургундский цвет), что согласуется с наличием малоинтенсивной полосы поглощения в видимой области спектра с максимумом при 515 нм (рис. 5). Наблюдающаяся в УФ-диапазоне полоса с максимумом при 309 нм имеет гораздо большую интенсивность и, также, как и полоса в видимой области спектра, не имеет разрешенной колебательной подструктуры.
Кристаллы TD3 винно-коричневого цвета, имеющие форму параллелепипедов, получены при медленном испарении раствора бирадикала TD3 в смеси гептан–хлористый метилен. По данным рентгеноструктурного анализа бирадикал TD3 кристаллизуется в нецентросимметричной пространственной группе Pn. Длины связей N–O нитронилнитроксильного фрагмента равны 1.279(2) и 1.270(2) Å, длина связи другой нитроксильной группы (O(3)–N(3)) больше и составляет 1.280(2) Å (рис. 6), что является типичным для парамагнетиков данного типа [36]. В бирадикале TD3 угол между плоскостями ПONCNO и ПHet (табл. 1), характеризующий перегиб имидазолинового цикла, равен 18.1°. Торсионный угол O3–N3–C1–N1 равен 56.2(3)°, тогда как в родственном бирадикале TD1 или в его сольвате с н-гептаном величина данного угла заметно больше и составляет 65.3(3) или 67.1(2)° соответственно (по данным работ [28, 29]). В бирадикале TD2, закристаллизованном в виде сольвата с циклогексаном и содержащем ароматический заместитель при атоме азота нитроксильной группы, величина аналогичного торсионного угла составляет всего 15.1(2)° [34]. Таким образом, в молекуле TD3 величина торсионного угла, определяющего эффективность кросс-сопряжения магнитных орбиталей, оказывается между величинами соответствующих углов в бирадикалах TD1 и TD2. Данное обстоятельство и в целом вся совокупность данных о строении TD1–TD3, являющихся аналогами триметиленметана, высокоценны для построения обсуждаемых ниже магнитно-структурных корреляций.
Рис. 6.
Молекулярная структура бирадикала TD3. Тепловые колебания неводородных атомов показаны в анизотропном приближении (p = 50%).
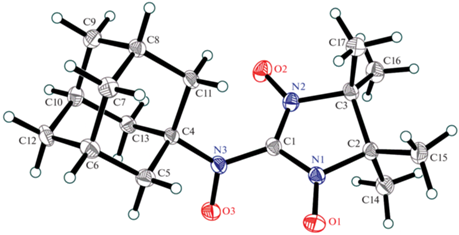
Таблица 1.
Избранные длины связей (l, Å) и углы (∠, град.) в TD3
| Параметр | l, Å | Параметр | ∠, град. |
|---|---|---|---|
| O(1)–N(1) | 1.279(2) | C4–N3–O3 | 120.0(1) |
| O(2)–N(2) | 1.270(2) | O3–N3–C1 | 116.1(2) |
| O(3)–N(3) | 1.280(2) | C4–N3–C1 | 123.2(2) |
| N(1)–C(1) | 1.343(2) | N1–C1–N2 | 111.1(2) |
| N(1)–C(2) | 1.505(2) | C1–N2–O2 | 126.2(2) |
| N(2)–C(1) | 1.344(2) | C1–N1–O1 | 126.2(2) |
| N(2)–C(3) | 1.508(2) | N1–C1–N3–O3 | 56.2(2) |
| N(3)–C(1) | 1.398(2) | ПСNO–ПONCNOа | 60.1 |
| N(3)–C(4) | 1.493(2) | ПONCNO–ПHetб | 18.1 |
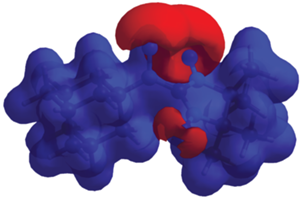
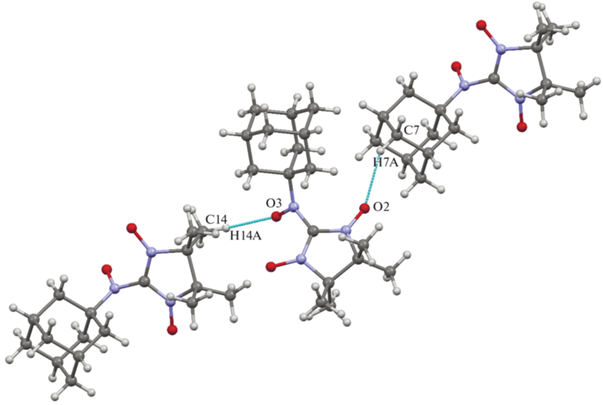
Распределение электронной плотности в молекуле TD3 определяет рассчитанное с помощью программы CrystalExplorer [37] поле электростатического потенциала (рис. 7). Видно, что атомы кислорода, особенно атомы O3 и O1, создают значительное отрицательное электростатическое поле, которое во многом предопределяет характер формирующихся коротких контактов и упаковки бирадикалов в кристалле. Так, с участием атомов кислорода O образуются короткие контакты типа N–O···H–C. Последние включают в себя кон-такты типа O3···H14A–C14 с параметрами l(O3···H14A) = 2.488 Å, ∠(O3···H14A–C14) = 157.47° и короткие контакты типа O2···H7A–C7 с геометрическими параметрами l(O2···H7A) = 2.516 Å, ∠(O2···H7A–C7) = 150.53° (рис. 8).
Рис. 7.
Поле электростатического потенциала молекулы TD3 в кристалле (изоповерхность с потенциалом 3.642 В).
По данным магнитных измерений, для бирадикала TD3 величина произведения мольной магнитной восприимчивости на температуру (χТ) равна 0.95 см3 K моль–1 при 300 K, а при температурах ниже 180 K несколько возрастает, выходя на плато 0.97 см3 K моль–1 (рис. 9). Высокотемпературное значение χТ выше теоретической чисто спиновой величины 0.75 см3 K моль–1 для двух невзаимодействующих парамагнитных центров со спинами S = 1/2 при g-факторе, равном 2. Характер температурной зависимости и величина χТ при температуре ниже 180 K, близкая к теоретической величине 1.0 см3 K моль–1 для триплетного бирадикала с S = 1 и g = 2, указывают на наличие сильных обменных взаимодействий ферромагнитного характера между неспаренными электронами.
Рис. 9.
Температурная зависимость мольной магнитной восприимчивости (χ) для бирадикала TD3 (в координатах χ(Т) × Т от Т).

Для выбора модели анализа температурной зависимости χT была проведена оценка эффективности межмолекулярных обменных каналов с использованием квантово-химических расчетов. Фрагмент кристаллической решетки, представляющий собой пару молекул, между которыми реализуются кратчайшие расстояния между атомами с высокой спиновой плотностью (рис. 10а), показан на рис. 10б. Расчеты обменного взаимодействия между бирадикалами внутри данной пары методом неограниченной по спину теории функционала плотности нарушенной симметрии (BS-DFT) дали очень низкое значение обменного параметра J = –0.13 см–1 (J/kB = –0.19 K). Расчеты обменного взаимодействия в данной паре с использованием более точного и затратного метода CASSCF(4,4)/NEVPT2 с активным пространством, включающим в себя четыре электрона на четырех орбиталях, предсказывают очень слабое антиферромагнитное взаимодействие бирадикалов с J/kB = –0.14 K.
Рис. 10.
Распределение спиновой плотности в основном триплетном состоянии бирадикала TD3 (а) и структура обменно-связанного димера {TD3}2 (б).
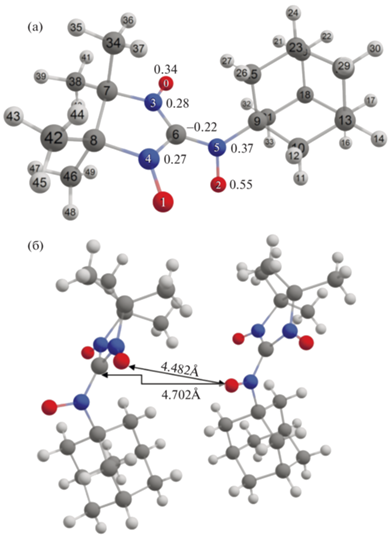
Квантово-химические расчеты показали, что энергия межмолекулярных обменных взаимодействий существенно уступает по своей величине энергии внутримолекулярного обмена. Это позволяет при анализе зависимости χТ(Т) учитывать только обменные взаимодействия внутри бирадикала TD3 и использовать модель Блини–Бауэрса [38] для обменно-связанного димера (H = = –2JS1/2·S1/2). Экспериментальная зависимость χТ(T ) хорошо описывается моделью обменно-связанного димера с оптимальным значением параметра обменного взаимодействия J = 306 ± 5 см–1 (J/kB = 440 ± 7 К).
Известно, что для бирадикалов, однократно заполненные орбитали которых пространственно не разделены, параметры обменных взаимодействий с удовлетворительной точностью удается рассчитывать методом BS-DFT [7]. К такого типа бирадикалам относятся, например, триметилметан и м-ксилилен, а также их гетероатомные производные, в том числе нитроксильные [7, 29]. Действительно, расчет методом BS-DFT (табл. 2) предсказал для TD3 основное триплетное состояние и сильное ферромагнитное взаимодействие с J = 615 см–1 (J/kB = 885 K). Расчет методом CASSCF с достаточно большим активным пространством (10 электронов на семи молекулярных орбиталях, 5 из которых приведены на рис. 11) предсказывает существенно меньшее значение: J = 266 cм–1. Учет динамической электронной корреляции методом NEVPT2 приводит к росту параметра J (табл. 2). Из табл. 2 видно, что оцененное из эксперимента значение параметра J хорошо согласуется с высокоуровневыми расчетами, попадая как раз в промежуток между результатами расчетов CASSCF и NEVPT2. Как это часто бывает [7], метод BS-DFT заметно завышает предсказанное значение J. табл. 3
Таблица 2.
Экспериментальное и рассчитанные различными методами значения параметра обменного взаимодействия J для TD3
| Метод | J, см–1 | J/kB, K |
|---|---|---|
| BS-UB3LYP/def2-TZVP | 615 | 885 |
| CASSCF(10,7)/def2-TZVP | 266 | 383 |
| CASSCF(10,7)/NEVPT2 | 413 | 594 |
| Эксперимент | 306 ± 5 | 440 ± 7 |
Рис. 11.
Пять из семи молекулярных орбиталей активного пространства (МО82–МО88) с указанием их заселенности, использованных в расчетах CASSCF/NEVPT2 для TD3.

Таблица 3.
Кристаллографические данные
| Брутто-формула | C17H27N3O3 | |
|---|---|---|
| Молекулярная масса | 321.41 | |
| Температура эксперимента, К | 100.0(1) | |
| Длина волны, Å | 1.54184 | |
| Сингония | Моноклинная | |
| Пространственная группа | Pn | |
| Параметры кристаллической решетки | a = 6.29632(5) Å | α = 90° |
| b = 12.33545(11) Å | β = 93.3234(7)° | |
| c = 10.97899(8) Å | γ = 90° | |
| Объем, Å3 | 851.282(12) | |
| Z | 2 | |
| Рассчитанная плотность, г · см–3 | 1.254 | |
| Коэффициент поглощения, мм–1 | 0.699 | |
| F(000) | 348 | |
| Диапазон hkl-индексов | –8 ≤ h ≤ 6, –15 ≤ k ≤ 15, –13 ≤ l ≤ 13 | |
| Кол-во собранных отражений | 11500 | |
| Кол-во независимых отражений | 2888 [R(int) = 0.0244] | |
| Кол-во наблюдаемых отражений, [I > 2σ(I)] | 2880 | |
| GOOF | 1.043 | |
| Итоговые значения R-факторов [I > 2σ(I)] | R1 = 0.0313, wR2 = 0.0828 | |
| R-факторы (с учетом всего массива) | R1 = 0.0313, wR2 = 0.0829 | |
| Остаточная электронная плотность, е Å–3 | 0.266 and –0.218 | |
ЗАКЛЮЧЕНИЕ
Таким образом, в ходе настоящего исследования получен и выделен в свободном виде первый адамантил-замещенный триплетный бирадикал с большой величиной энергии синглет-триплетного расщепления. Бирадикал подвергается обратимому электрохимическому окислению при потенциале 0.79 В и квазиобратимому восстановлению при –1.18 В. По данным РСА, в бирадикале значение угла между плоскостями парамагнитных фрагментов составляет ~60°, что обусловливает реализацию сильного внутримолекулярного обменного взаимодействия ферромагнитного характера J/kB = 440 K (H = –2JS1/2·S1/2). Высокая стабильность, обратимое электрохимическое окисление и сильный ферромагнитный обмен открывают возможность применения синтезированного триплетного бирадикала в разнообразных областях наук о материалах и медицины.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
4,4,5,5-Тетраметил-4,5-дигидро-1H-имидазол-3-оксид-1-оксил NN-H синтезировали по известной методике [24]. Коммерческие реактивы и растворители использовали без дополнительной очистки. Ход реакции контролировали с помощью ТСХ на пластинках “Sorblif” (алюминиевая основа). Для колоночной хроматографии использовали силикагель “Silica 60 0.04–0.63 мм для колоночной хроматографии” фирмы “Macherey Nagel”. ИК-спектры образцов записывали на спектрофотометрах “BRUKER Vertex-70 FTIR” и “Vector-22”. Измерение спектров поглощения в УФ и видимом диапазоне проводили на спектрометре Agilent 8453 для 1 × 10–4 М раствора TD3 в ацетонитриле, используя 10 мм кварцевую кювету с тефлоновой крышкой. Растворы готовили в сухом перчаточном боксе. Масс-спектры высокого разрешения регистрировали на приборе “Bruker micrOTOF II” и “DFS Thermo scientific instrument”.
1-Нитрозоадамантан получали по методике, близкой к ранее описанной [39]. Раствор мета-хлорнадбензойной кислоты (70%, 5.6 г, 33.1 ммоль) в этилацетате (10 мл) прибавляли по каплям в течение 30 мин к перемешиваемой смеси 1-аминоадамантана (5.0 г, 33.1 ммоль), эфира (20 мл) и воды (15 мл). По окончании прибавления мета-хлорнадбензойной кислоты реакционную смесь, приобретшую голубой цвет, перемешивали в течение 30 мин, затем переносили в делительную воронку. Органический слой отделяли, водный экстрагировали этилацетатом (2 × 10 мл). Объединенные органические вытяжки промывали 20%-м водным раствором Na2CO3 (3 × 10 мл), 8 N водным раствором HCI (3 × 10 мл) и водой (3 × 10 мл), затем сушили MgSO4 и фильтровали. Раствор упаривали, остаток перекристаллизовывали из смеси эфира с гептаном. Выход 1-нитрозоадамантана 1.6 г (30%), бесцветные кристаллы, т. пл. 150–151°C. Объединенные кислые экстракты подщелачивали NaOH до pH 9–10, экстрагировали эфиром (2 × 10 мл). Экстракты упаривали, сушили MgSO4, фильтровали и упаривали. Остаток перекристаллизовывали из смеси хлористого метилена с гептаном и получали 2.9 г (58%) исходного 1-аминоадамантана.
2-{N-[(3s,5s,7s)-Адамантил-1]-N-оксиламино}-4,4,5,5-тетраметил-4,5-дигидро-1H-имидазол-3-оксид-1-оксил TD3. К интенсивно перемешиваемому при –70– –80°C раствору 4,4,5,5-тетраметил-4,5-дигидро-1H-имидазол-3-оксид-1-оксила (314 мг, 2.0 ммоля) в сухом ТГФ (20 мл) прибавляли 1.0 M раствор LiN(SiMe3)2 в гептане (2.2 мл, 2.2 ммоль) в атмосфере аргона. Реакционную смесь перемешивали при –80°C в течение 25 мин, после чего к ней прибавляли раствор 1-нитрозоадамантана (363 мг, 2.2 ммоль) в сухом ТГФ (5 мл). Охлаждение прекращали и, после того как температура реакционной смеси достигала комнатной, к ней последовательно прибавляли концентрированный водный раствор NH4Cl (5 мл), воду (20 мл) и CH2Cl2 (50 мл). Органический слой отделяли, водный экстрагировали CH2Cl2 (3 × 20 мл). Объединенные органические растворы сушили MgSO4 и фильтровали. Фильтрат охлаждали, прибавляли к нему MnO2 (400 мг, 4.6 ммоль). Реакционную смесь перемешивали в течение 10 мин, фильтровали, упаривали в вакууме. Полученный остаток хроматографировали на колонке с SiO2 (элюент – этилацетат). Фракцию винного цвета упаривали, остаток перекристаллизовывали из смеси гептана с CH2Cl2. Выход 160 мг (25%), кристаллы винно-коричневого цвета, имеющие форму параллелепипеда, разлагаются при нагревании выше 130–140°С; Rf = 0.85 (этилацетат), Rf= 0.17 (CHCl3). ИК (KBr, ν, см–1): 2928, 2912, 2888, 2858, 1730, 1457, 1422, 1376, 1290, 1275, 1221, 1177, 1134, 1111, 1074, 1044, 985, 971, 937, 868, 816, 736, 670, 541. Масс-спектр, m/z: 322.2130 [M + H]+; вычислено для C17H28N3O$_{3}^{ + }$: 322.2125. Найдено, %: C, 63.9; H, 8.4; N, 13.0. Вычислено для C17H27N3O3, %: C, 63.5; H, 8.5; N, 13.1.
Циклическая вольтамперометрия. Приготовление раствора соединения TD3 и все измерения проводили в сухом перчаточном боксе в атмосфере аргона и показателями влажности и кислорода, не превышающими 2 м. д. Электрохимические испытания бирадикала, растворенного в фоновом электролите, проводили в стандартной трехэлектродной стеклянной ячейке при скоростях развертки потенциала 0.05–1 В с–1. Рабочим электродом служил стеклоуглеродный дисковый электрод с диаметром диска 1.7 мм. Перед использованием поверхность электрода полировали наждачной бумагой и затем пастой ГОИ до достижения зеркального блеска. В качестве вспомогательного электрода выступала платиновая проволока, прокаленная в пламени газовой горелки для удаления с поверхности оксидов и других возможных загрязнений. Потенциалы изучаемых процессов измеряли относительно электрода сравнения, представляющего собой серебряную проволоку, покрытую слоем хлорида серебра, отделенную от основного объема электролита электролитическим мостиком, заполненным раствором фонового электролита. Электрод сравнения калибровали относительно пары ферроцен/ферроцений (Е0 = 0.400 В относительно нормального водородного электрода). Фоновый электролит представлял собой 0.1 М раствор Bu4NBF4 (99%, Sigma Aldrich) в ацетонитриле (≥99.9%, HPLC Gradient grade, Fisher Chemical) c содержанием воды не более 20 м. д., согласно титрованию по Карлу Фишеру при использовании титратора Mettler-Toledo Titrator C10SD.
СКВИД-магнетометрия. Магнетохимические измерения проводили на магнетометре MPMSXL (Quantum Design) в диапазоне 2–300 K, парамагнитную составляющую магнитной восприимчивости χ вычисляли с использованием констант Паскаля [40].
Рентгеноструктурное исследование. Рентгеноструктурный анализ монокристаллов бирадикала TD3 выполнен при 100 K на четырехкружном дифрактометре Rigaku Synergy S, оборудованном детектором HyPix6000HE (каппа-геометрия, метод ω-сканирования) с использованием монохроматизированного CuKα-излучения (табл. 2). Интенсивности отражений были установлены и полуэмпирически скорректированы с учетом поглощения излучения кристаллом в программе CrysAlisPro [41]. Структура расшифрована прямыми методами с помощью программы SHELXT [42] и уточнена МНК в полноматричном приближении по F 2 в программном пакете OLEX2 [43] с использованием программы SHELXL-2018 [44]. Все неводородные атомы уточнены в анизотропном приближении. Все атомы водорода были помещены в идеальные расчетные положения и уточнены с учетом относительных изотропных смещений.
Кристаллографические параметры и детали уточнения структур бирадикала TD3 приведены в табл. 2. Структурные данные депонированы в Кембриджском банке структурных данных (CCDC № 2191144, их можно получить по адресу deposit@ccdc.cam.ac.uk или http://www.ccdc.cam. ac.uk/data_request/cif).
ЭПР исследования. Спектр стационарного ЭПР бирадикала TD3 был получен в стекле толуола при 80 К на коммерческом спектрометре Bruker Elexsys E580 в Х-диапазоне (9.73 ГГц) в условиях отсутствия микроволнового насыщения и модуляционного уширения. Моделирование спектра проводилось в среде EasySpin [45].
Квантово-химические расчеты. Для расчета параметров обменного взаимодействия ($\hat {H} = - 2J{{\hat {\vec {S}}}_{1}}{{\hat {\vec {S}}}_{2}}$) использовали неограниченную по спину теорию функционала плотности нарушенной симметрии (BS-DFT) [46]. Параметр обменного взаимодействия J рассчитывали по формуле
Список литературы
Hicks R.G. (Ed.) Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds. Chi-chester: John Wiley and Sons, 2010. 606 p. https://doi.org/10.1002/9780470666975
Ouari O., Gigmes D. (Eds.) Nitroxides: Synthesis, Pro-perties and Applications. London: Royal Society of Chemistry, 2021. 592 p. https://doi.org/10.1039/9781788019651
Likhtenshtein G.I. Nitroxides. Brief History, Fundamentals, and Recent Developments. In: Springer Series in Materials Science. Hull R., Jagadish C., Kawazoe Yo., Kruzic J., Osgood R.M., Parisi J., Pohl U.W., Seong T.-Ye., Uchida S., Wang Z.M. (Eds.). Cham: Springer, 2020. V. 292. 316 p. https://doi.org/10.1007/978-3-030-34822-9
Kumar S., Kumar Yo., Keshri S.K., Mukhopadhyay P. // Magnetochemistry 2016. V. 2. № 4. P. 42. https://doi.org/10.3390/magnetochemistry2040042
Studer A., Tebben L. // Angew. Chem., Int. Ed. 2011. V. 50. № 22. P. 5034–5068. https://doi.org/10.1002/anie.201002547
Veciana J., Arčon D., Deumal M., Inoue K., Kinoshita M., Novoa J.J., Palacio F., Prassides K., Rawson J.M., Rovira C. (Eds.) Structure and Bonding in π-Electron Magnetism. Heidelberg: Springer Berlin, 2001. 208 p. https://doi.org/10.1007/3-540-44684-2
Tretyakov E.V., Ovcharenko V.I., Terent’ev A.O., Kry-lov I.B., Magdesieva T.V., Mazhukin D.G., Gritsan N.P. // Russ. Chem. Rev. 2022. V. 91. № 2. https://doi.org/10.1070/RCR5025
Baumgarten M. High Spin Molecules Directed Towards Molecular Magnets. In: EPR of Free Radicals in Solids. Trends in Methods and Applications. Lund A., Shiotani M. (Eds.). MA: Springer, 2003. P. 491–528. https://doi.org/10.1007/978-1-4757-5166-6_12
Rajca A. Magnetism of Nitroxides. In: Nitroxides: Synthesis, Properties and Applications. Ouari O., Gigmes D. (Eds.). London: Royal Society of Chemistry, 2021. P. 359–391. https://doi.org/10.1039/9781788019651-00359
Abe M. // Chem. Rev. 2013. V. 113. № 9. P. 7011–7088. https://doi.org/10.1021/cr400056a
Baumgarten M. High Spin Organic Molecules. In: Materials and Energy. World Scientific Reference on Spin in Organics. Miller J.S. (Ed.). Singapore: World Scientific Publishing Co Pte Ltd, 2018. V. 4. P. 1–93. https://doi.org/10.1142/9789813230200_0001
Chupakhin O.N., Utepova I.A., Varaksin M.V., Tretya-kov E.V., Romanenko G.V., Stass D.V., Ovcharenko V.I. // J. Org. Chem. 2009. V. 74. № 7. P. 2870–2872. https://doi.org/10.1021/jo900085s
Suzuki S., Nakamura F., Naota T. // Org. Lett. 2020. V. 22. № 4. P. 1350–1354. https://doi.org/10.1021/acs.orglett.9b04655
Suzuki S., Nakamura F., Naota T. // Mater. Chem. Front. 2018. V. 2. № 3. P. 591–596. https://doi.org/10.1039/C7QM00565B
Tanimoto R., Suzuki S., Kozaki M., Okada K. // Chem. Lett. 2014. V. 43. № 5. P. 678–680. https://doi.org/10.1246/cl.131162
Tretyakov E.V., Tolstikov S.E., Romanenko G.V., Bogomyakov A.S., Cherkasov V.K., Stass D.V., Ovcharen-ko V.I. // Russ. Chem. Bull. 2011. V. 60. № 11. P. 2325–2330. https://doi.org/10.1007/s11172-011-0356-8
Zhivetyeva S.I., Zayakin I.A., Bagryanskaya I.Yu., Zay-tseva E.V., Bagryanskaya E.G., Tretyakov E.V. // Tetrahedron. 2018. V. 74. № 28. P. 3924–3930. https://doi.org/10.1016/j.tet.2018.05.075
Tanimoto R., Suzuki S., Kozaki M., Kanzaki Y., Shiomi D., Sato K., Takui T., Tanaka R., Okada K. // ChemistrySelect. 2020. V. 5. № 36. P. 11170–11176. https://doi.org/10.1002/slct.202002927
Tolstikov S.E., Tretyakov E.V., Gorbunov D.E., Zhurko I.F., Fedin M.V., Romanenko G.V., Bogomyakov A.S., Gri-tsan N.P., Mazhukin D.G. // Chem. Eur. J. 2016. V. 22. № 41. P. 14598–14604. https://doi.org/10.1002/chem.201602049
Varaksin M.V., Tretyakov E.V., Utepova I.A., Romanen-ko G.V., Bogomyakov A.S., Stass D.V., Sagdeev R.Z., Ovcharenko V.I., Chupakhin O.N. // Russ. Chem. Bull. 2012. V. 61. № 7. P. 1469–1473. https://doi.org/10.1007/s11172-012-0190-7
Ovcharenko V.I., Chupakhin O.N., Kovalev I.S., Tretyakov E.V., Romanenko G.V., Stass D.V. // Russ. Chem. Bull. 2008. V. 57. № 10. P. 2227–2229. https://doi.org/10.1007/s11172-008-0309-z
Chupakhin O.N., Tretyakov E.V., Utepova I.A., Varak-sin M.V., Romanenko G.V., Bogomyakov A.S., Veber S.L., Ovcharenko V.I. // Polyhedron. 2011. V. 30. № 4. P. 647–653. https://doi.org/10.1016/j.poly.2010.11.029
Gurskaya L., Rybalova T., Beregovaya I., Zaytseva E., Kazantseva M., Tretyakov E.V. // J. Fluor. Chem. 2020. V. 237. P. 109613. https://doi.org/10.1016/j.jfluchem.2020.109613
Tretyakov E.V., Utepova I.A., Varaksin M.V., Tolstikov S.E., Romanenko G.V., Bogomyakov A.S., Stass D.V., Ovcha-renko V.I., Chupakhin O.N. // Arcivoc. 2011. № 8. P. 76–98. https://doi.org/10.3998/ark.5550190.0012.806
Tretyakov E.V., Fedyushin P.A., Panteleeva E.V., Stass D.V., Bagryanskaya I.Yu., Beregovaya I.V., Bogomyakov A.S. // J. Org. Chem. 2017. V. 82. № 8. P. 4179–4185. https://doi.org/10.1021/acs.joc.7b00144
Fedyushin P., Panteleeva E., Bagryanskaya I., Maryunina K., Inoue K., Stass D., Tretyakov E. // J. Fluor. Chem. 2019. V. 217. P. 1–7. https://doi.org/10.1016/j.jfluchem.2018.10.016
Fedyushin P., Gurskaya L., Panteleeva E., Koshcheev B., Maksimov A., Rybalova T.V., Zaytseva E., Tretyakov E. // Fluorine Notes. 2019. V. 123. P. 7–8. https://doi.org/10.17677/fn20714807.2019.02.04
Suzuki S., Furui T., Kuratsu M., Kozaki M., Shiomi D., Sato K., Takui T., Okada K. // J. Am. Chem. Soc. 2010. V. 132. № 45. P. 15908–15910. https://doi.org/10.1021/ja107769z
Tretyakov E.V., Tolstikov S.E., Romanenko G.V., Bogomyakov A.S., Stass D.V., Maryasov A.G., Gritsan N.P., Ovcharenko V.I. // Russ. Chem. Bull. 2011. V. 60. № 12. P. 2608–2612. https://doi.org/10.1007/s11172-011-0400-8
Misochko E.Ya., Korchagin D.V., Akimov A.V., Masitov A.A., Tolstikov S.E., Tretyakov E.V., Ovcharenko V.I. // J. Phys. Chem. A. 2013. V. 117. № 33. P. 8065–8072. https://doi.org/10.1021/jp405572n
Tsujimoto H., Suzuki S., Kozaki M., Shiomi D., Sato K., Takui T., Okada K. // Chem. Asian J. 2019. V. 14. № 10. P. 1801–1806. https://doi.org/10.1002/asia.201801615
Tolstikov S., Tretyakov E., Fokin S., Suturina E., Romanenko G., Bogomyakov A., Stass D., Maryasov A., Fedin M., Gritsan N., Ovcharenko V. // Chem. Eur. J. 2014. V. 20. № 10. P. 2793–2803. https://doi.org/10.1002/chem.201302681
Shundrin L.A., Irtegova I.G., Vasilieva N.V., Tretyakov E.V., Zueva E.M., Ovcharenko V.I. // Tetrahedron Lett. 2015. V. 56. № 10. P. 1207–1210. https://doi.org/10.1016/j.tetlet.2015.01.134
Kumagai T., Suzuki S., Kanzaki Yu., Shiomi D., Sato K., Takui T., Tanaka R., Okada K., Kozaki M. // Chemistry Lett. 2022. V. 51. № 4. P. 458–460. https://doi.org/10.1246/cl.220021
Bard A.J., Faulkner L.R. Electrochemical Methods: Fundamentals and Applications, 2nd Edition. New York: John Wiley and Sons, NY, 2001. 864 p.
Tretyakov E.V., Ovcharenko V.I. // Russ. Chem. Rev. 2009. V. 78. № 11. P. 971–1012. https://doi.org/10.1070/rc2009v078n11abeh004093
Spackman P.R., Turner M.J., McKinnon J.J., Wolff S.K., Grimwood D.J., Jayatilaka D., Spackman M.A. // J. Appl. Cryst. 2021. V. 54. № 3. P. 1006–1011. https://doi.org/10.1107/S1600576721002910
Bleaney B., Bowers K.D. // Proc. Roy. Soc. 1952. V. 214. P. 451–465. https://doi.org/10.1098/rspa.1952.0181
Greer M.L., Sarker H., Mendicino M.E., Blackstock S.C. // J. Am. Chem. Soc. 1995. V. 117. № 42. P. 10460–10467. https://doi.org/10.1021/ja00147a007
Carlin R.L. Magnetochemistry. Berlin: Springer-Verlag, 1986. 339 p. https://doi.org/10.1007/978-3-642-70733-9
CrysAlisPro. Version 1.171.41. Rigaku Oxford Diffraction, 2021.
Sheldrick G.M. // Acta Crystallogr., Sect. A: Found. Crystallogr. 2015. V. 71. P. 3–8. https://doi.org/10.1107/S2053273314026370
Dolomanov O.V., Bourhis L.J., Gildea R.J., Howard J.A.K., Puschmann H. // J. Appl. Cryst. 2009. V. 42. № 2. P. 339–341. https://doi.org/10.1107/S0021889808042726
Sheldrick G.M. // Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2015. V. 71. P. 3–8. https://doi.org/10.1107/S2053229614024218
Stoll S., Schweiger A. // J. Magn. Reson. 2006. V. 178. P. 42–55. https://doi.org/10.1016/j.jmr.2005.08.013
Nudlemann L., Case D.A., Mouesca J.M. // Coord. Chem. Rev. 1995. V. 144. P. 199–244. https://doi.org/10.1016/0010-8545(95)07011-L
Soda T., Kitagawa Y., Onishi T., Takano Y., Shigeta Y., Nagao H., Yoshioka Y., Yamaguchi K. // Chem. Phys. Lett. 2000. V. 319. № 3–4. P. 223–230. https://doi.org/10.1016/S0009-2614(00)00166-4
Becke A.D. // J. Chem. Phys. 1993. V. 98. № 7. P. 5648–5652. https://doi.org/10.1063/1.464913
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. № 2. P. 785–789. https://doi.org/10.1103/PhysRevB.37.785
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. № 18. P. 3297–3305. https://doi.org/10.1039/B508541A
Frisch M., Ragazos I.N., Robb M.A., Schlegel B.H. // Chem. Phys. Lett. 1992. V. 189. № 6. P. 524–528. https://doi.org/10.1016/0009-2614(92)85244-5
Andersson K., Malmqvist P.A., Roos B.O. // J. Chem. Phys. 1992. V. 96. № 2. P. 1218–1226. https://doi.org/10.1063/1.462209
Angeli C., Cimiraglia R., Evangelisti S., Leininger T., Malrieu J.P. // J. Chem. Phys. 2001. V. 114. № 23. P. 10252–10264. https://doi.org/10.1063/1.1361246
Neese F. // Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017. V. 8. e1327. https://doi.org/10.1002/wcms.1327
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Химия, науки о материалах