

Доклады Российской академии наук. Науки о жизни, 2020, T. 491, № 1, стр. 141-145
НОВЫЙ ФУНКЦИОНАЛЬНЫЙ АНТАГОНИСТ РЕЦЕПТОРА ТИРЕОТРОПНОГО ГОРМОНА НА ОСНОВЕ ТИЕНО-[2,3-d] ПИРИМИДИНА
К. В. Деркач 1, А. А. Бахтюков 1, В. Н. Сорокоумов 2, А. О. Шпаков 1, *
1 Федеральное государственное бюджетное учреждение науки Институт эволюционной физиологии и биохимии им. И.М. Сеченова Российской академии наук
Санкт-Петербург, Россия
2 Федеральное государственное бюджетное образовательное учреждение высшего образования “Санкт-Петербургский государственный университет”
Санкт-Петербург, Россия
* E-mail: alex_shpakov@list.ru
Поступила в редакцию 06.02.2020
После доработки 06.02.2020
Принята к публикации 06.02.2020
Аннотация
Антагонисты рецептора тиреотропного гормона (ТТГ) необходимы для лечения ТТГ-зависимых опухолей и болезни Грейвса. Нами разработано соединение 5-амино-N-(трет-бутил)-4-(4-иодфенил)-2-(метилтио)тиено[2,3-d]пиримидин-6-карбоксамид (TP48) и показано, что оно снижает стимулированную ТТГ активность аденилатциклазы в тироидальных мембранах крысы. Предварительная обработка крыс соединением TP48 (в/б, 40 мг/кг) снижает повышение уровней общего и свободного тироксина в крови и повышение экспрессии генов тиреоглобулина и D2-дейодиназы в щитовидной железе, ответственных за синтез тиреоидных гормонов, которые вызываются интраназальным введением животным тиролиберина (300 мкг/кг). Эти данные указывают на то, что соединение TP48 является функциональным антагонистом рецептора ТТГ и может быть использовано для коррекции тиреоидного статуса при гипертиреозе.
Разработка препаратов, позволяющих снизить активность рецептора тиреотропного гормона (ТТГ) и предотвратить развитие ТТГ-зависимых опухолей и гипертиреоидных состояний, включая болезнь Грейвса, представляет собой одну из актуальных проблем современной эндокринологии и онкологии. Основными причинами ТТГ-зависимых опухолей являются активирующие мутации в рецепторе ТТГ и сопряженных с ним Gs-белках и длительное повышение уровня ТТГ в крови, характерное для гипотиреоза [1], в то время как болезнь Грейвса вызывается гиперактивацией рецептора ТТГ специфичными к нему стимулирующими антителами [2]. Наибольшие перспективы при создании антагонистов и инверсионных агонистов рецептора ТТГ связывают с низкомолекулярными лигандами аллостерического сайта рецептора, локализованного внутри образованного семью гидрофобными спиралями трансмембранного канала [3–5]. Низкомолекулярные лиганды способны ингибировать не только стимулированную ТТГ или стимулирующими антителами активность рецептора, но и снижать его базальную активность, перманентно повышенную при некоторых типах ТТГ-зависимых опухолей.
В отличие от низкомолекулярных лигандов, ТТГ с высокой аффинностью связывается с ортостерическим сайтом, расположенным во внеклеточном домене рецептора ТТГ [6]. Связывание ТТГ с рецептором в тироцитах приводит к стимуляции фермента аденилатциклазы (АЦ) через посредство Gs-белков и повышению внутриклеточного уровня цАМФ, а также к активации фосфолипазы Сβ и кальций-зависимых путей через посредство Gq/11-белков. Результатом этого является стимуляция продукции тироксина и повышение экспрессии генов белков, вовлеченных в синтез тиреоидных гормонов [4, 6]. Среди таких белков тиреоглобулин, являющийся поставщиком тирозина для синтеза тиреоидных гормонов, Na+/I–-котранспортер, доставляющий необходимый для синтеза тиреоидных гормонов йод в тироциты, и ферменты тиреопероксидаза, катализирующая йодирование тирозина в тиреоглобулине, и D2-дейодиназа, осуществляющая конверсию тироксина в 3,5,3'-трийодтиронин, эффекторный гормон тиреоидной оси.
Ранее на основе структуры тиено-[2,3-d]пиримидина нами и другими авторами были разработаны регуляторы рецептора лютеинизирующего гормона (ЛГ) [7–9], чей аллостерический сайт сходен с таковым рецептора ТТГ [10]. Группой Марвина Гершенгорна на основе тиено-[2,3-d]пиримидина был создан антагонист рецептора ТТГ с52, подавляющий ТТГ-индуцированную продукцию тиреоидных гормонов в тироцитах, но при этом влияющий на активность рецептора ЛГ [11]. Цель исследования состояла в разработке и изучении нового тиено-[2,3-d]пиримидинового производного 5-амино-N-(трет-бутил)-4-(4-йодфенил)-2-(метилтио)тиено[2,3-d]пиримидин-6-карбоксамида (TP48) c активностью функционального антагониста рецептора ТТГ, способного ингибировать базальную и стимулированную тиролиберином продукцию тиреоидных гормонов у самцов крыс.
TP48 получали добавлением к раствору 5-амино-4-(4-йодфенил)-(метилтио)тиено[2,3-d]пиримидин-6-карбоновой кислоты (2 ммоль) в диметилформамиде (10 мл) раствора трет-бутиламина (4 ммоль) и диизопропилэтиламина (4 ммоль) в дихлорметане (10 мл) с последующей обработкой 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламмоний-тетрафторборатом (2.4 ммоль). После перекристаллизации TP48 очищали на силикагеле в системе дихлорметан–ацетон (20:1). Выход составил 75%, т. пл. 193–195°С. Спектр ЯМР 1H, δ (400 MHz, СDCl3): 7.91 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 5.94 (s, 2H), 5.25 (s, 1H), 2.66 (s, 3H), 1.47 (s, 9H). По данным масс-спектрометрии масса иона [M + K+] для TP48 составила 536.9868 (вычислено 536.9886; C18H19IN4OS2K). Спектры 1H-ЯМР регистрировали с помощью спектрометра “Bruker Avance III 400” (Германия), масс-спектры высокого разрешения – с помощью спектрометра “Bruker micrOTOF” (Германия).
Для экспериментов использовали трехмесячных самцов крыс Wistar, все процедуры проводили в соответствии с правилами, разработанными Комитетом по биоэтике ИЭФБ РАН (15.02.2018 г.), и правилами и требованиями “European Communities Council Directive 1986” (86/609/EEC).
Выделение фракций плазматических мембран из семенников и щитовидной железы крыс и определение в них активности АЦ (АТФ-пирофосфатлиазы циклизующей, НФ 4.6.1.1) проводили, как описано ранее [9, 12]. Мембраны (50–100 мкг белка) инкубировали в течение 12 мин при 37°С в смеси, содержащей 50 мМ Tris-HCl (pH 7.5), 5 мМ MgCl2, 0.1 мМ цАМФ, 1 мМ АТФ, 37 КБк [α-32P]АТФ, 20 мМ креатинфосфата, 0.2 мг/мл креатинфосфокиназы. Радиоактивность измеряли на счетчике “LKB 1209/1215 RackBeta” (Швеция). Активность АЦ выражали в пмоль цАМФ/мин/мг белка. TP48 растворяли в диметилсульфоксиде (ДМСО), в контрольные пробы добавляли растворитель. Преинкубацию мембран с TP48 проводили в течение 10 мин при +4°С для обеспечения его связывания с рецептором ТТГ.
Для изучения активности TP48 в условиях in vivo его растворяли в ДМСО и вводили крысам (в/б, 40 мг/кг). Контрольным крысам вводили ДМСО. Тиролиберин («Sigma», США) вводили интраназально в дозе 300 мкг/кг, как описано ранее [13]. Первый забор крови осуществляли в 10.00, в 10.30 крысам групп TP и TP-TRH в/б вводили TP48 (группам К и TRH – ДМСО), в 11.00 крысам групп TRH и TP-TRH и/н вводили тиролиберин (группам К и TP – физиологический раствор), в 12.30 и 14.00 проводили второй и третий заборы крови (в каждой группе n = 6). Образцы крови получали из хвостовой вены, используя местный наркоз (анестезия 2% раствором лидокаина, 2–4 мг/кг). Уровни свободного (fТ4) и общего (tТ4) тироксина и свободного (fТ3) и общего (tТ3) трийодтиронина в крови определяли с помощью наборов “Иммунотех” (Россия), уровень тестостерона – с помощью набора “Алкор-Био” (Россия).
Для проведения ПЦР в реальном времени из щитовидной железы крыс выделяли тотальную РНК, используя TRI-реагент (“Molecular Research Center, Inc.”, США). Обратную транскрипцию проводили, используя набор “RevertAid H Minus First Strand cDNA Synthesis Kit”. Амплификацию проводили в смеси, содержащей 10 нг ПЦР-продукта, по 0.4 мкМ прямого и обратного праймеров, qPCRmix-HS SYBR+LowROX (“Евроген”, Россия). Сигнал детектировали с помощью амплификатора “7500 Real-Time PCR System” (“Thermo Fisher Scientific Inc.”, США). В качестве референсных использовали гены 18S-рРНК (18S rRNA) и β-актина (Actb). Полученные данные рассчитывали с помощью метода delta-delta Ct.
Статистический анализ данных осуществляли, используя программу “Microsoft Office Excel 2007”, нормальность распределения проверяли с помощью критерия Шапиро–Уилка. Для сравнения двух выборок с нормальным распределением использовали t-критерий Стьюдента, для сравнения трех групп – дисперсионный анализ с поправкой Бонферрони. Статистически значимыми считали различия при p < 0.05, данные представлены как M ± SD.
В экспериментах in vitro показано, что преинкубация с TP48 (10–6 и 10–4 М) фракций мембран, выделенных из щитовидной железы крыс, не влияла на базальную активность АЦ, но ингибировала стимулирующий эффект ТТГ (10–8 М). В отсутствие TP48 прирост активности АЦ, вызываемый ТТГ, составил 57.7 ± 1.6 пмоль цАМФ/мин/мг белка, после преинкубации с 10–6 и 10–4 М TP48 снижался до 42.3 ± 1.2 и 14.1 ± 0.8 пмоль цАМФ/мин/мг белка соответственно (P < 0.05). Соединение TP48 не влияло на базальную и стимулированную хорионическим гонадотропином (10–8 М), агонистом рецептора ЛГ, активность АЦ в тестикулярных мембранах крыс (данные не представлены). Таким образом, TP48 ингибирует АЦ эффект ТТГ в тироидальных мембранах, препятствуя гормональной активации рецептора ТТГ и функционируя, тем самым, как его антагонист, но не влияет на активность рецептора ЛГ в тестикулярных мембранах, что указывает на специфичность его действия.
Введение самцам крыс TP48 через 3.5 ч приводило к снижению уровней fT4 и fT3, что указывает на его способность подавлять базальную продукцию тиреоидных гормонов, стимулированную циркулирующим в крови животных эндогенным ТТГ (табл. 1). Наряду со снижением базального уровня тиреоидных гормонов, через 3.5 ч после обработки TP48 в ткани щитовидной железы снижалась экспрессия гена Nis, кодирующего Na+/I–-котранспортер, и подавлялась экспрессия гена Tshr, кодирующего рецептор ТТГ (рис. 1).
Таблица 1.
Влияние обработки крыс соединением TP48 на базальные и стимулированные тиролиберином уровни свободного (fT4) и общего (tT4) тироксина и свободного (fT3) и общего (tT3) трийодтиронина в крови
| Группа | fT4, пмоль/л | tT4, нмоль/л | fT3, пмоль/л | tT3, нмоль/л |
|---|---|---|---|---|
| До начала обработки | ||||
| К | 40.7 ± 1.6 | 80.2 ± 4.1 | 4.2 ± 0.2 | 4.6 ± 0.3 |
| TP | 39.3 ± 1.0 | 82.7 ± 2.9 | 3.9 ± 0.2 | 4.2 ± 0.3 |
| TRH | 41.5 ± 1.8 | 83.8 ± 4.5 | 4.3 ± 0.2 | 4.0 ± 0.2 |
| TP+TRH | 40.1 ± 1.4 | 82.5 ± 3.0 | 4.1 ± 0.3 | 4.0 ± 0.2 |
| Через 2 ч после введения ДМСО (K, TRH) или TP48 (TP, TP+TRH) и через 1.5 ч после введения физиологического раствора (K, TP) или тиролиберина (TRH, TP+TRH) | ||||
| К | 39.4 ± 1.4 | 81.7 ± 2.9 | 4.0 ± 0.3 | 4.3 ± 0.3 |
| TP | 36.4 ± 1.3 | 72.8 ± 3.9 | 3.1 ± 0.3 | 2.9 ± 0.3* |
| TRH | 50.4 ± 2.0* | 110.7 ± 5.0* | 5.7 ± 0.4* | 6.1 ± 0.3* |
| TP+TRH | 45.3 ± 1.9 | 99.3 ± 5.2* | 5.3 ± 0.3* | 5.7 ± 0.3* |
| Через 3.5 ч после введения ДМСО (K, TRH) или TP48 (TP, TP+TRH) и через 3 ч после введения физиологического раствора (K, TP) или тиролиберина (TRH, TP+TRH) | ||||
| К | 38.8 ± 1.5 | 79.3 ± 3.9 | 4.4 ± 0.2 | 4.2 ± 0.3 |
| TP | 32.2 ± 1.2* | 66.2 ± 5.7 | 2.2 ± 0.2* | 3.2 ± 0.4 |
| TRH | 57.0 ± 1.8* | 128.3 ± 6.3* | 6.3 ± 0.3* | 6.2 ± 0.2* |
| TP+TRH | 45.9 ± 1.8*# | 105.0 ± 5.9*# | 5.2 ± 0.4 | 5.3 ± 0.4 |
Рис. 1.
Влияние соединения TP48 на экспрессию генов белков, вовлеченных в синтез тиреоидных гормонов, в щитовидной железе крыс в отсутствие и при их обработке тиролиберином. 1 – контроль (К), 2 – обработка TP48, в/б, 40 мг/кг (TP), 3 – обработка тиролиберином, и/н, 300 мкг/кг (TRH), 4 – последовательная обработка TP48 и тиролиберином (TP+TRH).
Гены Tg, TPO, Dio2, Nis и TshR кодируют тиреоглобулин, тиреопероксидазу, D2-дейодиназу, Na+/I–-котранспортер и рецептор ТТГ. Уровень экспрессии мРНК генов нормировали по экспрессии мРНК генов 18S rRNA и Actb. Значения RQ рассчитывали по отношению к группе K. * – различия между контролем и группами TP, TRH и TP+TRH статистически значимы при P < 0.05; # – различия между группами TRH и TP+TRH статистически значимы при P < 0.05. M ± SEM. n = 6.
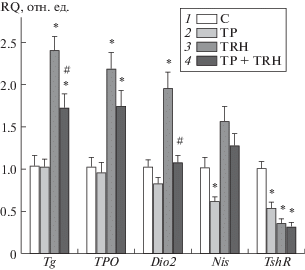
Обработка крыс тиролиберином, стимулирующим продукцию ТТГ тиреотрофами гипофиза, повышала уровни всех функционально важных форм тиреоидных гормонов, в наибольшей степени через 3 ч, и вызывала значительное повышение экспрессии генов Tg, TPO и Dio2, кодирующих тиреоглобулин, тиреопероксидазу и D2-дейодиназу, и снижение экспрессии гена Tshr в щитовидной железе. Отмечали также тенденцию к повышению экспрессии гена Nis, но различия с контролем не были статистически значимыми (табл. 1, рис. 1). Предварительная обработка крыс с помощью TP48 снижала стимулированные тиролиберином уровни тироксина и экспрессию генов Tg и Dio2 при сохранении низкого уровня экспрессии гена Tshr (табл. 1, рис. 1). Ослабление экспрессии ряда генов тиреоидогенеза при обработке крыс ТP48 указывает на то, что чувствительность тироцитов к стимулирующему влиянию ТТГ снижается, поскольку ТТГ является основным позитивным регулятором экспрессии этих генов [14]. Снижение ответа на ТТГ обусловлено как присущей TP48 активностью функционального антагониста рецептора ТТГ, так и вызываемым им снижением экспрессии гена Tshr. Уровень тестостерона при обработке TP48 не менялся, что указывает на отсутствие его влияния на рецепторы ЛГ в семенниках крыс (данные не представлены).
Таким образом, на основе структуры тиено-[2,3-d]пиримидина разработано новое соединение TP48 с активностью функционального антагониста рецептора ТТГ, которое in vitro снижало стимулированную ТТГ активность АЦ в тироидальных мембранах, при введении крысам снижало у них базальные и стимулированные тиролиберином уровни тиреоидных гормонов, а также подавляло экспрессию генов тироидогенеза в щитовидной железе.
Список литературы
Rowe C.W., Paul J.W., Gedye C., et al. // Endocr. Relat. Cancer. 2017. V. 24. P. R191–R202. https://doi.org/10.1530/ERC-17-0010
Rapoport B., McLachlan S.M. // Thyroid. 2007. V. 17. P. 911–922.
Neumann S., Gershengorn M.C. // Ann. Endocrinol. (Paris). 2011. V. 72. P. 74–76. https://doi.org/10.1016/j.ando.2011.03.002
Nunez Miguel R., Sanders J., et al. // Auto Immun. Highlights. 2017. V. 8. P. 2. https://doi.org/10.1007/s13317-016-0090-1
Marcinkowski P., Hoyer I., Specker E., et al. // Thyroid. 2019. V. 29. P. 111–123. https://doi.org/10.1089/thy.2018.0349
Kleinau G., Worth C.L., Kreuchwig A., et al. // Front. Endocrinol. (Lausanne). 2017. V. 8. P. 86. https://doi.org/10.3389/fendo.2017.00086
van Koppen C.J., Zaman G.J., Timmers C.M., et al. // Naunyn Schmiedebergs Arch. Pharmacol. 2008. V. 378. P. 503–514. https://doi.org/10.1007/s00210-008-0318-3
Nataraja S.G., Yu H.N., Palmer S.S. // Front. Endocrinol. (Lausanne). 2015. V. 6. P. 142. https://doi.org/10.3389/fendo.2015.00142
Derkach K.V., Dar’in D.V., Bakhtyukov A.A., et al. // Biochemistry (Moscow). Suppl. Ser. A: Memb. Cell. Biol. 2016. V. 10. P. 294–300. https://doi.org/10.1134/S1990747816030132
Hoyer I., Haas A.K., Kreuchwig A., et al. // Biochem. Soc. Trans. 2013. V. 41. P. 213–217. https://doi.org/10.1042/BST20120319
Neumann S., Kleinau G., Costanzi S., et al. // Endocrinology. 2008. V. 149. P. 5945–5950. https://doi.org/10.1210/en.2008-0836
Shpakov A.O., Shpakova E.A., Tarasenko I.I., et al. // Cell Tissue Biol. 2014. V. 8. P. 488–498. https://doi.org/10.1134/S1990519X1406008X
Derkach K.V., Bogush I.V., Berstein L.M., et al. // Horm. Metab. Res. 2015. V. 47. P. 916–924. https://doi.org/10.1055/s-0035-1547236
Carvalho D.P., Dupuy C. // Mol. Cell. Endocrinol. 2017. V. 458. P. 6–15. https://doi.org/10.1016/j.mce.2017.01.038
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Науки о жизни