

Доклады Российской академии наук. Науки о жизни, 2021, T. 500, № 1, стр. 455-459
ИНГИБИРОВАНИЕ ОНКОГЕНА с-MYC АНТИБИОТИКАМИ-ПРОИЗВОДНЫМИ АУРЕОЛОВОЙ КИСЛОТЫ
А. К. Исагулиева 1, 2, *, Н. В. Сошникова 1, А. А. Штиль 3
1 ФГБУН Институт биологии гена
Российской академии наук
Москва, Россия
2 ФГБНУ Научно-исследовательский институт по изысканию новых антибиотиков им. Г.Ф. Гаузе
Москва, Россия
3 ФГБНУ Национальный медицинский исследовательский центр онкологии
им. Н.Н. Блохина
Москва, Россия
* E-mail: kia2303@ya.ru
Поступила в редакцию 27.04.2021
После доработки 06.05.2021
Принята к публикации 07.05.2021
Аннотация
Внутриклеточные мишени антибиотиков-производных ауреоловой кислоты – оливомицина А и его полусинтетического производного оливамида – GC-богатые участки в малой бороздке ДНК. Нами установлено, что оба соединения в наномолярных концентрациях ингибируют транскрипцию онкогена c-Myc в опухолевых клетках человека. Механизм ингибирования не требует взаимодействия антибиотиков с полноразмерным сайтом связывания GC-специфического транскрипционного фактора Sp1, а обусловлен присутствием GC-квартетов с оптимальной константой комплексообразования.
Оливомицин А (I; рис. 1) и другие антибиотики-производные ауреоловой кислоты активно исследовались и некоторое время применялись в качестве противоопухолевых агентов. Мишень этих соединений – малая бороздка ДНК [1, 2]. Образование стабильного комплекса I с мишенью приводит к ингибированию транскрипции и репликации; гибель опухолевых клеток достигается в наномолярных концентрациях [3]. Общерезорбтивная токсичность ограничивает клиническое использование столь активного природного соединения. Требуется исследование молекулярных механизмов действия I для создания его эффективных и менее токсичных аналогов.
Предпочтительные сайты связывания I содержат GC-квартеты; наибольшую константу связывания обеспечивают центральные динуклеотиды 5'-GG-3' или 5'-GC-3' [4]. Подобные последовательности часто встречаются в регуляторных областях генов и могут полностью или частично перекрываться участками узнавания транскрипционных факторов (ТФ), например, Sp1. Аффинность к ДНК и выраженная GC-специфичность объясняют способность I и других соединений этого класса ингибировать транскрипцию генов. Детальное изучение комплексов с ДНК позволило создать полусинтетический аналог I – оливамид (II; рис.1) с уменьшенной константой комплексообразования, терапевтической активностью и переносимостью в моделях in vivo [5].
Антибиотик I подавляет экспрессию c-Myc – онкогена [6, 7], экспрессированного во многих опухолях [8]. Существенная часть транскрибируемого участка гена c-Myc, а также его 5'-UTR область представлены CpG-богатыми участками и содержат GC-консенсусы, с которыми способны связываться I и II [9]. Промоторы P0-P3 гена c-Myc также содержат GC-богатые области. Правомерно предположить, что подавление транскрипции c-Myc может происходить из-за связывания I и II с регуляторными участками гена или с сайтами связывания ТФ. Взаимодействуют ли I и II только с определенными регуляторными областями, в частности, с консенсусными участками для GC-зависимых ТФ, или и с другими GC-участками?
В настоящей работе ингибирование c-Myc соединениями I и II исследовали на культивируемых опухолевых клетках человека: родительской линии НСТ116 (аденокарцинома кишки) и сублиниях HCT116TRE-CMV-Fl-cMyc и A375-TRE-CMV-Fl-cMyc (меланома) с соответствующими индуцибельными генетическими конструкциями.
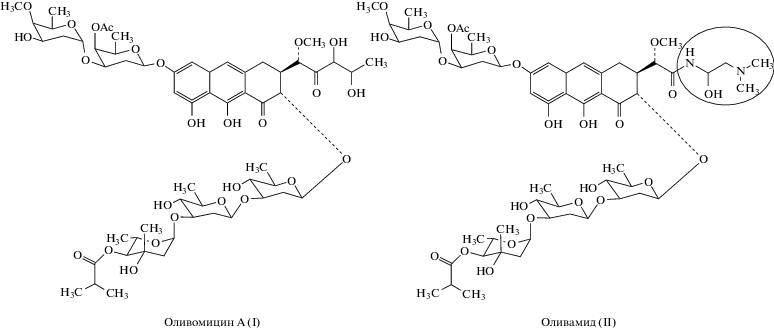
Относительное содержание мРНК эндогенного c-Myc определяли на клетках НСТ116. После добавления I или II (100 нМ) клетки HCT116 инкубировали в течение 3–24 ч и оценивали уровень мРНК c-Myc с помощью количественной ПЦР после обратной транскрипции. Интенсивность сигналов рассчитывали относительно уровня мРНК гена RPLP0. Соединения I и II после 6 ч инкубации снижали количество мРНК c-Myc на ~25% по сравнению с интактными клетками (рис. 2); к 24 ч транскрипты c-Myc почти не определялись.
Рис. 2.
Уровень мРНК c-Myc в клетках HCT116 после инкубации с соединениями I и II (100 нМ) в течение 3–24 ч. Уровень мРНК c-Myc в необработанных клетках принят за 1. Представлены средние величины и стандартные отклонения по результатам трех экспериментов.
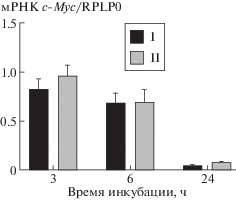
Известно, что производные ауреоловой кислоты ингибируют транскрипцию Sp1-зависимых генов [10]. Консенсусный участок связывания ТФ Sp1 (G/T)GGGCGG(G/A)(G/A)(C/T)) содержит GC-участки – вероятные сайты связывания I. Показано, что производные ауреоловой кислоты специфичны именно для Sр1-зависимых промоторов [10, 11]. Однако экспрессия самого Sp1 также ингибируется производными ауреоловой кислоты [11, 12]; 5'-нетранслируемый район и область старта транскрипции этого гена содержат большое число CpG-островков. Для решения вопроса о том, обязательно ли мишень I и II должна включать сайт взаимодействия указанного ТФ, получена сублиния клеток HCT116-TRE-CMV-Fl-cMyc с возможностью индукции экзогенного c-Myc доксициклином (Dox). Генетическая конструкция, использованная для получения сублинии, включала последовательность TRE-CMV, не имеющую GC-участков связывания Sp1 (рис. 3а, верхняя и средняя панели).
Рис. 3.
Ингибирование активации c-Myc под действием I и II.
а – Схема фрагмента плазмидной конструкции TRE-CMV-Fl-cMyc и экспрессии Fl-cMyc при добавлении I (100 нМ) и Dox (1 мкг/мл); б – иммуноблоттинг лизатов клеток HCT116-TRE-CMV-Fl-cMYC при добавлении I и доксициклина (Dox), стрелками обозначены эндогенная (○) и экзогенная (●) формы белка cMyc; в – иммуноблоттинг лизатов клеток A375-TRE-CMV-Fl-cMyc при действии Dox, I и II: в дорожках 4 и 7 вначале добавляли I/II, через 1 ч вносили Dox; в дорожках 5 и 8 – обратный порядок добавления модуляторов. Бета-актин использовали для нормализации – контроля внесения белков в полиакриламидный гель.
Принцип функционирования индуцибельной системы Tet-On: в отсутствие Dox элемент rtTA блокирует инициацию транскрипции (верхняя панель); Dox cвязывает элемент rtTA, активируя транскрипцию (средняя панель). Связывание I или II с GC-участками возможно только в CMV-промоторе (нижняя панель), поэтому действие антибиотиков на экспрессию репортерного гена c-Myc обусловлено их комплексообразованием только с этими участками.
Экспрессию рекомбинантного белка Fl-cMyc активировали добавлением Dox (рис. 3б). Соединение I (100 нМ, 24 ч) полностью подавляло экспрессию эндогенного c-Myc. В комбинации с Dox I ингибировал и эндогенный белок c-Myc, и экзогенную форму, активируемую Dox. Схожие эффекты наблюдали при добавлении I и II к клеткам сублинии A375-TRE-CMV-Fl-cMyc (конструкция Fl-cMyc под контролем Tet-On элемента и CMV-промотора; рис. 3в). Соединения I и II оказывали выраженный ингибирующий эффект на обе формы белка cMyc (рис. 3в, дорожки 3, 4 и 6, 7 по сравнению с контролями). Если клетки предварительно инкубировали 1 ч с Dox, подавление cMyc было менее выраженным (рис. 3в, дорожки 5 и 8). Сравнивая эффективность I и II для разных линий клеток, можно предположить, что степень ингибирования эндогенного и экзогенного cMyc может быть тканеспецифичной и определяться молекулярным окружением генов-мишеней и отдельных сайтов связывания I и II.
Полученные результаты подтверждают предположение о том, что для связывания с ДНК и ингибирования транскрипции производными ауреоловой кислоты наличие полноразмерного сайта связывания Sp1 не обязательно. Определяющим для формирования комплекса с ДНК и, следовательно, ингибирования транскрипции являются предпочтительные для I или II GC-участки. Таким образом, производные ауреоловой кислоты ингибируют транскрипцию не сайт-специфическим, а сиквенс-специфическим образом. Для I нуклеотидные последовательности в GC-квартетах, предпочтительные для формирования стабильных комплексов, установлены [4]; требуется выявить закономерности предпочтения для II. Важно, что активность II как ингибитора онкогена с-Мус почти не уступает I. Наряду со способностью вызывать гибель опухолевых клеток разного тканевого происхождения и приемлемыми характеристиками in vivo [5], оливамид II приобретает значение противоопухолевого лекарственного “кандидата”.
Список литературы
Брикенштейн В.Х., Питина Л.Р., Баренбойм Г.М., и др. Стереохимия и кинетика взаимодействия с ДНК противоопухолевого антибиотика оливомицина // Мол. биология. 1984. Т. 18, № 4. С. 1606–1616.
Lombó F., Menéndez N., Salas J.A., et al. The aureolic acid family of antitumor compounds: structure, mode of action, biosynthesis, and novel derivatives // Applied Microbiology and Biotechnology. 2006. V. 73. № 1. P. 1–14.
Sabín J.G., Morís F. Exploring Novel Opportunities for Aureolic Acids as Anticancer Drugs // Biochemistry & Pharmacology: Open Access. 2013. V. 2. № 1. P. 1–3.
Beniaminov A.D., Chashchina G.V., Livshits M.A., et al. Discrimination between G/C Binding Sites by Olivomycin A Is Determined by Kinetics of the Drug-DNA Interaction // International Journal of Molecular Sciences. 2020. V. 21. № 15. P. 5299.
Tevyashova A.N., Shtil A.A., Olsufyeva E.N., et al. Modification of olivomycin A at the side chain of the aglycon yields the derivative with perspective antitumor characteristics // Bioorganic & Medicinal Chemistry. 2011. V. 19. № 24. P. 7387–7393.
Самусенко А.В. Механизмы гибели клеток при действии оливомицина и его производных: Дисс. канд. мед. наук. Москва; 2009.
Isagulieva A.K., Beniaminov A.D., Tatarskiy V.V., et al. Targeting gene transcription: mechanisms of antitumor potency of Olivomycin A and its preclinical derivative [letter] // FEBS Open Bio. 2019. V. 9. S1. P. 153.
Vita M., Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer // Seminars in Cancer Biology. 2006. V. 16. № 4. P. 318–330.
Mognol G.P., de Araujo-Souza P.S., Robbs B.K., et al. Transcriptional regulation of the c-Myc promoter by NFAT1 involves negative and positive NFAT-responsive elements // Cell Cycle. 2012. V. 11. № 5. P. 1014–1028.
Previdi S., Malek A., Albertini V., et al. Inhibition of Sp1-dependent transcription and antitumor activity of the new aureolic acid analogues mithramycin SDK and SK in human ovarian cancer xenografts // Gynecologic Oncology. 2010. V. 118. № 2. P. 182–188.
Sleiman S.F., Langley B.C., Basso M., et al. Mithramycin Is a Gene-Selective Sp1 Inhibitor That Identifies a Biological Intersection between Cancer and Neurodegeneration // Journal of Neuroscience. 2011. V. 31. № 18. P. 6858–6870.
Choi E.-S., Nam J.-S., Jung J.-Y., et al. Modulation of specificity protein 1 by mithramycin A as a novel therapeutic strategy for cervical cancer // Scientific Report. 2014. V. 4. № 1. P. 1–8.
Дополнительные материалы отсутствуют.
Инструменты
Доклады Российской академии наук. Науки о жизни