

Доклады Российской академии наук. Науки о жизни, 2020, T. 492, № 1, стр. 287-292
СТРУКТУРНО-ФУНКЦИОНАЛЬНЫЕ ПОВРЕЖДЕНИЯ ФИБРИНОГЕНА В РЕЗУЛЬТАТЕ ПЕРОКСИД-ИНДУЦИРОВАННОГО ОКИСЛЕНИЯ
Л. В. Юрина 1, *, А. Д. Васильева 1, В. Л. Кононенко 1, А. Е. Бугрова 1, М. И. Индейкина 1, 3, А. С. Кононихин 2, член-корреспондент РАН Е. Н. Николаев 4, М. А. Розенфельд 1
1 ФГБУН Институт биохимической физики
им. Н.М. Эмануэля РАН
Москва, Россия
2 ФИЦ ИХФ им. Семенова РАН, Институт энергетических проблем химической физики
им. В.Л. Тальрозе РАН
Москва, Россия
3 ФГАОУ ВО “Московский физико-технический институт (государственный университет)”
Москва, Россия
4 АНОО ВПО “Сколковский институт науки
и технологий”
Сколково, Московская обл., Россия
* E-mail: lyu.yurina@gmail.com
Поступила в редакцию 11.02.2020
После доработки 11.02.2020
Принята к публикации 11.02.2020
Аннотация
Исследовано влияние пероксид-индуцированного окисления фибриногена на модификацию его первичной структуры и функциональных свойств. Показано, что сайтами окисления являются остатки Met, Trp и His. Методом ДРС установлено, что окислительная модификация фибриногена вызывает изменение микрореологических характеристик фибриновой сетки. Окисление фибриногена обусловливает снижение его толерантности к плазминовому гидролизу и нарушает способность фактора XIIIa к стабилизации фибринового геля.
Фибриноген (ФГ) является одним из белков плазмы крови, наиболее подверженных окислительной модификации. ФГ играет важную роль в процессе свертывания крови, фибринолизе, клеточных и матричных взаимодействиях, воспалительных процессах, заживлении ран и неоплазии.
Окисление ФГ повреждает его структуру, а также влияет на функцию белка. Известно, что окислительная модификация молекул ФГ человека вызывает изменение процесса самосборки фибрина, что в конечном итоге приводит к аномальным сгусткам. Они характеризуются тонкими индивидуальными фибриновыми фибриллами, малой пористостью и низкой проницаемостью, то есть считается, что окисление влияет на самосборку фибрина преимущественно за счет ингибирования латеральной ассоциации протофибрилл [1].
Ранее нами были исследованы окислительные модификации молекулы при её озон- и гипохлорит-индуцированном окислении [2, 3]. Полученные данные демонстрировали возможность структуры ФГ сохранять функционально важные аминокислотные остатки при окислении, что является следствием, как мы полагаем, большого количества остатков метионинов, рассматриваемых в белках, как перехватчики АФК [4]. Это позволило нам сделать вывод о том, что структура ФГ адаптирована к действию АФК. Чтобы получить дополнительные доказательства этой гипотезы, в данной работе, исследовалась окислительная модификация ФГ под действием перекиси водорода (Н2О2), являющейся одним из важнейших окислителей, вырабатывающихся в организме человека.
ФГ был выделен из цитратной плазмы крови доноров методом глицинового осаждения [5]. Окисление ФГ индуцировали раствором H2O2 (“Sigma-Aldrich”, США) в концентрации 150, 300, 600 и 1000 мкМ. После окисления ФГ проводили электрофорез восстановленных образцов белка, а также его ковалентносшитых полипептидных цепей под действием активированного коагуляционного фактора XIII (FXIIIа). Окислительные повреждения молекулы ФГ также оценивались по изменению накопления продуктов деградации плазминового гидролиза [6].
Окислительные сайты выявляли с по-мощью хромато-масс-спектрометрического анализа (ВЭЖХ-МС/МС) на системе, состоящей из хроматографа Agilent 1100 с устройством автоматического отбора проб (Agilent Technologies Inc., Santa Clara, USA) и тандемного масс-спектрометра 7T LTQ-FT Ultra (Thermo, Bremen, Germany) [7]. При подготовке проб образцы обрабатывались дитиотреитолом (DTT) для восстановления дисульфидных связей c последующим алкилированием йодоацетоамидом и гидролизом трипсином (Promega, USA). Триптические пептиды были идентифицированы с помощью программного обеспечения PEAKS Studio (V. 8.5, Bioinformatics Solutions Inc., Waterloo, On, Canada). Все эксперименты повторялись трижды. При сравнении результатов учитывались аминокислоты, не окисленные в контроле, а также те, уровень окисления которых по сравнению с контролем, возрастал более чем на 1%.
Микрореологические характеристики фибриновых гелей, образованных под действием тромбина из нативного и окисленного ФГ изучали методом динамического рассеяния света (ДРС) на приборе Malvern Zetasizer Nano S. Регистрировали корреляционные функции (КФ) g(2)(τ) флуктуаций интенсивности излучения 633 нм, рассеиваемого фибриновым гелем под углом 173°, в диапазоне времени задержки τ = 0.5–4 × 106 мксек с помощью программы Zetasizer в форме [g(2)(τ) – 1].
На рис. 1а представлены результаты электрофореза полипептидных цепей нативного и окисленного ФГ, которые свидетельствуют о том, что, независимо от концентрации окислителя, не наблюдалось ни фрагментации белка, ни образования ковалентных сшивок его цепей.
Рис. 1.
Электрофорез различных образцов ФГ.
(а) Электрофорез полипептидных цепей ФГ в полиакриламидном геле (5% – концентрирующий гель, 12% – разделяющий гель) в присутствии додецилсульфата натрия. 1 – неокисленный ФГ, 2 – окисленный 150 мкМ H2O2, 3 – окисленный 300 мкМ H2O2, 4 – окисленный 600 мкМ H2O2, 5 – окисленный 1000 мкМ H2O2.
(б) Электрофорез продуктов реакции сшивания ФГ FXIIIa в полиакриламидном геле (4% – концентрирующий гель, 8% – разделяющий гель) в присутствии додецилсульфата натрия: 1 – продукты реакции FXIIIa с неокисленным ФГ, ФГ, окисленным 150 мкМ H2O2 (2); 300 мкМ H2O2 (3), 600 мкМ H2O2 (4), 1000 мкМ H2O2 (5).
(в) Электрофорез продуктов гидролиза молекулы ФГ под действием плазмина с помощью электрофореза в полиакриламидном геле (4% – концентрирующий гель, 8% – разделяющий гель) в присутствии додецилсульфата натрия (SDS): 1 – негидролизованный ФГ; продукты гидролиза неокисленного ФГ (2), окисленного 150 мкМ H2O2 (3), окисленного 300 мкМ H2O2 (4), окисленного 600 мкМ H2O2 (5), окисленного 1000 мкМ H2O2 (6).

В присутствии FXIIIa полипептидные цепи фибрина участвуют в ковалентном сшивании, что проявляется в образовании γ–γ-димеров и α–α полимеров [8]. С повышением концентрации окислителя количество образующихся α–α полимеров и γ–γ-димеров снижается, о чем свидетельствует также увеличение содержания исходных Аα- и γ-цепей (рис. 1б). Очевидно, что это является следствием окислительной модификации структуры молекулы ФГ.
При оценке подверженности молекулы ФГ плазминовому гидролизу при окислении хорошо заметно, что даже при минимальном количестве окислителя (150 мкМ) значительно возрастает количество продуктов деградации (рис. 1в).
Методом масс-спектрометрии были проанализированы образцы неокисленного и обработанного 300 мкМ Н2О2 ФГ. Под воздействием Н2О2 в молекуле ФГ наиболее подверженными окислению оказались остатки метионина, триптофана и гистидина. Среди модификаций, обнаруженных в этих аминокислотных остатках, имеются случаи образования метионин сульфоксида, 2-оксогистидина и гидрокситриптофана вследствие присоединения одного атома кислорода к боковой цепи (+15.99), окисления триптофана до кинуренина (+3.99) и отщепления метантиола от боковой цепи Met (–48.00). Как видно из рис. 2, модифицированные в результате индуцированного окисления аминокислотные остатки были обнаружены во всех трех полипептидных цепях и всех структурных областях молекулы ФГ, за исключением Е области.
Рис. 2.
Первичная структура (последовательность аминокислотных остатков соответствует UniProt P02671 (FIBA_HUMAN), P02675 (FIBB _HUMAN) и P02679 (FIBG _HUMAN), http://www.uniprot.org) и экспериментально полученные методом ВЭЖХ-МС/МС последовательности пептидных фрагментов полипептидных цепей ФГ. Двойной линией подчеркнуты участки полипептидных цепей, детектированные и в контрольном, и в окисленном (300 мкМ Н2О2) образцах, одинарное подчеркивание снизу – участки, детектированные только в окисленном образце (300 мкМ Н2О2), одинарное подчеркивание сверху – участки, детектированные только в контрольном образце. Сайты модификации отмечены черными треугольниками сверху. Аминокислотные остатки на NH2 – концевых участках полипептидных цепей, обозначенные серым цветом, являются сигнальным пептидом.

В неокисленном образце также были обнаружены окислительные модификации следующих аминокислотных остатков: АαMet91, АαMet207, АαMet240, АαTrp276, АαMet476, АαMet517, АαMet584, BβMet118, BβMet190, BβMet305, BβMet314, BβMet367, γMet78, γTrp227, что может быть объяснено базовым окислением молекулы ФГ в плазме крови, а также дополнительным окислением в процессе его препаративного выделения, хранения и ввода пробы. Практически все аминокислотные остатки, модифицированные в контроле, демонстрируют умеренную степень окисления, которая существенно возрастает при индуцированном окислении.
Локализованные в Е области аминокислотные остатки, которые участвуют в связывании тромбина (AαTrp33, AαPhe35, AαAsp38, AαGlu39, BβAla68, BβAsp69, γAsp27 и γSer30) [9], не были подвержены окислительной модификации, что указывает на сохранение тромбин-связывающих сайтов ФГ при окислении. Содержащей наибольшее количество окислительных сайтов является αС-область (АαMet240, АαTrp276, АαTrp341, АαTrp391, АαMet476, АαMet517, АαHis544, АαHis545, АαMet584), что согласуется с ранее полученными нами ранее данными [2, 3] и подтверждает гипотезу о возможности данной области служить ловушкой для молекул АФК [10].
Для окисленного белка наблюдается замедление накопления α–α полимеров и γ–γ димеров, о чем свидетельствует также увеличение количества исходных Аα и γ цепей (рис. 1б). FXIIIa образует ковалентные сшивки между γGin398/399 и γLys406 [11], продуцируя γ–γ димеры и, с более медленной скоростью, α-α полимеры между Аα-цепями в нескольких сайтах: АαGin328, АαGin336, АαLys508, АαLys556 и АαLys562 [12]. Все детектированные аминокислотные остатки (γGin398/399 и γLys406, АαGin336, АαLys508) оставались в нативной форме при окислении. Это может служить дополнительным доказательством того, что ингибирование реакции ковалентного сшивания цепей ФГ при окислении не является следствием нарушения структуры каталитических сайтов, а обусловлено конформационными перестройками в окисленном белке, делающие эти каталитические сайты менее доступными для FXIIIa. Среди участков молекулы, подверженных плазминовому гидролизу, также не обнаружены сайты окислительных модификаций. Это подтверждает ранее высказанное предположение, что окисленные белки, благодаря увеличению своей гидрофобности, становятся более восприимчивыми к протеолитической деградации [13].
Полученные данные по перекисному окислению ФГ согласуются с полученными ранее результатами исследования озон- и гипохлорит-индуцированного окисления молекулы ФГ [2, 3, 10]. Гипохлорит и озон вызывали значительно большее повреждение молекулы ФГ, способствуя модификации более широкого ряда аминокислотных остатков и разнообразия типов окислительных модификаций [2, 3] по сравнению с Н2О2, однако, в целом, не затрагивали функционально ключевых участков ФГ. Из 24 идентифицированных модифицированных аминокислотных остатков 18 являются метионинами, 12 из которых оказались окисленными даже в контрольном образце. В их числе находится и АαMet476, окисление которого в ряде исследований [1, 5] рассматривается как основной фактор, ответственный за нарушение способности αC-областей участвовать в латеральной агрегации протофибрилл. Однако преимущественное окисление AaMet476 (проявляющееся даже в контрольном образце в результате автоокисления молекулы ФГ) указывает на то, что остаток, по-видимому, экспонирован на поверхности молекулы. Известно, что экспонированные метионины в белках легко окисляемы, наделены антиоксидантной функцией и не оказывают влияния на биологическую активность [4].
Окисление исходного ФГ изменяет геометрические и механические характеристики формируемых фибрилл и, в конечном итоге, структуру и механические свойства фибриновых сгустков. В контексте протекающих в организме окислительных процессов изучение этих изменений имеет фундаментальное медико-биологическое значение. На рис. 3 показаны нормированные зависимости [g(2)(τ) – 1] для гелей из нативного и окисленного ФГ.
Рис. 3.
Нормированные коррелограммы динамического светорассеяния в гелях из нативного (сплошная линия) и окисленного ФГ: пунктир – 300, штрих-пунктир – 500 мкМ Н2О2 на 1 мкМ белка.
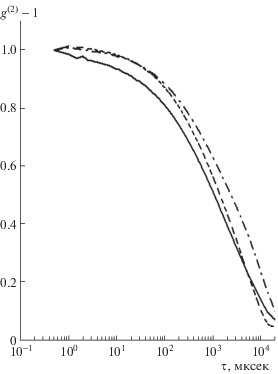
Регистрация проведена через 43, 73 и 22 мин, соответственно, после приготовления геля. Форма КФ отражает динамику сверхдемпфированных колебаний упругой сетки из фибрилл в геле, характеризуемую, в первом приближении, временем затухания τeff ∝ f/G, где f и G – эффективные величины коэффициента трения фибрилл в окружающей жидкости и модуля упругости фибриновой сетки соответственно [14]. Величину τeff можно оценить интегрально по формуле: ${{\tau }_{{eff}}} = \int {{{{[{{g}^{{(2)}}}(\tau ) - 1]}}^{{1/2}}}d\tau } $. Это дает величины τeff = = 8.5, 7.8 и 10.5 мсек для гелей из нативного ФГ и окисленного 300 и 500 мкМ Н2О2 на 1 мкМ белка соответственно. Форма зарегистрированных КФ и полученные оценки τeff позволяют заключить, что окисление ФГ перекисью водорода в дозах порядка 500 мкМ Н2О2 на 1 мкМ белка приводит к заметному повышению жесткости фибриновой сетки свежеобразованного геля. Это может быть обусловлено, как было показано ранее [15], следствием изменений структуры в связи с образованием массивных агрегатов фибрилл.
БЛАГОДАРНОСТИ
В работе использовалось оборудование ЦКП ИБХФ РАН.
ИСТОЧНИКИ ФИНАНСИРОВАНИЯ
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № 18-04-01313. Масс-спектрометрические данные получены при поддержке гранта Российского Научного фонда, № 14-24-00114.
Список литературы
Weigandt K.M., White N., Chung D., Ellingson E., Wang Y., Fu X., Pozzo D.C. // Biophys. J. 2012. V. 103. № 11. P. 2399–2407.
Юрина Л.И., Васильева А.Д., Бугрова А.Е., Индейкина М.И., Кононихин А.С., Николаев Е.Н., Розен-фельд М.А. // ДАН. 2019. Т. 484. № 3. С. 37–41.
Yurina L., Vasilyeva A., Indeykina M., Bugrova A., Biryukova M., Kononikhin A., Nikolaev E., Rosen-feld M. // Free Radical Research. 2019. V. 53. № 4. P. 430–455.
Luo S., Levine R.L. // FASEB J. 2009. V. 23. № 2. P. 464–472.
White N.J., Wang Y., Fu X., Cardenas J.C., Martin E.J., Brophy D.F., Wade C.E., Wang X., St John A.E., Lim E.B., Stern S.A., Ward K.R., López A., Chung D. // Free Radic. Biol. Med. 2016. V. 96. P. 181–189.
Rosenfeld M.A., Shchegolikhin A.N., Bychkova A.V., Leonova V.B., Biryukova M.I., Kostanova E.A. // Free Radic. Biol. Med. 2014. V. 77. P. 106–120.
Galetskiy D., Lohscheider J.N., Kononikhin A.S., Po-pov I.A., Nikolaev E.N., Adamska I. // Rapid Commun. Mass Spectrom. 2011. V. 25. № 1. P. 184–190.
Lorand L., Chenoweth D. // Proc. Natl. Acad. Sci. USA.1969. V. 63. № 4. P. 1247–1252.
Pechik I., Madrazo J., Mosesson M.W., Hernandez I., Gilliland G.L., Medved L. // Proc. Nat. Acad. Sci. USA. 2004. V. 101. № 1. P. 2718–2723.
Rosenfeld M.A., Vasilyeva A.D., Yurina L.V., Bych-kova A.V. // Free Radic. Res. 2018. V. 52. № 1. P. 14–38.
Chen R., Doolittle R.F. // Biochemistry 1971. V. 10. № 24. P. 4487–4491.
Sobel J.H., Gawinowicz M.A. // J. Biol. Chem. 1996. V. 271. № 32. P. 19288–19297.
Davies M.J. // Biochem. J. 2016. V. 473. № 7. P. 805–825.
Tanaka T., Hocker L.O., Benedek G.B. // J. Chem. Phys. 1973. V. 59. № 9. P. 5151–5159.
Wang L., Li L., Wang H., Liu J. // Biochem. J. 2016. V. 473. № 23. P. 4373–4384.
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Науки о жизни