

Электрохимия, 2019, T. 55, № 1, стр. 3-38
Прогресс в понимании механизмов электроокисления метанола и муравьиной кислоты на платиновых металлах (обзор)О. А. Петрий
О. А. Петрий *
Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: petrii@elch.chem.msu.ru
Поступила в редакцию 15.06.2018
После доработки 15.10.2018
Принята к публикации 02.11.2018
Аннотация
Pеакции электроокисления метанола и муравьиной кислоты относятся к числу важнейших модельных электрокаталитических процессов и используются в низкотемпературных прямых топливных элементах. Механизмы электроокисления данных веществ интенсивно исследуются уже в течение многих десятилетий. Значительный прогресс в этой области достигнут благодаря комбинированному применению электрохимических методов, методов in situ инфракрасной спектроскопии и дифференциальной электрохимической масс-спектрометрии, изотопно-кинетического метода, ab initio расчетов с использованием теории функционала плотности и сопоставлению с результатами газофазных исследований. Принципиально важную роль в понимании механизма процессов сыграло использование граней монокристаллов и поверхностей с заданным соотношением террас, ступеней и кинков. Это позволило интерпретировать информацию, накопленную для катализаторов, представленных наночастицами металлов, и выявить роль структурных и размерных факторов в электрокатализе. Наибольшее внимание уделено природе адсорбатов и интермедиатов, детализации маршрутов реакций, механизму возможных медленных стадий, рН эффектам, роли природы аниона в кислых и катиона в щелочных растворах. Подчеркнуто влияние загрузки катализатора и стадийности процессов электроокисления на эффективность работы практических топливных элементов. Анализируются механизмы Лангмюра–Гиншельвуда и Илеа–Риделя в применении к процессам электроокисления и некоторые особенности электроокисления СО-адсорбата. Обсуждаются данные о механизме взаимодействия между адсорбированным кислородом и С1-соединениями. Рассматриваются особенности процессов на биметаллических поверхностях и cтратегии дизайна катализаторов на основе представлений о механизме процессов, контроля структуры/состава поверхности и специфической декорации адатомами металлов. Сформулированы некоторые актуальные направления исследований для дальнейшего углубления в механизмы электроокисления С1-соединений.
СОДЕРЖАНИЕ
I. Введение
II. Основные методы исследования механизмов электроокисления метанола и муравьиной кислоты
IIa.Электрохимические методы
IIб. Дифференциальная электрохимическая масс-спектрометрия (ДЭМС)
IIв. Методы in situ вибрационной спектроскопии
IIг. Теоретические методы
III. Схемы электроокисления метанола. Теоретический и экспериментальный подходы
IV. Представления о механизме электроокисления муравьиной кислоты/формиата
V. Влияние рН и состава раствора на электроокисление муравьиной кислоты/формиата
VI. Энергии активации реакций электроокисления метанола и муравьиной кислоты.
VII. Механизм электроокисления СО-адсорбата
VIII. Взаимодействие метанола и муравьиной кислоты с адсорбированным кислородом
IX. Влияние структуры поверхности и размерных факторов на механизм электроокисления
X. Электроокисление метанола и муравьиной кислоты на бинарных катализаторах
XI. Заключение
Список литературы
I. ВВЕДЕНИЕ
Процессы электроокисления метанола и муравьиной кислоты относятся к важнейшим электрокаталитическим процессам [1, 2]. Механизмы этих процессов исследуются уже в течение почти ста лет.
В ходе интенсивных исследований 1960–1970 гг. была создана феноменологическая картина процессов, высказаны предположения о природе адсорбатов и медленных стадий электроокисления. Значительный вклад на этом этапе был внесен исследованиями, выполненными на кафедре электрохимии Московского университета им. М.В. Ломоносова и в Институте электрохимии АН СССР (см., например, [3–8] и ссылки в них). В дальнейшем, начиная с 80-х годов, развитие in situ инфракрасной (ИК) спектроскопии и техники работ с гранями монокристаллов привело к накоплению данных, позволяющих понимание природы процессов на молекулярном уровне. Одновременно были предприняты интенсивные разработки практических топливных элементов, использующих метанол и муравьиную кислоту в качестве топлива. Прямые метанольный и муравьинокислый топливные элементы достигли стадии коммерциализации. Переход от фундаментальных аспектов к технологическим разработкам был стимулирован осознанием реальной возможности применения этих элементов для питания портативных электронных устройств и гибридных электромобилей [2, 9–13]. Было опубликовано несколько прекрасных стимулирующих обзоров, обобщающих достижения на разных этапах развития проблемы (см., например, [14–23]).
В начале 90-х годов сильным стимулом к развитию прямых кислотных топливных элементов послужила замена сернокислотного электролита твердым протонпроводящим полимерным электролитом – нафионом [20]. Появление полимерных анионообменных мемран, мало чувствительных к карбонизации, привело к активной разработке щелочных топливных элементов, в том числе использующих формиаты в качестве топлива [24–26]. Использование щелочных электролитов позволяет перейти к менее дорогим эффективным катализаторам окисления С1-соединений. Помимо этого, в щелочных электролитах открываются новые возможности для создания более эффективного кислородного электрода.
Полное окисление метанола в кислых и щелочных средах является 6-электронным процессом в соответствии с уравнениями:
(1)
$\begin{gathered} {\text{С }}{{{\text{Н }}}_{{\text{3}}}}{\text{OH}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{C}}{{{\text{O}}}_{2}} + 6{{{\text{H}}}^{ + }} + 6{\text{e;}} \\ E_{{{\text{а н о д }}}}^{0} = 0.02\,\,{\text{В }};\,\,{\text{pH}}\,\,0, \\ \end{gathered} $(2)
$\begin{gathered} {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}} + 6{\text{ O}}{{{\text{H}}}^{--}} \to {\text{C}}{{{\text{O}}}_{2}} + {\text{5}}{{{\text{H}}}_{{\text{2}}}}{\text{O}} + 6{\text{e;}} \\ E_{{{\text{а н о д }}}}^{0} = - 0.81\,\,{\text{В }};\,\,{\text{pH}}\,\,14, \\ \end{gathered} $(3)
$\begin{gathered} {\text{HCOOH}} \to {\text{C}}{{{\text{O}}}_{2}} + {\text{2}}{{{\text{H}}}^{ + }} + 2{\text{e}}; \\ E_{{{\text{а н о д }}}}^{0} = - 0.22\,\,{\text{В }};\,\,{\text{pH}}\,\,0, \\ \end{gathered} $(4)
$\begin{gathered} {\text{Н С О }}{{{\text{О }}}^{{\text{--}}}}\,\,{\text{ + }}\,\,{\text{3О }}{{{\text{Н }}}^{{\text{--}}}} \to {\text{С О }}_{{\text{3}}}^{{{\text{2}} - }}\,\,{\text{ + }}\,\,{\text{2}}{{{\text{Н }}}_{{\text{2}}}}{\text{О }}\,\,{\text{ + }}\,\,{\text{2е ;}} \\ E_{{{\text{а н о д }}}}^{{\text{0}}}{\text{ = }} - {\text{1}}{\text{.05}}\,\,{\text{В ;}}\,\,\,{\text{ р Н }}\,\,{\text{14}}{\text{.}} \\ \end{gathered} $Давно было установлено, что при электроокислении метанола на Pt в кислой среде в качестве стабильных интермедиатов образуются формальдегид и муравьиная кислота. Это обстоятельство, а также высокое число участвующих в процессе электронов и необходимость участия дополнительного кислорода (в виде Н2О или ОН) приводят к предположению, что электроокисление муравьиной кислоты должно быть более простым процессом для установления его механизма. Однако мы убедимся в дальнейшем, что эта простота лишь кажущаяся.
Теоретическая объемная плотность энергии муравьиной кислоты (1725 Вт ч л–1) составляет только 1/3 соответствующей величины для метанола (4780 Вт ч л–1). Однако более низкий кроссовер муравьиной кислоты через мембрану по сравнению с метанолом позволяет использовать существенно более высокие концентрации топлива (>10–15 М против 1–3 М для метанола). Кроме того, скорости электроокисления муравьиной кислоты выше, чем метанола. Эти обстоятельства привели к росту интереса к использованию муравьиной кислоты в топливных элементах и, соответственно, к высоким темпам изучения механизма электроокисления этого вещества.
Для практических целей важно, чтобы потенциал анода при рабочих плотностях тока не превышал 0.2–0.4 В по обратимому водородному электроду в том же растворе. При этом скорость (или частота) конвертации (turnover rate or frequency) должна быть более 0.1 с–1 [1]. Эта величина характеризует число полных актов электрокаталитической реакции на одном центре поверхности за одну секунду. Считается, что среди монометаллических катализаторов наиболее активны Pt для окисления метанола и Pd для окисления муравьиной кислоты. Однако скорости окисления метанола и муравьиной кислоты на этих металлах при таких потенциалах и низких температурах малы, что стимулировало многочисленные попытки нахождения условий для обеспечения более высоких скоростей реакций и, прежде всего, поиски более активных катализаторов. Это направление остается доминирующим и в настоящее время [27]. Вместе с тем очевидно, что поиск эффективных катализаторов невозможен без более глубокого проникновения в механизм электрокаталитических процессов.
Необходимо подчеркнуть, что исследование электроокисления малых органических молекул принципиально важно и для других областей, прежде всего, электросинтеза, электрогидрирования, электрополимеризации.
II. ОСНОВНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ ЭЛЕКТРООКИСЛЕНИЯ МЕТАНОЛА И МУРАВЬИНОЙ КИСЛОТЫ
В исследовании электрокаталитических процессов используется большой арсенал методов, которые описаны в многочисленных обзорах и монографиях (см., например, [1, 2]). Ниже кратко представлены методы, позволившие получить основной объем информации о механизме процессов электроокисления метанола и муравьиной кислоты. При этом внимание акцентировано на методических достижениях последнего десятилетия.
IIa. Электрохимические методы
Для изучения электрокаталитических процессов наиболее широко используется метод линейной и циклической вольтамперометрии. Aнализ некоторых возможностей этого метода, в том числе в комбинации с методом вращающегося дискового электрода, дан в работе Абруны и сотр. [28]. С помощью циклической вольтамперометрии был получен, в частности, большой объем информации о влиянии структуры поверхности, размера и формы наночастиц и других факторов на электроокисление метанола и муравьиной кислоты.
Для выяснения механизма реакций и вклада параллельных путей в общий процесс привлекается хроноамперометрия в сочетании с моделированием кинетики реакции. Так, в работе Хусманса и Копера [29] дан анализ предшествующих исследований с использованием хроноамперометрии, предложена новая модельная интерпретация транзиентов тока и получены тафелевские зависимости для окисления метанола.
Фелью и сотр. [30] для выяснения механизма реакций электроокисления метанола и муравьиной кислоты использовали импульсную вольтамперометрию. В этом методе, предложенном Клавилье [31], на электрод периодически накладываются короткие импульсы потенциала до значений, при которых полностью окисляется адсорбированный интермедиат, и начальный ток, наблюдаемый при переключении к более низким потенциалам, обусловлен стадией дегидрирования реактанта на чистой поверхности. При этом самые начальные участки транзиентов, относящиеся к токам адсорбции водорода и заряжения двойного слоя, исключаются из рассмотрения, поскольку эти процессы имеют значительно более короткие постоянные времени, чем процессы дегидрирования. Начальные токи характеризуют собственную (intrinsic) активность материала электрода, не искаженную адсорбированными интермедиатами.
В [32] после электроокисления и десорбции интермедиата потенциал электрода переключался к исходному значению, а затем измерялась вольтамперограмма с высокой скоростью наложения потенциала (30–40 В с–1). Предполагалось, что при этом удается изучить стадию дегидрирования молекул метанола. Далее на основании теории стандартной вольтамперометрии для многоэлектронного процесса находились константа скорости и коэффициент переноса. В более поздней работе [33] был выполнен конвуляционный анализ быстрых вольтамперограмм электроокисления метанола до растворимых продуктов на поликристаллической платине. В обеих работах получены низкие константы скорости реакции, свидетельствующие о ее необратимости. Однако оценка коэффициента диффузии молекул метанола приводила к непонятно низким значениям (порядка 10–12 см2 с–1).
Большое внимание по-прежнему привлекают данные, получаемые при сканировании потенциала в анодном и катодном направлениях в различных интервалах потенциалов и скоростей развертки на разных электродных материалах либо в некоторых случаях для разных С1-соединений (например, [34]). Особое внимание уделяется пику окисления на катодной развертке и отношению высот анодного и катодного пиков (рис. 1).
Рис. 1.
Циклические вольтамперограммы электроокисления метанола на Pt/C (1) и PtRu/C (2) в растворе 0.1 М HClO4 + 0.5 M CH3OH при скорости развертки потенциала 50 мВ с–1 (по данным [35]).

Во многих работах это отношение рассматривается в качестве критерия СО-толерантности и каталитической активности (см., например, [36]). Однако в работах [35, 37] убедительно доказана неадекватность этого критерия. При этом в [35] привлекался метод спектроскопии импеданса, а в [37] – метод вибрационной спектроскопии. Более однозначные выводы об относительной активности катализаторов и СО-толерантности можно сделать из сопоставления высот пиков на анодной развертке в корреляции с соотношением квазистационарных токов, найденных из хроноамперометрических измерений, а также из сопоставления потенциалов, при которых начинаются процессы электроокисления метанола или муравьиной кислоты.
Измерения при очень медленных развертках потенциала часто рассматриваются как квазистационарные (см., например, [38]). Вместе с тем измерения при более жестких критериях стационарности [39] показывают, что по крайней мере для катализаторов с высоко развитой поверхностью наклоны тафелевских кривых электроокисления метанола при низких анодных потенциалах оказываются более низкими. Причины очень медленного установления стационарных токов требуют дополнительного изучения. Это явление вызывает трудности при трактовке механизма процесса на основании наклонов тафелевских зависимостей, если последние относятся к разным критериям стационарности. Вполне возможно, что при длительной поляризации постепенно накапливается некоторый снижающий активность катализатора яд, природа которого не установлена [21].
Метод спектроскопии импеданса используется как для изучения механизма электроокисления (как, например, в [36, 40]), так и для диагностики топливных элементов [41]. Измерения импеданса указывают на зависимость механизма электроокисления метанола от потенциала и на участие в процессе, по крайней мере, одного адсорбированного интермедиата.
IIб. Дифференциальная электрохимическая масс-спектрометрия (ДЭМС)
Meтод ДЭМС получил широкое распространение как высокочувствительный метод количественного анализа продуктов электроокисления метанола и муравьиной кислоты в реальном времени in situ.
Идея использования масс-спектрометра в сочетании с электрохимической ячейкой была впервые реализована Брукенштейном и Гадде [42]. Существенное развитие метода началось после того, как Хайтбаум и сотр. [43, 44] предложили использовать полупроницаемую тефлоновую мембрану как для разделения электрохимической ячейки и камеры спектрометра, так и одновременно в качестве носителя для дисперсного катализатора. В дальнейшем Хартунг и Балтрушат [45] показали, что в сочетании с тонкослойной ячейкой можно проводить определение продуктов окисления и на гладких электродах, в том числе на гранях монокристаллов. В [46] данные ДЭМС были использованы для подтверждения двухмаршрутного механизма окисления метанола и выявления факторов, которые определяют относительный вклад каждого из маршрутов. Копер и сотр. [47] предложили оригинальную оn-line масс-спектрометрическую систему для измерений в менисковой конфигурации. В этой работе и в [48] можно найти анализ различных подходов к использованию ДЭМС на электродах с небольшой поверхностью.
Метод ДЭМС позволяет характеризовать субмонослойные количества прочно хемосорбированных адсорбатов при их электроокислении до СО2. Так как постоянная времени метода достаточно небольшая (~0.1 с), то можно определять продукты реакции в ходе измерения циклических вольтамперограмм (вольт-амперная масс-спектрометрия), при ступенчатой хроноамперометрии или в ходе измерения транзиентов бестокового потенциала. Дополнительная конкретная информация о механизме процесса окисления может быть получена с помощью соединений, меченных изотопами (например, СD3OH, CH3OD, H13COOH) [49, 50]. Юзис и Бем [51] адаптировали ДЭМС систему для измерений при повышенных температурах и давлениях.
Одной из ключевых проблем в методе ДЭМС является дизайн злектрохимической ячейки, сопрягаемой с масс-спектрометром [52]. Конструкция ячейки должна, прежде всего, обеспечивать контролируемый перенос реактанта к электроду и продуктов реакции в камеру спектрометра. Были сконструированы специальные тонкослойные ячейки для комбинирования метода ДЭМС с методом кварцевого микровзвешивания [52] и методом in situ инфракрасной спектроскопии (см., например, [53]).
В классическом виде ДЭМС предназначена для определения газообразных и летучих продуктов реакции. Чтобы преодолеть это ограничение, Абруна и сотр. [54] разработали специальную проточную ячейку с двумя электродами в канале (double-band-electrode channel flow cell). Один электрод служит для сбора газообразных/летучих продуктов окисления, а второй – для электрохимического обнаружения нелетучего продукта (муравьиной кислоты). Бем и сотр. [55] предложили новый подход к полному количественному анализу продуктов электроокисления метанола в кислых растворах, использующий комбинацию масс-спектрометрии с электронным импактом и с ионизацией методом электроспрея после процедуры дериватизации–экстракции–сепарации. Этот подход был распространен на электроокисление метанола в щелочных растворах [56]. Возможности метода масс-спектрометрии использованы еще не в полном объеме [48].
Cопоставление результатов ДЭМС, полученных в разных условиях, приводит к выводу, что соотношение выходов промежуточных и конечных продуктов зависит от потенциала электрода, времени электролиза, степени шероховатости и структуры поверхности, а для дисперсных катализаторов и катализаторов на носителях – от загрузки катализатора, концентрации реактанта, состава электролита и условий его размешивания, температуры и некоторых других факторов. Это соотношение отличается от наблюдаемого при объемном электролизе. Причины такого различия и роль указанных выше многочисленных факторов в соотношении продуктов нуждаются в дальнейшем выяснении.
IIв. Методы in situ вибрационной спектроскопии
Вибрационная спектроскопия позволяет получить информацию о химическом составе и структуре адсорбатов и интермедиатов в процессах электроокисления малых органических молекул.
Использование in situ ИК-спектроскопии началось после пионерского вклада Бьюика, Бедена, Лами и Кунимацу [57]. Для успешного применения метода необходимо было преодолеть ряд проблем, обусловленных прежде всего сильной абсорбцией инфракрасного излучения раствором (молекулами растворителя, ионами электролита, молекулами растворенных веществ) и малой величиной сигнала от адсорбатов из-за их небольшой концентрации (обычно порядка 1015 частиц на см2 поверхности) по сравнению с концентрацией молекул в объеме раствора. Решение этих проблем возможно с использованием тонкослойных, в том числе проточных, ячеек, снабженных специальным окном, пропускающим инфракрасное излучение, и применением различных методик и приемов выделения слабых сигналов от адсорбатов [1, 2]. Вначале был использован метод с модуляцией электрического потенциала электрода (EMIRS). Затем появились такие модификации метода, как ИК-спектроскопия с фурье-преобразованием (FTIRS), ИК-спектроскопия с нормализацией вычитанием (SNIFTIRS), спектроскопия с модуляцией поляризации ИК-фотона (PM-IRRAS) и др.
Существенный прогресс в использовании ИК-спектроскопии в электрохимических системах начался после того, как Осава [58] предложил методику усиленной поверхностью ИК-спектроскопии (SEIRAS) в конфигурации внутреннего нарушенного полного отражения (ATR). Эта методика в англоязычной литературе обозначается аббревиатурой ATR-SEIRAS. Ее особенность состоит в использовании в качестве электрода тонких SEIRAS-активных слоев катализатора, нанесенных на призму (или полусферу) из Si или Ge. ИК-излучение не проходит непосредственно через раствор электролита, а достигает границы раздела через призму, проникая при этом в слой электролита всего на несколько нм. Схема одного из современных вариантов ячейки показана на рис. 2.
Рис. 2.
Схематическое изображение тонкослойной проточной электрохимической ячейки для комбинированных ДЭМС- и in situ ATR-FTIRS-измерений. In-1, In-2 – два ввода для электролита; CE-1, CE-2 – два ввода для вспомогательных электродов; WE, RE – каналы для связи с рабочим электродом и электродами сравнения; Out – выход для электролита. Стрелками показаны направления потоков раствора через капиллярные отверстия внутри ячейки (по данным [59]).
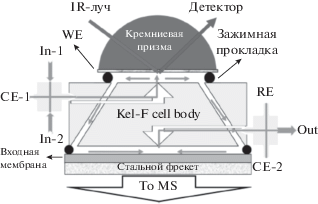
Группа Бема предложила совершенную систему для комбинированной on-line ДЭМС и in situ ИК-спектроскопии в ATR-конфигурации (DEMS/ATR-FTIRS) [60] с возможностями менять скорость протока раствора и замены одного раствора другим.
В работах Вандловского и сотр. [61–63] была показана возможность фабрикации хорошо охарактеризованных электрокаталитически и SEIRAS-активных модельных электродов из Pt, Pd и Rh с использованием квазимонокристаллических SEIRAS-активных темплатов.
Метод ATR-SEIRAS позволяет проводить мониторинг электрокаталитических реакций в реальном времени при развертке потенциала со скоростями 50 мВ/с и даже более.
Эффективным методом изучения электрохимических процессов является усиленная поверхностью рамановская спектроскопия – SERS (см. обзоры [64–66]). Достоинствами этого метода, помимо высокой чувствительности и возможности использования в водной среде, служат также возможность характеризовать частицы по колебаниям, параллельным поверхности электрода, и более простые правила отбора. Однако платиновые металлы обладают низкой SERS-активностью. Вивер и сотр. [67, 68] предложили стратегию электролитического нанесения ультратонких (~3–10 монослоев) пленок платиновых металлов на SERS-активное золото. Такие слои обладают электрокаталитическим поведением, свойственным платиновым металлам, и повышенной SERS-активностью, позволяющей надежно характеризовать адсорбаты и интермедиаты. В дальнейшем получила развитие электрохимическая рамановская спектроскопия, усиленная наночастицами типа ядро–оболочка, в которых ядро составляет SERS-активный материал [69]. Существенный вклад в разработку этого метода (в англоязычной литературе – electrochemical shell-isolated nanoparticle-enhanced Raman spectroscopy – EC-SHINSERS) внесла группа Тиана. Недавно электроокисление муравьиной кислоты было изучено методом SERS на электролитических осадках платины, нанесенных на золотые наностержни [70]. Природа эффекта усиления рассеивания излучения адсорбированными на различных материалах частицами еще требует дальнейшего выяснения.
При трактовке спектральных данных, относящихся к электрохимическим адсорбатам, надо дополнительно учитывать Штарк-эффект – сдвиг спектральных линий адсорбата при изменении потенциала на границе фаз. Эта проблема обсуждалась, например, в [60, 71, 72]. Для более надежной интерпретации спектров привлекаются расчеты в рамках теории функционала плотности (см. например, [72, 73]). В работах Смоткина и сотр. (см. обзор [74]) operando Штарк-эффект используется для характеризации электрокаталитических интерфейсов в реальных метанольных топливных элементах.
Новые возможности предоставляет метод генерации суммарной частоты (sum frequency generation). Так, Вьецковский и сотр. [75] с помощью этого метода нашли различие в состоянии СО, адсобированного из растворов СО и образовавшегося из НСООН. В последнем случае, кроме линейной формы СО, обнаружены еще три мультисвязанных формы и наблюдалась динамика их взаимных превращений. В [76] предпринята попытка регистрации слабо связанных молекул НСООН, обеспечивающих новый путь электроокисления муравьиной кислоты на Pt.
IIг. Теоретические методы
Процессы электроокисления метанола и муравьиной кислоты оказываются привлекательными объектами для применения методов теоретической химии [2, 77].
Стабильность и химическая природа интермедиатов, возникающих при дегидрировании метанола на платиновых металлах и сплавах Pt с Ru и Sn, были анализированы посредством расчетов из первых принципов в [78, 79] на основе различных моделей поверхности и различных модификаций теории функционала плотности (в английской транскрипции – DFT). Этот анализ показал перспективность развиваемых подходов.
C помощью DFT-расчетов находят энергии адсорбции интермедиатов, которые могут образовываться в соответствии с предполагаемой схемой процесса и моделью поверхности, и строят диаграммы потенциальной энергии для дегидрирования органического вещества, диссоциации воды с образованием ОНадс и реакции СОадс + + ОНадс. Далее оценивают энергии активации отдельных стадий, что позволяет высказать предположения о лимитирующей стадии процесса.
Увеличение быстродействия вычислительных машин, рост их мощности и объема памяти вызывает возрастающий интерес к применению методов квантовой химии, что уже привело к появлению большого числа теоретических работ, посвященных анализу механизмов окисления метанола и муравьиной кислоты (см., например, [80–113 ]).
Применение DFT-расчетов в электрохимических условиях сталкивается с трудностями, вызванными необходимостью моделирования границы раздела электрод–раствор, роли взаимодействия адсорбатов с молекулами растворителя, роли ионов раствора и скачка потенциала на границе фаз, который может изменяться в значительно больших интервалах, чем достижимый в ультравысоком вакууме.
III. СХЕМЫ ЭЛЕКТРООКИСЛЕНИЯ МЕТАНОЛА. ТЕОРЕТИЧЕСКИЙ И ЭКСПЕРИМЕНТАЛЬНЫЙ ПОДХОДЫ
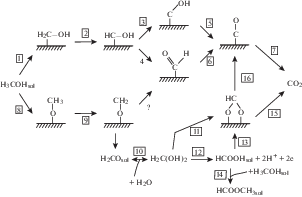
В.С. Багоцкий, Ю.Б. Васильев и О.А. Хазова предложили [5] общую схему процессов электроокисления С1-соединений в виде треугольной матрицы (лестнично-матричный механизм по [114]). В упрощенном виде эта схема показана на рис. 3.

Стабильные продукты окисления расположены на гипотенузе матрицы, продукты дегидрирования – в направлении слева направо, а продукты окисления с участием ОН (или Н2О) – по вертикали. Адсорбаты непосредственно связаны с субстратом. Эта схема не учитывает все возможные реакции. Так, для метанола и муравьиной кислоты возможны процессы с отщеплением кислотного водорода и образованием соответственно частиц НСО и НСОО. Вместе с тем, схема использовалась в качестве исходной в ряде работ для более детального анализа возможного механизма процессов окисления С1-соединений.
В случае метанола первой стадией электроокисления является адсорбция молекулы метанола на поверхности катализатора. Далее в принципе возможны три сценария: 1) дегидрирование молекулы с разрывом одной из связей С–Н; 2) дегидрирование с отщеплением кислотного водорода и образованием адсорбированного метоксила СН3О; 3) разрыв С–О-связи с отщеплением ОН-группы и образованием адсорбированного метила СН3. Последний процесс энергетически маловероятен (см. например, [98]), во всяком случае при низких температурах. Поэтому рассматривают процессы 1 и 2 как два возможных наиболее вероятных исходных маршрута реакции электроокисления.
Отметим, что предположение о двухмаршрутном механизме электроокисления С1-соединений было сформулировано впервые Брайтером [115, 116].
Схемы электроокисления метанола предлагались и обсуждались в очень большом числе работ. Основываясь на расчетах ab initio теории функционала плотности и на экспериментальных данных, группы Вьецковского и Неурока [84, 91] предположили, что двухмаршрутный механизм (dual paths mechanism) заложен уже в первой стадии дегидрирования молекулы метанола. Расщепление С–Н-связи ведет к гидроксиметилу (СН2О)адс и, в конце концов, к СОадс (СО-маршрут), который устойчив при низких потенциалах и, блокируя места на поверхности, тормозит процесс. В то же время расщепление О–Н-связи приводит к образованию метоксила (СН3О)адс, который связан с Pt через кислород. При дегидрировании метоксила образуется формальдегид, который может частично десорбироваться в раствор, а частично окисляется дальше. Этот маршрут называется часто прямым.
Копер и сотр. [77, 117], проведя одновременные электрохимические и масс- спектрометрические измерения на Pt(111), Pt(110), Pt(100), предложили более детальную схему двухмаршрутного электроокисления метанола (рис. 4). Они учли полученный Осавой и сотр. [118] вывод о том, что активным интермедиатом электроокисления может быть формиат.
Абруна и сотр. [28] предприняли попытку суммировать результаты предыдущих и собственных исследований, что привело к схеме, представленной на рис. 5.
Рис. 5.
Детализированная схема окисления метанола в кислом (слева) и щелочном (справа) электролитах. Пунктир – маршруты, которые предполагались в литературе, однако вряд ли были убедительно подтверждены экспериментально. Штрих-пунктирная линия представляет маршрут, который соответствует начальным стадиям дегидрирования, установленным измерениями в ультравысоком вакууме. +Pt* – минимальное число Pt-мест, требуемое для соответствующей реакции; – Pt – освобождаемые атомы платины после осуществления стадии (cогласно [28]).

Эта схема основана на измерениях на Pt в растворах H2SO4 или HClO4 концентраций 0.1–1 М и в растворах NaOH или KOH. В схеме указано также минимальное число атомов Pt, которое необходимо для реализации той или иной стадии, и число атомов Pt, которое освобождается после соответствующей стадии. Оценка этих чисел рассматривалась в ряде работ (например, [119, 120]), однако остается не вполне однозначной.
Hедавно Саконг и Гросс [111] предложили схемы интермедиатов окисления метанола в гетерогенном катализе и электрокатализе (рис. 6) на основе проведенных ими расчетов в рамках теории функционала плотности. Согласно этой работе, основное отличие между электрокаталитическим и каталитическим (газофазным) окислением состоит в том, что в электрокатализе участвует ОН-группа, а в катализе – атомарный кислород. Кроме того, диольные молекулы могут легко образовываться в водных растворах, но отсутствуют в газофазном процессе. В предложенной схеме указаны энтальпии процессов образования интермедиатов (в эВ), рассчитанные с помощью теории функционала плотности.

Следует отметить, что представление о формиате как активном интермедиате было подвергнуто сомнению в работе Куниматцу и коллег [121]. Эти авторы нашли, что величина адсорбции формиата при Е = 0.6 В (по ОВЭ в том же растворе) мало изменяется с ростом концентрации метанола в ряду 0.04–1 М, тогда как плотность тока окисления при этом существенно возрастает.
Наряду с попытками идентификации интермедиатов электроокисления метанола и детализацией общей схемы процесса были предприняты и предпринимаются усилия для оценки вклада отдельных маршрутов реакции в общий процесс, выявления факторов, влияющих на относительный вклад каждого маршрута, и количественное описание кинетики с учетом двухмаршрутного механизма.
Уравнение для скорости процесса, протекающего по двум параллельным путям, было предложено Брайтером [116]. В дальнейшем был предпринят еще ряд попыток кинетического анализа двухмаршрутного механизма (например, [122, 123]). Один из наиболее детализированных подходов дан Штуве и сотр. [124].
Как указывалось выше, Хусманс и Копер [29] развили модельный подход к описанию хроноамперометрических данных с учетом реакций разложения метанола, окисления СО и прямого пути окисления:
(5)
${\text{С }}{{{\text{Н }}}_{{\text{3}}}}{\text{О Н }} \to {\text{С }}{{{\text{О }}}_{{{\text{а д с }}}}} + 4{{{\text{Н }}}^{ + }} + 4{\text{е }}\,\,\,\,\,{\text{CO - м а р ш р у т ,}}\,\,\,{{i}_{{{\text{р а з л }}}}},$(6)
${{{\text{Н }}}_{{\text{2}}}}{\text{О }} + * \leftrightarrow {\text{О }}{{{\text{Н }}}_{{{\text{а д с }}}}} + {{{\text{Н }}}^{ + }} + {\text{е ,}}$(7)
${\text{С }}{{{\text{О }}}_{{{\text{а д с }}}}} + {\text{О }}{{{\text{Н }}}_{{{\text{а д с }}}}} \to {\text{С }}{{{\text{О }}}_{{\text{2}}}} + {{{\text{Н }}}^{ + }} + {\text{е }} + 2{\text{*}}\quad\quad\quad\,\,\,{\text{С О - о к и с л е н и е ,}}\,\,\,{{i}_{{{\text{о к }}}}},$(8)
$\begin{gathered}{CH_3}{OH} \to {\text{и н т е р м е д и а т ы }}\to {CO_2}{\;п р я м о й м а р ш р у т },\,\,{{i}_{{{\text{п р }}}}}, \\ \mathop{\,\,\,\,\,\,{\text{ч а с т и ч н а я д и ф ф у з и я в о б ъ е м р а с т в о р а }}}\limits^\downarrow \\ \end{gathered} $Общий ток i в цепи, согласно этой схеме, равен
Решение дифференциального уравнения, описывающего кинетику образования и окисления СО,
(10)
${{{{d{{\theta }_{{{\text{CO}}}}}} \mathord{\left/ {\vphantom {{d{{\theta }_{{{\text{CO}}}}}} {dt = {{k}_{{{\text{р а з л }}}}}\left( {1 - {{\theta }_{{{\text{CO}}}}}} \right)}}} \right. \kern-0em} {dt = {{k}_{{{\text{р а з л }}}}}\left( {1 - {{\theta }_{{{\text{CO}}}}}} \right)}}}^{2}} - {{k}_{{{\text{о к }}}}}{{\theta }_{{{\text{CO}}}}}\left( {1 - {{\theta }_{{{\text{CO}}}}}} \right),$(11)
$\begin{gathered} i\left( t \right) = {{i}_{{{\text{р а з л }}}}}\left[ {1 - {{\theta }_{{{\text{CO}}}}}\left( t \right)} \right] + 4{\text{e}}{{N}_{{{\text{Pt}}}}}{{k}_{{{\text{р а з л }}}}}{{\left[ {1 - {{\theta }_{{{\text{CO}}}}}\left( t \right)} \right]}^{2}} + \\ + \,\,2{\text{e}}{{N}_{{{\text{Pt}}}}}{{k}_{{{\text{о к }}}}}{{\theta }_{{{\text{CO}}}}}\left( t \right)\left[ {1 - {{\theta }_{{{\text{CO}}}}}\left( t \right)} \right]. \\ \end{gathered} $В этих уравнениях iпр – ток прямого маршрута, iразл и kразл – ток и константа скорости разложения метанола, iок и kок – ток и константа скорости окисления СОадс , 4 и 2 – числа электронов, участвующих в соответствующих реакциях, NPt – поверхностная плотность атомов Pt, е – элементарный заряд. Второй порядок реакции разложения метанола ранее был найден Франащуком и коллегами [122], а второй порядок реакции окисления СОадс, который соответствует механизму Лангмюра–Хиншельвуда, был установлен Лебедевой и коллегами [123]. Согласно полученному уравнению, форма хроноамперограмм зависит от соотношения kок/kразл. Если это отношение больше или равно 4, ток должен возрастать во времени, так как скорость удаления СОадс выше скорости его образования. В противоположном случае ток падает во времени, вследствие заполнения поверхности СОадс. Таким образом, сама форма транзиентов указывает, в принципе, на соотношение скоростей различных маршрутов реакции.
Уравнение (11) описывает хроноамперограммы электроокисления метанола на Pt[n(111) × (110)] в сернокислом растворе. Транзиенты тока измерялись при ступенчатом изменении потенциала от 0.855 В, при котором поверхность освобождается от предварительно адсорбированных продуктов, к более низким значениям. Было установлено, что окисление самого метанола, а также продуктов его распада протекает с более высокой скоростью на ступенчатых поверхностях и возрастает с ростом плотности ступеней. В дальнейшем группа Копера использовала метод on-line электрохимической масс-спектрометрии (OLEMS) c анализом отношения выходов метилформиата (природа процессов, приводящих к образованию метилформиата, обсуждается ниже) и СО2 для прояснения селективности и структурной чувствительности образования интермедиатов (формальдегида и муравьиной кислоты) и роли природы аниона электролита [117]. Учитывая особенности использованной методики, авторы характеризуют свои результаты как качественные.
Идея определения вклада различных маршрутов в суммарный процесс электроокисления метанола по отношению выходов СО2, НСНО и НСООН была реализована Иваситой и сотр. [125] с помощью метода HPLC.
Ванг и Балтрушат [126] методом ДЭМС изучили продукты электроокисления метанола в растворах серной и хлорной кислот на пoликристаллической Pt, Pt(111), Pt(332) и при декорировании поверхности рутением при различных температурах и потенциалах. С учетом различий в условиях опытов полученные данные в целом согласуются с ранее опубликованными группами Иваситы и Копера и с представлением о двухмаршрутном механизме. В [127] определение соотношения выходов СО2 и метилформиата, а следовательно, соотношения скоростей параллельных маршрутов, было проведено на грани Pt(311).
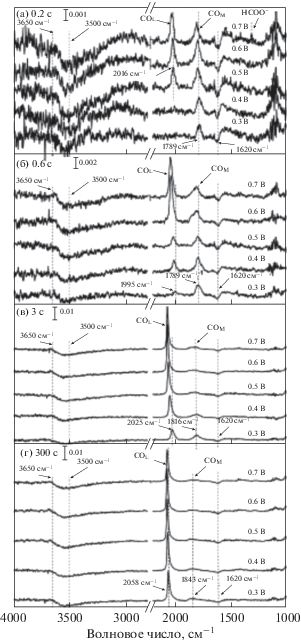
Используя метод ATR-FTIRS в сочетании с проточной тонкослойной ячейкой, группа Чена предложила методику нахождения вкладов СО- и прямого маршрутов в электроокисление метанола [59, 128–130]. Особенность использованной методики связана, прежде всего, с измерением и анализом зависимости ИК-полосы СОадс от времени при постоянном потенциале, наряду с DEMS-анализом. Репрезентативные спектры (рис. 7) позволяют проследить появление линейной формы СОL, мультисвязанной формы СОМ и формиата НСОО. Кинетика дегидрирования метанола до СОадс и кинетика окисления СОадс до СО2 анализировались путем записи транзиентов тока, а также на основе изменения ИК-сигнала СОадс при замене раствора 12СН3ОН на раствор 13СН3ОН и наоборот. Было найдено, что при фиксированном покрытии поверхности СО скорости дегидрирования СН3ОН до СОадс и окисления СОадс возрастают в интервале потенциалов 0.3–0.7 В. Тафелевский наклон для процесса дегидрирования метанола составляет 440 ± 40 мВ и не зависит от степени покрытия поверхности СО. Тафелевская зависимость для электроокисления СОадс имеет наклон 39 мВ при Е < 0.55 В, а при Е > 0.55 В – ~149 мВ. При увеличении потенциала от 0.6 до 0.75 В фарадеевский ток возрастает в 5 раз, выход СО2 уменьшается с 25 до 8%, а вклад СО-маршрута уменьшается с 33 до 18%. Основным продуктом при Е > 0.6 В оказывается формальдегид.
Рис. 7.
Некоторые репрезентативные ИК-спектры Pt-поверхности, записанные через 0.2; 0.6; 3; 300 с после замены раствора 0.1 М НСlO4 на раствор 0.1 М НСlO4 + 2 M CH3OH при потенциалах 0.3; 0.4; 0.5; 0.6; 0.7 В (по данным [129]).
Группа Бема [131, 132] предложила подход для анализа маршрутов окисления метанола путем введения в раствор добавок побочных продуктов (формальдегида или муравьиной кислоты, в том числе меченых 13С) в сочетании с ИК-спектроскопией и on-line масс-спектрометрией. Было отмечено сильное подавление скорости адсорбции/окисления метанола в интервале потенциалов 0.05–0.3 В при концентрациях добавок, существенно меньших, чем концентрация метанола. Этот эффект является результатом, главным образом, различных тенденций образования СОадс в присутствии разных С1-молекул и сильным эффектом адсорбции СО на электроокисление метанола при низких потенциалах.
Образование стабильных промежуточных продуктов окисления влияет на эффективность прямого метанольного топливного элемента, причем это влияние зависит от величины загрузки катализатора, скорости протекания электролита, температуры. Первые два эффекта можно понять в рамках модели “десорбция–реадсорбция–дальнейшая реакция”, которая была предложена в работе Бема и сотр. [133]. В этой модели растворимый интермедиат, возникающий на предшествующей стадии, далее выключается из процесса при низкой загрузке катализатора или высокой скорости потока электролита. При высокой загрузке и низкой скорости потока он успевает реадсорбироваться и подвергается дальнейшему окислению. Эта проблема была обсуждена в недавней работе Пикапа и сотр. [134], в которой показано, что образование стабильных продуктов и их диффузия в объем раствора фактически уменьшают эффективное число электронов, участвующих в процессе окисления.
Отдельного внимания заслуживает обсуждение механизма образования метилформиата при электроокислении метанола. Метилформиат НСООСН3 был обнаружен методом ДЭМС среди продуктов электроокисления метанола в работе Иваситы и Фильштиха [135]. Авторы предположили, что метилформиат появляется в результате взаимодействия между метанолом и муравьиной кислотой, диффундирующей в объем раствора. Такое предположение использовалось и в ряде последующих работ (например, [77, 136]).
Попытка уточнить механизм образования метилформиата при электроокислении метанола была предпринята Абд-Эль-Латифом и Балтрушатом в [137]. Авторы нашли, что константа скорости реакции взаимодействия метанола и муравьиной кислоты слишком низка, чтобы обеспечить наблюдаемые выходы метилформиата (порядка 1% [77, 137, 138]). На основании этих и ряда других данных был сделан вывод, что метилформиат образуется вследствие нуклеофильной атаки молекул метанола на адсорбированный интермедиат в соответствии со схемой, представленной на рис. 8 [137]. Это предположение согласуется с данными об увеличении выхода метилформиата при больших концентрациях метанола в растворе [139].
Рис. 8.
Схема образования метилформиата на поликристаллической платине в ходе электроокисления метанола (согласно [137]).

IV. ПРЕДСТАВЛЕНИЯ О МЕХАНИЗМЕ ЭЛЕКТРООКИСЛЕНИЯ МУРАВЬИНОЙ КИСЛОТЫ И ФОРМИАТА
В случае муравьиной кислоты представление о двухмаршрутном механизме окисления обычно связывают с работами Капона и Парсонса [140] и Парсонса и ВандерНута [16]. В отличие от электроокисления метанола, причиной двухмаршрутного механизма служит возможность дегидратации молекулы муравьиной кислоты на поверхности катализатора. Поэтому двухмаршрутный механизм может быть представлен сочетанием реакций:
прямой путь (дегидрирование молекулы)
(12)
$\begin{gathered} {\text{Н С О О Н }} \to {\text{а к т и в н ы й и н т е р м е д и а т }} \to \\ \to {\text{С }}{{{\text{О }}}_{2}} + 2{{{\text{Н }}}^{ + }} + 2{\text{е }}, \\ \end{gathered} $СО-маршрут (дегидратация молекулы)
(13)
${\text{Н С О О Н }} \to {\text{С }}{{{\text{О }}}_{{{\text{а д с }}}}} + {{{\text{Н }}}_{{\text{2}}}}{\text{О }}.$СО-маршрут приводит к образованию СО-адсорбата, который блокирует места для дегидрирования молекул муравьиной кислоты и тем самым тормозит процесс электроокисления при низких потенциалах.
В отличие от платины, на палладии реакция дегидратации НСООН не происходит либо протекает с очень низкой скоростью, что и объясняет более высокую скорость окисления муравьиной кислоты на этом металле. Однако в определенных условиях поляризации СОадс постепенно накапливается, что приводит к торможению процесса.
В последние 10–15 лет изучение механизма электроокисления муравьиной кислоты на Pt и Pd было направлено, главным образом, на решение следующих проблем: 1) выяснение природы активного интермедиата в прямом пути; 2) анализ факторов, позволяющих устранить СО-маршрут либо, по крайней мере, снизить его эффективность; 3) понимание природы процессов, приводящих к образованию СОадс на Pd при низких потенциалах; 4) влияние рН раствора на электроокисление муравьиной кислоты/формиата. При этом активно применялись и вычислительные методы. Обзор вычислительных аспектов изучения дегидрирования муравьиной кислоты на поверхностях различных материалов представлен в [97].
Первоначально утверждалось, что скорость-определяющим интермедиатом прямого пути является формил –СООН, адсорбированный через атом С (cм., например, [1, 141]) и возникающий при дегидрировании молекулы. Однако позднее Осава и его группа методом ATR-SEIRAS обнаружили [142] на химически осажденном на Si тонкослойном Pt-электроде адсорбированный в мостиковой форме формиат НСОО. При достаточно высоких потенциалах формиат был обнаружен и на Pd [143]. Обнаружение адсорбированного формиата вызвало взрывной интерес нескольких групп исследователей к выяснению его роли в электроокислении муравьиной кислоты.
Следует отметить, что формиат является активным интермедиатом окисления муравьиной кислоты в ультравысоком вакууме [144], причем последующее разложение НСОО оказывается лимитирующей стадией на Pt, тогда как на Au процесс лимитируется скоростью образования НСОО.
В предположении, что формиат является активным интермедиатом прямого пути окисления муравьиной кислоты, Осава и сотр. предложили уравнение для скорости электроокисления в виде
(14)
$i = {{d{{\theta }_{{{\text{formate}}}}}} \mathord{\left/ {\vphantom {{d{{\theta }_{{{\text{formate}}}}}} {dt}}} \right. \kern-0em} {dt}} = k{{\theta }_{{{\text{formate}}}}}\left( {1 - {{\theta }_{{{\text{CO}}}}} - 2{{\theta }_{{{\text{formate}}}}}} \right).$Это уравнение означает, что формиат адсорбирован на платине в мостиковой форме и его дальнейшее окисление требует наличия соседнего вакантного места для разрыва связи С–Н [145–147].
Позднее Куеста с соавторами [148, 149] пришли к выводу, что формиат является ключевым интермедиатом как в прямом, так и в СО-маршруте электроокисления (рис. 9) и, кроме того, участвует в бимолекулярном разложении двух соседних адсорбированных частиц с последующим переносом электрона [148]. При этом наличие соседних свободных мест не является обязательным. Таким образом, НСООадс в мостиковой форме можно рассматривать и как точку бифуркации для двухмаршрутного механизма, и как общий прекурсор для образованиия СО2 и СО.
Рис. 9.
Двухмаршрутные механизмы электроокисления муравьиной кислоты на платине: (а) обычно предполагаемый механизм, в котором СОадс и активный интермедиат образуются из адсорбированных молекул НСООН; (б) модифицированный механизм, согласно которому мостиковая форма адсорбированного формиата является точкой бифуркации двух параллельных маршрутов (согласно [148]).
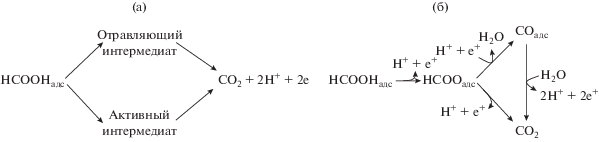
Выводы [148] оказываются в соответствии с результатами Грозовского и др. [150], согласно которым ток окисления муравьиной кислоты прямо пропорционален степени заполнения поверхности формиатом. В [148] проведено cопоставление вывода о природе интермедиата с ранее предлагавшимися на основании электрохимических измерений [140], в том числе в сочетании с радиохимическими [151].
Однако группы Бема и Чена , используя такую же технику, как и в [146], но проточную тонкослойную ячейку, пришли к выводу, что формиат является скорее частицей-статистом (spectator в англоязычной литературе), чем активным интермедиатом [152–156]. К такому же выводу пришли Окамото и коллеги [157], анализируя интенсивность полосы поглощения формиата и токи окисления муравьиной кислоты. DFT-расчеты [91] показали сравнительно высокую стабильность формиата к окислению, что косвенно подтверждает представление о формиате как частице-статисте.
В [152] была предложена схема трехмаршрутного механизма электроокисления муравьиной кислоты, в которой слабо адсорбированные молекулы НСООН рассматриваются как активный интермедиат (рис. 10).
Рис. 10.
Трехмаршрутный механизм электроокисления метанола на платине, включающий СО-маршрут; формиатный маршрут; прямой маршрут с участием слабо адсорбированной молекулы муравьиной кислоты (согласно [152]).

Ванг и Лиу [92] на основании расчетов в рамках теории периодического функционала плотности в сочетании с континуальной моделью раствора показали, что формиат не является ни интермедиатом, ни спектатором, но скорее темплатом, который промотирует адсорбцию молекул НСООН в СН-конфигурации, выступающих в качестве активного прекурсора образования СО2.
В работах Якоба и сотр. [158, 159] на основе расчетов из первых принципов был предложен двухмаршрутный механизм, включающий процесс с промежуточной мостиковой формой НСООадс и прямой путь с участием НСООНадс и короткоживущей формой СОО в качестве интермедиата.
Осава и Копер с коллегами [160–162] предложили модифицированный механизм, в котором формиат-ионы (а не молекулы муравьиной кислоты) окисляются через слабо адсорбированный формиатный прекурсор, причем как ОНадс, так и мостиковая форма НСООадс, блокируя активные места поверхности, тормозят окисление НСОО–.
Группа Чена [163] высказала предположение, что адсорбция молекулы муравьиной кислоты с переносом заряда является лимитирующей стадией, а активный интермедиат прямого пути – некоторая частица Х–, точная природа которой пока остается неясной.
В [164] комбинация формиат-темплатной модели [85] и кислотно-основного равновесия [161, 162] была применена для объяснения эффекта рН и природы аниона на электроокисление муравьиной кислоты. Было высказано предположение, что на Pt(111) в ряду рН 0–2 в растворах, содержащих хлорную, серную или уксусную кислоты, лимитирующая стадия окисления муравьиной кислоты включает, по крайней мере, две адсорбированные частицы – адсорбированный анион (сульфат, ацетат или формиат в мостиковой форме) и формиат-ион из раствора.
Бримауд и коллеги [165] на основе детального изучения влияния рН раствора и природы аниона фона на электроокисление муравьиной кислоты на Pt и Au предположили, что димер-подобные частицы типа НСООН (или НСООадс) и НСОО– являются активным интермедиатом процесса. Позднее [166] влияние рН раствора и природы аниона фона было обсуждено в более широком интервале рН, в том числе в растворах, содержащих хлорид- и фосфат-анионы.
Цай и сотр. [167] привели сводку предложенных схем окисления НСООН, отметив, что окончательные выводы о природе активного интермедиата пока еще сделать невозможно.
В [168] в результате DFT-анализа окисления муравьиной кислоты на Pt и Au в газовой фазе и в электрохимических условиях сделан вывод, что на платине процесс протекает, в зависимости от потенциала, через формиатный и/или прямой путь.
DFT-расчеты показывают, что образование СО из НСООН на Pt(111) через межмолекулярную дегидратацию НСООН димеров (например, по реакции 2НСООН → НСООН + СО + Н2О) имеет более низкий энергетический барьер, чем дегидратация одиночных молекул НСООН [169]. Следует отметить, что образование димеров в объеме раствора за счет водородных связей типично для карбоксильных кислот.
Заслуживает внимания работа [170], посвященная теоретическому анализу окисления муравьиной кислоты. Используя ab initio расчеты модифицированным методом, авторы пришли к выводу, что формиатные частицы при приближении к поверхности в СН-конфигурации безбарьерно распадаются на СО2 и Надс.
В работе [75] обнаружены слабо адсорбированные молекулы НСООН, которые могут быть интермедиатом электроокисления. Формиат был зарегистрирован Ксу и коллегами [63] в качестве активного интермедиата на Rh-модифицированной пленке Au(111-25 нм). На золотом электроде не образуется СОадс и очень четко фиксируется формиат, как показали Куэста с соавторами [171]. Они дали кинетический анализ процесса с учетом образования ассоциатов. В [172] спектроскопические доказательства образования НСООадс представлены на примере коммерческого Pt/C-катализатора. Сравнение с данными [157] для платинового тонкопленочного электрода указывает на возможное различие в СО-маршрутах на пленке и наночастицах платины.
Электроокисление формиата в сильно щелочной среде, когда он является единственным реактантом, изучалось в относительно небольшом числе работ. Кристенсен и сотр. [173] методом in situ FTIR обнаружили образование на поликристаллической платине СОадс в линейной и мостиковой формах. В постулированной модели процесса интермедиат связан с поверхностью скорее через О-атом, чем через атом углерода. Абруна и сотр. [174] опубликовали результаты исследования электроокисления НСОО– при рН 14 на поликристаллической платине методами циклической вольтамперометрии в сочетании с потенцио-статическими импульсами и методом ДЭМС в интервале потенциалов 0.2–0.7 В по ОВЭ в том же растворе. Метод ДЭМС был видоизменен в связи с тем, что единственный продукт реакции – СО2 – в щелочи быстро конвертируется в ${\text{С О }}_{{\text{3}}}^{{{\text{2}} - }}{\text{.}}$ В указанном интервале потенциалов скорость окисления формиата существенно ниже скорости окисления муравьиной кислоты в кислых растворах. Данные по окислению адсорбатов в ходе анодной развертки привели авторов к выводу, что помимо СОадс, присутствует еще один стабильный адсорбат, который может подвергаться окислению и трансформироваться в СОадс в присутствии Надс.
Вьецковский и сотр. [175] изучили электроокисление формиата на платиновой черни с высокой поверхностью при различных температурах (20–80°С). При повышении температуры скорость окисления формиата при низких потенциалах достигает величин, представляющих практический интерес. На основании полученных электрохимических данных и опубликованных ранее результатов был предложен модифицированный трехмаршрутный механизм (рис. 11).
Рис. 11.
Схема трехмаршрутного механизма электроокисления формиата на платине в щелочной среде (по данным [175]).

Детальное исследование динамики взаимодействия муравьиной кислоты с поликристаллическим Pt-пленочным электродом методом разрешенной во времени ATR-FТIR-спектроскопии и изотопным методом при низких потенциалах (0–0.4 В) и температурах 3 и 23°С было опубликовано недавно Юзисом и Бемом [176]. На рис. 12 в качестве примера приведены данные in situ спектроскопии, полученные в этой работе.
Рис. 12.
Эволюция во времени ATR-FTIR-спектров при введении 0.1 М НСООН или DCOOH в контакт с Pt-пленочным электродом в 0.5 М НСlO4 при 0 В (верхняя панель) и при 0.4 В ( нижняя панель) при различных температурах (по данным [176]).

Аппаратура позволяла измерять ИК-спектры с временным разрешением 25 мс. Измерения при 3°С были проведены с целью замедлить скорость реакции и таким образом стабилизировать короткоживущие частицы интермедиата. Кинетический изотопный эффект, найденный благодаря использованию DCOOH, позволил оценить вклад расщепления С–Н-связи в стадию дегидратации муравьиной кислоты. При потенциалах 0–0.1 В обнаружены адсорбированные молекулы НСООН. Никакой временной задержки между появлением адсорбированного в мостиковой форме формиатом и появлением СОадс при потенциалах >0.1 В обнаружено не было. В то же время при низких потенциалах (<0.1В) формиатный сигнал отсутствовал, тогда как СОадс был найден даже при 0 В. Начальная скорость дегидратациии НСООН с образованием СОадс проходит через максимум при 0.2 В и зависит от изотопного состава молекул муравьиной кислоты. Детальное обсуждение совокупности полученных данных привело авторов к выводу о том, что однозначные заключения о механизме взаимодействия муравьиной кислоты с поверхностью платины пока еще невозможны.
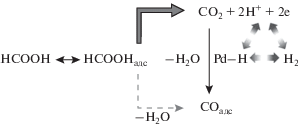
Значительные усилия были направлены на выяснение причин дезактивации палладия в процессе электроокисления муравьиной кислоты [169, 177–182]. Высокая активность палладия в этом процессе связана с тем, что на палладии практически отсутствует СО-маршрут (реакция (13)). Предположение об отсутствии маршрута образования каталитического яда при электроокислении НСООН на палладии было сформулировано Капоном и Парсонсом [183].
Это предположение получило прямое спектроскопическое доказательство в работе Марковича и сотр. [184]. Анализ возможных путей разложения НСООН на Pd был выполнен в ряде работ с помощью DFT-расчетов, в том числе с целью обоснования на молекулярном уровне причин низкой скорости СО-маршрута (см. например, [101, 102, 180, 182]). Ю и Пикап [178] на основании электрохимических измерений пришли к выводу, что деградация каталитических свойств Pd/C-катализатора во времени вероятнее всего связана с накоплением СОадс. Как показано группой Цай [179] методом ATR-SEIRAS, при потенциале разомкнутой цепи и при рабочих потенциалах, действительно, происходит накопление СОадс на Pd, нанесенном на Au-пленку. Однако это является результатом взаимодействия СО2 – продукта прямого пути – с сорбированным палладием водородом. Таким образом, на палладии, вероятно, реализуется схема, представленная на рис. 13.
Рис. 13.
Схема процессов образования СОадс при восстановлении продукта дегидрирования (СО2) на палладии (по [179]).
Этот механизм был подтвержден на примере палладиевой черни [185] и для тонких Pd-слоев на Au [186]. Обрадович и Гойкович [186] характеризуют явление как самоотравление катализатора вследствие последующей реакции с участием продукта реакции.
Заслуживает внимания тот факт, что при проведении измерений на палладии в условиях, когда практически отсутствует СО-маршрут, между анодной и катодной вольтамперограммами не наблюдается гистерезиса.
Некоторые примеры, иллюстрирующие этот эффект, представлены на рис. 14 и 15.
Рис. 14.
Циклические вольтамперограммы на кубических наночастицах палладия в 0.1 М НСlО4 (черные кривые) и в 0.5 М Н2SO4 (серые кривые) в отсутствие (б) и в присутствии (а) 0.5 М НСООН. Скорость развертки потенциала 20 и 50 мВ с–1 (по данным [187]).

Рис. 15.
Циклические вольтамперограммы окисления НСООН на Pd-черни (а), после ее модификации Pt (б–д) и на массивной Pt (е) в растворе 0.1 М H2SO4 + + 0.5 M HCOOH. Сплошные кривые – анодная развертка; пунктир – катодная развертка. Скорость развертки потенциала – 50 мВ с–1 (по данным [188]).
Чен и Копер [189] на Pd ML Pt(100) зарегистрировали токи окисления муравьиной кислоты на фоне хлорной кислоты, лимитируемые диффузией.
V. ВЛИЯНИЕ рН И СОСТАВА РАСТВОРА НА ЭЛЕКТРООКИСЛЕНИЕ МУРАВЬИНОЙ КИСЛОТЫ/ФОРМИАТА
Влияние рН и состава раствора на скорость электроокисления органических веществ может быть использовано для установления и детализации механизма процессов. В последние годы активный интерес вызвало изучение влияния рН на электроокисление муравьиной кислоты/формиата [160, 161, 164–166, 190–193]. Частично эти данные уже обсуждались выше.
Изучение эффекта рН в широком диапазоне его значений и, особенно, при средних значениях рН сопряжено с рядом трудностей, прежде всего вызванных тем, что процесс электроокисления сопровождается образованием или потреблением протонов в приэлектродном слое и возможным изменением рН этого слоя. Яркий пример возникающих при этом осложнений был продемонстрирован Мюллером и сотр. [194, 195]. Использование вращающихся электродов не всегда позволяет привести величины рН приэлектродного слоя к объемному значению рН. В [191] было обращено внимание на роль изменения рН приэлектродного слоя и его отличия от объемного значения и в случае использования анионообменной мембраны. Возможные следствия, возникающие в условиях низкой буферности раствора, на примере ряда электрокаталитических явлений были продемонстрированы в [196].
Наиболее подходящими для изучения эффекта рН в широком диапазоне являются фосфатные буферы. Однако анионы фосфорной кислоты могут адсорбироваться более сильно, чем реактанты или интермедиаты, и таким образом влиять на скорость процесса, маскируя собственно рН-эффект.
Хаан и Мозель [190] изучили влияние рН в интервале 1–5 на электроокисление муравьиной кислоты на 0.1–1 М фоне индифферентного электролита (перхлората, сульфата или фосфата) и нашли, что скорость процесса возрастает в этом интервале рН.
Осава и сотр. [160, 161] провели измерения на вращающемся дисковом Pt-электроде в интервале рН 0–12 в фосфатном буфере и обнаружили колоколообразную зависимость от рН с максимумом вблизи рКа муравьиной кислоты (рКа = 3.7). Авторы высказали предположение, что рост скорости процесса с ростом рН до максимума является следствием возрастания концентрации НСОО– в растворе. Спад тока при рН > pKa был объяснен конкуренцией между НСОО– и ОН– в поверхностном слое и блокированием активных центров ионами ОН– либо ОНадс. Была предложена модель процесса, основанная на предположении о НСОО– как реактанте даже при рН, близких к нулю, и о диссоциации молекул НСООН в приповерхностном слое. Для анализа были использованы величины максимума тока на катодной развертке, так как при катодной развертке поверхность свободна от СОадс, возникающего в соответствии с СО-маршрутом.
Практически в параллель с Осавой и сотр. детальное сопоставление влияния рН на электроокисление муравьиной кислоты на Au и Pt было выполнено Бемом и сотр. [165]. На золотом электроде была найдена колоколообразная зависимость скорости окисления от рН с максимумом при рКа муравьиной кислоты. На платине скорость процесса увеличивалась до рН ~ 5, а затем выходила на плато, протянувшееся вплоть до рН 10. Было высказано предположение, что отличие от данных Осавы и сотр. связано с использованием в [161] сульфатных и перхлоратных сред. Позднее Бем и группа Фелью [166] провели систематическое изучение эффекта рН на поликристаллическом платиновом электроде в широком интервале рН (0–12) на фоне различных, в том числе и комбинированных электролитов. В сульфатных и перхлоратных средах изменение скорости процесса с ростом рН было подобным: скорость возрастала до рН ~ 5, затем выходила на плато, простирающееся до рН 10, а при дальнейшем росте рН резко падала. В фосфатных и хлоридных средах найдена колоколообразная зависимость от рН. Отсюда был сделан вывод, что природа аниона фона является определяющим фактором в рН зависимости. Высказаны предположения о возможной роли взаимодействий между адсорбированными анионами и формиат-анионами, адсорбирующимися в С–Н-конфигурации.
Киблер и сотр. [192] сравнили рН-эффекты при электроокислении муравьиной кислоты/формиата на поли- и монокристаллических (конфигурации (111)) платиновых и золотых электродах в фосфатном буфере с рН в ряду 1–12. Были отмечены влияние структуры поверхности на рН-эффект и более сложный характер зависимости для платины вследствие наличия СО-маршрута. Выводы этой работы, в целом, находятся в согласии с [165].
Недавно Чен и сотр. [197] изучили одновременно рН-эффект и Н/D кинетический изотопный эффект при электроокислении муравьиной кислоты/формиата на вращающемся Pt(111) электроде в менисковой конфигурации (скорость вращения 1000 об/мин). В работе использовались растворы 0.1 М НСООН или DCOOH + + 0.1 M Н2SO4 + x M NaOH (x = 0–0.54), рН которых изменялся от 0 до 14. В отличие от данных [165] в сульфатном фоне получена колоколообразная зависимость скорости процесса от рН с максимумом при рКа. Кинетический изотопный эффект составлял ~5; 2; 1 в растворах с рН 1.1; 3.6; 13. Полученные результаты заставляют предположить, что прекурсором процесса являются ионы НСОО–, а лимитирующая стадия процесса НСОО– → НСООадс + е. Результаты этой работы ставят под сомнение полученные ранее выводы в небуферных растворах поверхностно-неактивных анионов, а также вывод об определяющей роли специфической адсорбции анионов в рН-эффекте. Кроме того, по-видимому, уменьшение скорости реакции при рН > 5 вряд ли связано с адсорбцией ОН на поверхности платины.
Резюмируя изложенное, следует признать, что эффект рН в окислении муравьиной кислоты/формиата на платине заслуживает дальнейшего исследования. Влияние рН на электроокисление муравьиной кислоты/формиата на Pd-электроде практически не изучено.
Адсорбция анионов раствора была использована для установления природы интермедиата окисления муравьиной кислоты Бемом и сотр. [154]. Согласно ИК-спектроскопическим измерениям в смеси НСООН и СН3COOH, было найдено подавление ИК-полосы адсорбированного формиата вследствие адсорбции ацетата. Однако торможение электроокисления муравьиной кислоты оказывалось при этом не столь значительным, чем можно было ожидать, исходя из модельных представлений о формиате как основном интермедиате. Отсюда авторы сделали вывод, что мостиковая форма адсорбированного формиата не является активным интермедиатом в прямом пути окисления и предложили третий путь окисления с участием слабо адсорбированных молекул НСООН, который представлен выше на рис. 10.
Анионы Сl– подавляют электроокисление НСООН на палладии в кислых растворах [189]. ATR-SEIRAS измерения показали при этом, что покрытие поверхности СО в мостиковой форме возрастает во всем интервале потенциалов, а адсорбция НСОО в мостиковой форме возрастает лишь в некотором интервале потенциалов. Таким образом, эффект Cl– не сводится только к блокировке поверхности.
В работе [198] была подчеркнута роль загрязнений при измерениях в щелочных растворах. Эти загрязнения попадают либо из стекла, если измерения проводятся в стеклянных ячейках, либо из электролитов, используемых для измерений. В качестве загрязнений, вероятнее всего, выступают следы катионов 3d-переходных металлов.
Маркович и сотр. [199] обнаружили влияние природы катиона щелочного металла на скорость ряда электрокаталитических процессов, в том числе и на электроокисление метанола, на платине в щелочных растворах. Они объяснили этот эффект наличием нековалентных взаимодействий в поверхностном слое. Явление привлекло внимание к изучению адсорбции катионов на платине в щелочных растворах и ее роли в электрокаталитических процессах [200–202].
VI. ЭНЕРГИИ АКТИВАЦИИ РЕАКЦИЙ ЭЛЕКТРООКИСЛЕНИЯ МЕТАНОЛА И МУРАВЬИНОЙ КИСЛОТЫ
Энергия активации может быть рассчитана по результатам изучения влияния температуры на скорость электродного процесса.
Для электроокисления метанола этот параметр определялся в большом числе работ на разных типах катализаторов. Краткий обзор более ранних исследований можно найти в [28]. В этой работе представлены результаты оригинальных измерений на поликристаллической платине в кислых и щелочных растворах в условиях линейной развертки потенциала в анодном и катодном направлениях в интервале температур 20–60°С и в широком интервале потенциалов, а также результаты метода хроноамперометрии. Отмечено, что рассчитываемые кажущиеся энергии активации являются сложными функциями таких факторов, как адсорбция интермедиатов, анионов фона и форм адсорбированного кислорода.
Ванг и Балтрушат [126] изучили влияние температуры на окисление метанола методами вольтамперометрии и хроноамперометрии в сочетании с он-лайн ДЭМС на поликристаллической платине, в том числе модифицированной рутением, и на гранях Pt(111) и Pt(332). Было отмечено, что с ростом температуры скорости обоих маршрутов окисления метанола возрастают в равной степени, что означает отсутствие изменений в механизме процесса и близкие величины энергий активации разных маршрутов.
Эффект температуры на электроокисление НСООН был изучен Варела и сотр. [203, 204] на гладкой поликристаллической платине [203] и на грани Pt(100) [204]. На грани Pt(100) при низких потенциалах токи не зависели от температуры, что свидетельствует о нулевой энергии активации. Это явление заслуживает специального исследования.
Изучение влияния температуры на электроокисление формиата [175] на платиновой черни привело к выводу, что скорость всех реакций, включенных в трехмаршрутный механизм (рис. 11), возрастает с температурой, но это возрастание отвечает различным энергиям активации. При низких потенциалах с ростом температуры вероятно изменение медленной стадии двухэлектронного процесса окисления – от медленного переноса первого электрона к медленному переносу второго электрона. Этот вывод следует из величин тафелевских наклонов процесса.
Из данных метода хроноамперометрии в ряде работ (см., например, [29, 129]) были построены тафелевские зависимости, отвечающие t = 0, при низких анодных потенциалах, когда ток определяется стадией дегидрирования молекулы метанола: СН3ОН → СН2ОНадс + Н+ + е. В интервале потенциалов 0.3–0.65 В на Pt(554) наклон составляет 110 мВ, а на Pt(553) – 95 мВ [29]. Однако по измерениям [129] наклон составил 440 ± 30 мВ. Согласно DFT-расчетам [107], тафелевский наклон для этой стадии должен быть равным 166 мВ. Причины столь большого разброса данных, а также эксперимента и расчета пока не ясны.
VII. МЕХАНИЗМ ЭЛЕКТРООКИСЛЕНИЯ СО-АДСОРБАТА
СО является устойчивым при низких потенциалах адсорбатом, который образуется на платине из метанола и муравьиной кислоты в СО-маршрутах и, блокируя места на поверхности, тормозит процессы при Е < 0.5 В и температурах 25–75°С. Знание механизмов образования и электроокисления СОадс чрезвычайно важно для конструирования катализаторов электроокисления, толерантных к СО. Поэтому изучению адсорбции и электроокисления СО посвящено большое количество работ.
Гильман [205] предложил механизм “реагирующих пар” для электроокисления СОадс в кислых растворах – взаимодействие СО-адсорбата с кислородсодержащими частицами, адсорбированными на соседних местах. Такие частицы образуются в результате адсорбции молекул воды и обычно представляются как ОНадс, хотя точная природа этих частиц еще не ясна. Таким образом, общая схема реакции окисления СОадс выглядит следующим образом:
(15)
${{{\text{Н }}}_{{\text{2}}}}{\text{О }} + {\text{* }} \leftrightarrow {\text{О }}{{{\text{Н }}}_{{{\text{а д с }}}}} + {{{\text{Н }}}^{ + }} + {\text{е }},$(16)
${\text{С }}{{{\text{О }}}_{{{\text{а д с }}}}} + {\text{О }}{{{\text{Н }}}_{{{\text{а д с }}}}} \to {\text{С О О }}{{{\text{Н }}}_{{{\text{а д с }}}}},$(17)
${\text{С О О }}{{{\text{Н }}}_{{{\text{а д с }}}}} \to {\text{С }}{{{\text{О }}}_{{\text{2}}}} + {{{\text{Н }}}^{ + }} + {\text{е }} + 2*,$Реакцию (16) можно рассматривать как разновидность механизма Лангмюра–Хиншельвуда, широко известного в катализе. Так как реакция (16) является медленной, а последующая реакция (17) – быстрой, шансы обнаружить карбоксигидроксил СООНадс в качестве интермедиата окисления СОадс невелики, что и подтверждается тщательным анализом результатов измерений методом ATR-SEIRAS (cм., например, [209, 210], хотя в более ранней работе [211] некоторые особенности спектра СОадс были истолкованы как следствие присутствия СООН в поверхностном слое).
Брайтер [212] гальваностатическим методом обнаружил, что хемосорбированный при разомкнутой цепи СО на платинированной платине окисляется в двух областях потенциалов. В настоящее время, когда основным методом изучения электрокаталитических процессов является вольтамперометрия, область низких анодных потенциалов (0.35–0.6 В) называется областью предпика (pre-ignition region) [22, 23, 213, 214]. Механизм окисления в области предпика представляет особый интерес, так как эта область относится к практически важному интервалу перенапряжений. Предпик обычно приписывают окислению СОадс на дефектных местах, природа которых остается предметом дискуссий (места на ступенях или, например, наноостровки адатомов Pt на террасах [215]). Условия воспроизводимого получения предпика были выяснены в [216]. В этой работе, а также в [217] обсуждаются некоторые концепции, объясняющие природу предпика.
В области более высоких перенапряжений обнаруживаются один или несколько пиков на вольтамперограммах, природа которых обсуждалась в большом числе работ [214, 218–222].
Для электроокисления СОадс было предложено два основных механизма: 1) модель “среднего поля”, согласно которой подвижность СОадс является быстрой в сравнении со скоростью его взаимодействия с ОНадс и обеспечивает быстрое смешивание СОадс и ОНадс [77, 123, 223]; 2) модель нуклеации–роста, согласно которой адсорбированные частицы неподвижны, а окисление происходит по границам контактов доменов/фаз ОНадс и СОадс [224, 225]. Во многих случаях модель среднего поля позволяет удовлетворительно описать транзиенты тока окисления СОадс при постоянном потенциале. В то же время in situ ИК спектроскопия предоставляет доказательства диполь-дипольного взаимодействия между адсорбированными молекулами СО [218, 226], что должно приводить к образованию островков СО. Кроме того, поверхностная диффузия СОадс оказывается, по-видимому, не столь быстрой, как предполагалось ранее [216, 217]. Майлард, Савинова и Штимминг [227] подчеркнули, что для наночастиц на носителях результаты вольтамперометрического и хроноамперометрического изучения электроокисления хемосорбированного СО зависят от размерного распределения наночастиц и их коалесценции, а сами электрохимические измерения могут служить для диагностики структуры катализатора.
Интерес представляет сопоставление электроокисления СО, адсорбированного из растворов СО, и СОадс, образовавшегося из метанола или муравьиной кислоты в СО-маршруте. Такое сравнение пока не было проведено в должном масштабе и достаточно детально. Сравнение ИК-спектров на наночастицах Pt в [129] показало, что в растворах метанола обнаруживается только СО в линейной форме и этот адсорбат более слабо связан с поверхностью наночастиц, чем возникающий в присутствии СО в растворе.
VIII. ВЗАИМОДЕЙСТВИЕ МЕТАНОЛА И МУРАВЬИНОЙ КИСЛОТЫ С АДСОРБИРОВАННЫМ КИСЛОРОДОМ
Несколько работ было выполнено с целью выяснения механизма взаимодействия метанола и муравьиной кислоты с предварительно адсорбированным кислородом на Pt при разомкнутой цепи.
Подловченко, Манжос и Максимов в серии работ [228–232] провели измерения транзиентов бестокового потенциала при введении Pt-электрода с предварительно адсорбированным кислородом в контакт с растворами С1-соединений. В ходе сдвига потенциала определялась степень заполнения поверхности адсорбированным кислородом с помощью быстрых катодных импульсов. В случае метанола был выполнен анализ раствора на муравьиную кислоту и формальдегид хроматографическим методом. Согласно предположениям авторов, в области высоких заполнений поверхности кислородом сдвиг потенциала вызван прямым взаимодействием молекул растворенного вещества с адсорбированными атомами кислорода (по механизму Илеа–Риделя). Для области средних заполнений Оадс было предложено уравнение для транзиентов бестокового потенциала в растворах HCOOH и CH3OH, полученное в предположении о протекании сопряженных реакций – электроокисления органических молекул на свободных от кислорода местах и электровосстановления адсорбированных атомов кислорода. Предложенное уравнение удовлетворительно описывает транзиенты для области средних заполнений поверхности Оадс. Уравнение учитывает различие в количестве атомов платины, необходимых для хемосорбции молекул HCOOH и CH3OH.
Варела и сотр. исследовали влияние времени предварительного окисления платины и природы аниона (${\text{С lO}}_{{\text{4}}}^{ - }{\text{,}}$ ${\text{SO}}_{{\text{4}}}^{{{\text{2}} - }},$ Cl–) на поведение потенциала после размыкания цепи в растворах метанола [233] и применили метод in situ ATR-FTIR для анализа продуктов взаимодействия метанола [234] и муравьиной кислоты [235] с хемосорбированным на Pt кислородом. Они пришли к выводу, что это взаимодействие носит автокаталитический характер. В [235] были предложены дифференциальные уравнения, описывающие прямое взаимодействие растворенных молекул НСООН с адсорбированным кислородом с учетом роли подповерхностного кислорода, прямое окисление HCOOH до СО2, реакцию окисления НСООН через промежуточное образование СОадс, окисление СОадс. Интегрирование этих уравнений позволило рассчитать транзиенты потенциала и зависимости выхода СО2 и СОадс от потенциала, которые удовлетворительно совпали с найденными на опыте.
Чен с сотр. и Юзис и Бем в совместной работе [236] изучили взаимодействие СО, HCOOH, H2CO, CH3OH c окисленной платиной методом измерения потенциала после размыкании цепи в сочетании с in situ ИК- и масс-спектроскопией. Сочетание этих методов позволило получить наиболее полную картину эволюции состава адсорбатов и продуктов реакций во время сдвига потенциала. Авторы выделяют три фазы процесса. Cразу после размыкания цепи при 1.2 В идет восстановление Оадс с образованием ОНадс и/или Pt и практически не наблюдается выделения СО2. После появления некоторого количества свободных атомов Pt начинается интенсивное выделение СО2 за счет окисления С1-молекул (по-видимому, по механизму Лангмюра–Гиншельвуда). В третьей фазе этот процесс тормозится вследствие накопления СОадс в линейной и мультисвязанной формах либо из раствора (в случае растворов СО), либо из-за разложения С1-молекул.
Изучение взаимодействия С1-соединений с адсорбированным кислородом представляет интерес с точки зрения выяснения влияния кроссовера метанола на эффективность топливного элемента. В действительности это влияние оказывается более сложным, чем в описанных выше экспериментах, и требует специального изучения в условиях, приближенных к реализующимся в практическом топливном элементе. Это, в частности, показано недавно Юрзинским и сотр. [237] с помощью метода ДЭМС и анализа продуктов реакции окисления метанола на катоде топливного элемента.
IX. ВЛИЯНИЕ СТРУКТУРЫ ПОВЕРХНОСТИ И РАЗМЕРНЫХ ФАКТОРОВ НА МЕХАНИЗМ ЭЛЕКТРООКИСЛЕНИЯ
Электроокисление С1-соединений является удобным модельным объектом для выяснения роли структурных и размерных эффектов в электрокатализе. В большинстве работ сопоставлялись скорости окисления С1-соединений на различных катализаторах, хотя в ряде исследований предпринимались попытки объяснить найденные эффекты с учетом маршрутов процессов.
Рассматриваемые процессы относятся к структурно-чувствительным как в катализе, так и электрокатализе. Модельными объектами для изучения структурных эффектов являются, прежде всего, грани монокристаллов платиновых металлов. Большой вклад в изучение структурных эффектов при электроокислении метанола и муравьиной кислоты внесла группа Фелью из Аликантe, а обзор [238] может служить прекрасным введением в электрохимию поверхностей монокристаллов. Отметим, что одно из первых исследований электроокисления муравьиной кислоты на 16 различных гранях платины было выполнено методом вольтамперометрии Мотоо и Фуруя [239].
Копер и сотр. [29, 117] использовали он-лайн ДЭМС для изучения структурной чувствительности маршрутов электроокисления метанола на Pt(111), Pt((110), Pt(100) и на ступенчатых поверхностях Pt(554) и Pt(553). Найдено, что максимальная активность увеличивается в ряду Pt(111) < < Pt(110) < Pt(100), причем прямой путь через растворенные интермедиаты наиболее сильно выражен на Pt(110) и Pt(111), тогда как на Pt(100) преобладающим оказывается СО-путь. Наличие на гранях [110] ступеней катализирует прямой маршрут.
Грозовский и коллеги использовали методы импульсной вольтамперометрии и хроноамперометрии для выяснения структурной чувствительности электроокисления муравьиной кислоты [240, 241] и метанола [30]. Измерения на Pt(100) и вицинальных поверхностях, содержащих моноатомные (111) ступени, (Pt(2n-1,1,1)) позволили, в рамках определенных модельных представлений, сделать вывод, что при электроокислении НСООН для поверхностей с широкими террасами экстраполированные на нулевое время токи окисления и константы скорости образования СОадс практически не зависят от плотности ступеней, т.е. активности ступеней и террас близки для обоих маршрутов. Для узких террас (n < 6) собственная активность платины снижается. Образование СО вследствие дегидратации НСООН имеет место в узком интервале потенциалов, близких к потенциалу нулевого полного заряда (~0.38 В). Авторы пришли к выводу, что на идеальной поверхности Pt(111) образования СО не происходит, т.е. реализуется лишь прямой путь окисления. Анализ транзиентов тока в ходе электроокисления метанола при различных потенциалах [30] был выполнен на основе упрощенной модели Хусманса–Копера [29]. Наивысшая скорость прямого пути была найдена на Pt(100). Присутствие (111) и (100) ступеней на (100) и (111) террасах не приводит к увеличению активности этих поверхностей, но (110) ступени повышают активность катализатора. В условиях измерений первой и медленной стадией процесса оказывается стадия дегидрирования. Грань (111) на порядок менее активна, чем (100), однако она менее подвержена отравлению.
Копер [242] классифицировал и обсудил наноразмерные эффекты в электрокатализе с особенным вниманием к двум видам особенностей: 1) ступени и дефекты на (111) террасах или нанофасетках; 2) протяженные (110) террасы или фасетки.
Куеста [119] продемонстрировал роль ансамбль-эффекта при дегидрировании метанола до СОадс на Pt(111). При модифицировании поверхности платины цианид-ионами можно создать условия, когда свободными оказываются только пары атомов платины. При этом образования СОадс не происходит и реализуется лишь прямой маршрут реакции. Отсюда был сделан вывод, что СО-маршрут требует по крайней мере трех соседних атомов платины. В дальнейшем этот вывод был подтвержден с применением, наряду с вольтамперометрией и ИК-спектроскопией, метода ДЭМС [120]. Ансамбль-эффект был найден и при электроокислении муравьиной кислоты. Прием с использованием модификации поверхности цианид-ионами для блокирования одного из маршрутов процесса был назван Куестой “site- knockout strategy” [243]. Он предполагает, что хемосорбция цианида не влияет на электронные свойства поверхности.
Изучение процессов на наночастицах, находящих широкое использование на практике, привело к многочисленным попыткам выяснения природы зависимости электрокаталитических свойств от размера наночастиц. Размерный эффект, в действительности, оказывается результатом комплексного действия многих факторов. Так, от размера частиц могут зависеть их электронные свойства. С размером частиц изменяется соотношение террас и ступеней, а также выход тех или иных граней на поверхность. Размер наночастиц влияет на возможность появления поверхностных дефектов, наличие которых чрезвычайно важно в электрокатализе.
Вивер и сотр. [244] предприняли попытку выяснения роли размерных эффектов в электроокислении метанола, муравьиной кислоты и формальдегида, проведя изучение электроокисления этих соединений на Pt-наночастицах разного диаметра в ряду 2–9 нм. Было найдено уменьшение скорости электроокисления метанола на частицах с диаметром менее 4 нм, тогда как скорость электроокисления муравьиной кислоты возрастала с уменьшением размера частиц платины. Это различие авторы связали с рядом факторов, но наиболее существенным является разный механизм окисления: для окисления метанола требуется кислородсодержащая частица, тогда как для муравьиной кислоты это не обязательно.
Копер и сотр. [245] исследовали электроокисление С1-соединений на электролитических осадках платины на Au-подложке, полученных гальваностатическим методом. Был сделан вывод, что осадки имитируют свойства наночастиц даже при больших размерах, чем в [244].
Солла-Гуллон и коллеги [246] исследовали электроокисление метанола и муравьиной кислоты на наночастицах платины контролируемой формы. Частицы с ориентацией (111) проявляют наиболее низкую активность, но и наиболее медленно отравляются в ходе процесса.
Шао-Хорн и сотр. [247] нашли практически линейное соотношение между активностью наночастиц платины размером ~2 нм, нанесенных на многостенные углеродные нанотрубки, и числом ступеней типа n((111) × (111)) на их поверхности в реакции электроокисления метанола.
В [248] показано, что при исследовании размерного эффекта необходимо учитывать величину загрузки катализатора.
Значительные усилия были предприняты для выяснения роли структурных и размерных факторов в электроокислении муравьиной кислоты на палладии как наиболее активном индивидуальном катализаторе для этого процесса (cм., например, [187, 249–255]). Изучение процессов на палладии осложнено растворением водорода в его решетке, более низкой коррозионной стойкостью палладия по сравнению с платиной и более легкой реконструкцией поверхности в ходе циклирования потенциала. Структурная чувствительность была продемонстрирована при измерениях на низко- и высокоиндексных гранях массивного палладия [249, 250]. В ряде работ изучалась размерная зависимость скорости процесса на дисперсных палладиевых катализаторах. Повышенная активность малых наночастиц была объяснена особенностями их электронных свойств [251]. Однако Видал-Иглесиаз и коллеги [187], сравнив свойства кубических и сферических наночастиц, сделали вывод, что главную роль играет не размер наночастиц, а их поверхностная структура. К подобному выводу пришли авторы [255]. В этой работе отмечено возможное влияние загрязнения наночастиц стабилизатором, использованным при синтезе наночастиц, на результаты in situ спектроскопии.
Cистематическое изучение окисления муравьиной кислоты на палладиевых наночастицах различной формы и двойниковых структурах (кубы, бипирамиды, октаэдры, тетраэдры, додекаэдры и икосаэдры) выполнено Чой и коллегами [254]. Нанокристаллы с {100} фасетками оказываются более активными, чем с {111} нанофасетками. Скорость окисления возрастает при наличии двойниковых дефектов.
Штимминг и сотр. [256] изучили электроокисление муравьиной кислоты на электролитических осадках палладия различных толщин в интервале 3–15 нм. Осадки были нанесены с помощью двухимпульсного потенциостатического метода на высокоориентированный пиролитический графит. Анодные вольтамперограммы на осадке толщиной 15 нм были подобны наблюдаемым на массивном поликристаллическом палладии. На более тонких осадках токи оказались меньше, причем величины токов падали с уменьшением толщины осадка, а максимум окисления муравьиной кислоты смещался в катодную сторону. Авторы предположили, что этот эффект связан с наличием на тонких осадках высокой доли ступеней и низкокоординированных атомов, которые оказываются активными в процессе восстановления продукта реакции СО2 с образованием СОадс, согласно схеме на рис. 13.
X. ЭЛЕКТРООКИСЛЕНИЕ МЕТАНОЛА И МУРАВЬИНОЙ КИСЛОТЫ НА БИНАРНЫХ КАТАЛИЗАТОРАХ
Электроокисление метанола и муравьиной кислоты изучалось на разнообразных каталитических материалах. Подробное рассмотрение этих работ выходит за рамки настоящего обзора. Ниже кратко обсуждены особенности электроокисления на некоторых репрезентативных биметаллических катализаторах.
Система Pt–Ru исследовалась наиболее широко как перспективная в прикладном плане (cм., например, обзоры [27, 39] и ссылки в них). Важную роль в исследовании этой системы сыграла работа Ватанабы и Мотоо [257], в которой были впервые изучены процессы в присутствии адатомов рутения и привлечена концепция бифункционального механизма для истолкования природы сокатализа. Гаштайгер, Mаркович и коллеги [258, 259], используя PtRu-катализаторы с хорошо охарактеризованной поверхностью, также пришли к выводу, что основную роль в ускорении электроокисления метанола играет бифункциональный механизм, т.е. адсорбция метанола с дегидрированием на платиновых ансамблях и окислительная десорбция СОадс при взаимодействии с ОНадс, возникающим на соседних рутениевых атомах:
(18)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{O}}{{{\text{H}}}_{{\text{р }}}}_{{{\text{--р }}}} \leftrightarrow {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{O}}{{{\text{H}}}_{{{\text{а д с }}}}} \to {\text{C}}{{{\text{O}}}_{{{\text{а д с }}}}} + 4{{{\text{H}}}^{ + }} + 4{\text{e}},$(19)
${\text{Ru}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \leftrightarrow {\text{RuO}}{{{\text{H}}}_{{{\text{а д с }}}}} + {{{\text{H}}}^{ + }} + {\text{e,}}$(20)
${\text{C}}{{{\text{O}}}_{{{\text{а д с }}}}} + {\text{O}}{{{\text{H}}}_{{{\text{а д с }}}}} \to {\text{C}}{{{\text{O}}}_{{\text{2}}}} + {{{\text{H}}}^{ + }} + {\text{e}}.$Ускорение процесса при практически важных потенциалах на сплаве вызвано тем, что образование ОНадс на рутении происходит при существенно более низких потенциалах (почти на 0.3 В), чем на платине.
Была предложена модель, которая предсказывает, что оптимальный состав поверхности должен отвечать ~10 ат. % Ru в предположении, что три атома платины необходимы для адсорбции молекулы метанола.
Анализ кинетики электроокисления метанола на PtRu/C с точки зрения бифункционального механизма в квазистационарных условиях был выполнен в [260].
Группа Вьецковского [261], основываясь на результатах, полученных с использованием твердофазного электрохимического ЯМР, нашла, что рутений ослабляет связь Pt–CO, т.е. имеет место также электронный (или лиганд-) эффект.
Изучение свойств модифицированных платиной монокристаллов рутения показывает, что наблюдаемое при модификации увеличение каталитической активности связано с электронным эффектом, вызванным сжатием решетки (уменьшением межатомного расстояния Pt–Pt), и увеличением концентрации поверхностных дефектов [262]. Особенности электрокаталитического поведения Pt–Ru-систем типа ядро–оболочка обсуждены в [27, 263]. Контролируемо декорированные платиной коммерческие рутениевые наночастицы были синтезированы в [264] и показали вулканообразную зависимость от степени покрытия поверхности платиной с максимумом при 0.31. Такое поведение было истолковано как результат конкуренции двух процессов: дегидрирования метанола с отщеплением трех атомов водорода на платиновых ансамблях и удаления СО на Ru/Pt-местах по бифункциональному механизму. Кузнецов и коллеги [265] детально изучили влияние концентрации границ зерен в PtRu-наночастицах на электроокисление метанола и хемосорбированного СО и нашли снижение энергии активации процессов с увеличением степени коалесценции наночастиц и плотности межзеренных границ. При этом тафелевские наклоны квази-стационарных поляризационных кривых оставались практически постоянными. Снижение энергии активации с ростом плотности межзеренных границ указывает, вероятно, на возрастание скорости СО-маршрута электроокисления метанола.
В [266] данные фотоэлектронной спектроскопии с использованием синхротронного излучения были привлечены для изучения роли электронной структуры наночастиц PtRu в их электрокаталитической активности в реакциях электроокисления метанола и муравьиной кислоты. В случае метанола основную роль играет бифункциональный механизм, тогда как для муравьиной кислоты вклад изменения электронной структуры значителен.
Использование метода ДЭМС позволяет более детально представить механизм электроокисления на сплавах Pt–Ru. Абруна и сотр. [138] показали, как основываясь на электрохимических измерениях и данных метода ДЭМС (выходов по току СО2 и метилформиата), можно найти оптимальный состав катализатора для электроокисления метанола. Они подчеркнули роль снижения скорости дегидрирования/адсорбции метанола при высоких концентрациях рутения в сплаве. Ускорение процесса в присутствии рутения связано с увеличением скорости удаления СО в СО-маршруте процесса.
Корженевский и сотр. [267] разработали методику определения выхода формальдегида в ходе электроокисления метанола. Наименьший выход формальдегида на массивных PtRu-сплавах при низких анодных потенциалах обнаружен при атомной доле рутения, равной 0.3. По мнению авторов, это объясняется балансом атомных платиновых ансамблей, активирующих связь С–Н, и рутениевых центров, генерирующих ОН-группы.
Лиу и коллеги [268] теоретически учли возможность кроссовера между О–Н- и С–Н-маршрутами дегидрирования метанола на ступенчатой поверхности Pt(211) и при ее модификации адатомами рутения. Модификация приводит к существенному снижению барьера как первой стадии дегидрирования с разрывом связи С–Н, так и ряда дальнейших стадий и стимулирует возможный переход к О–Н-маршруту. В результате наиболее вероятной оказывается последовательность: СН3ОН → СН2ОН → (СН2О и/или СНОН) → → СНО → СО.
Динг и сотр. [269] на основе DFT-анализа пришли к выводу, что СО-маршрут мало вероятен с кинетической точки зрения, и процесс на PtRu/Pt(111) предпочтительно должен протекать по схеме: СН3ОН → СН3О → СН2О → СНО → → СНООН → СООН → СО2, с медленной стадией дегидрирования метоксила.
Другая бинарная система, cодержащая оксофильный компонент и в принципе перспективная для электроокисления С1-соединений, – Pt‒Sn. Эта система позволяет получить высокие скорости окисления СО при низких потенциалах и в этом плане даже более эффективна, чем Pt‒Ru. Ванг и коллеги [270] на хорошо охарактеризованном сплаве PtSn наблюдали значительное ускорение электроокисления растворенного СО, но практическое отсутствие эффекта в случае электроокисления метанола. Они предположили, что СОадс из метанола отличается от адсорбата, образующегося из растворенного СО. Данные об активности синтезированных разными способами Pt–Sn-систем противоречивы. Краткий анализ ранних работ (однако недостаточно полный) можно найти в [271]. Сплавы Pt–Sn, синтезированные в этой работе по реакции прекурсоров с муравьиной кислотой, показали высокие характеристики, особенно после определенной температурной обработки. В целом же условия получения Pt–Sn-катализаторов с высокой каталитической активностью для электроокисления метанола требуют дальнейшего исследования. В [272] были показаны влияние адатомов Sn и синэргетический эффект от совместного присутствия адатомов Sn и Ru на электроокисление метанола.
Четыре системы: Pt(111), Pt3Sn(111), Ru(0001) и монослой Pt на Ru(0001)-субстрате – были рассмотрены в рамках теории функционала плотности Норсковым и сотр. [273] с целью объяснения влияния сплавления Pt с Ru и Sn на электроокисление СО и влияния СО на ионизацию молекулярного водорода.
Стеванович и коллеги [274] изучили влияние различных форм олова, в том числе подповерхностных атомов олова, на каталитическую активность платины.
Недавно Фариас и сотр. [275] сопоставили эффекты Sn- и Ru-адатомов на электроокисление СОадс на ступенчатых поверхностях платины. Они подчеркнули, что Sn и Ru проявляют высокое сродство к связыванию и диссоциации воды, но Sn инертно к адсорбции СО, тогда как Ru прочно адсорбирует СО. Эффект этих адатомов зависит от кристаллической структуры поверхности и от кристаллической ориентации платиновых мест. Не исключено, что важную роль в электрокатализе в присутствии Sn играет подвижность ОНадс, поскольку частицы СОадс на Pt-центрах практически неподвижны. Ризо и коллеги [276] изучили электроокисление СО на модифицированных оловом гранях Pt(100), Pt(110), Pt(111). На Sn/Pt((100) не найдено предпика окисления СО, в отличие от двух других граней. В области основного пика окисления предполагается классический бифункциональный механизм. Расчеты по теории функционала плотности и микрокинетическое моделирование показали [277], что наиболее вероятный путь разложения метанола на Pt3Sn(111) поверхности может быть представлен следующим образом: СН3ОН → СН3О → СН2О → → СНО → СО.
Россмейсл и коллеги [95] использовали в качестве дескрипторов при DFT-анализе электроокисления метанола на сплавах величины энергий связи ОН и СО с плотно упакованными поверхностями. Активные катализаторы для этого процесса должны обладать следующими тремя свойствами: 1) активировать молекулу метанола; 2) активировать молекулу воды; 3) обеспечивать взаимодействие поверхностных интермедиатов – СО и ОН. Согласно этой работе, таким условиям лучше всего отвечают системы Pt–Ru и Pt–Sn, а также поверхностные и объемные сплавы Pt–Cu, причем наиболее эффективный Pt–Cu-катализатор должен иметь поверхностный состав Cu3Pt1/Pt. Аналогичный вывод сделан Тритсарисом и Россмейслом [278], которые рассмотрели также и некоторые другие системы с плоскими и ступенчатами поверхностями.
На родии адсорбированный кислород появляется при более низких потенциалах по сравнению с платиной. Поэтому представляет интерес изучение электроокисления метанола на сплавах платина–родий. Как было показано Чоем и коллегами [279], электроокисление метанола ускоряется на сплаве Pt–Rh и особенно на сплаве Pt/Ru/Rh. Эффект был объяснен на основе классического бифункционального механизма. Однако Шенг и Сун [280] с помощью DFT-расчетов показали, что в присутствии Rh, наряду с классическим механизмом, вероятно ускорение процесса за счет снижения барьера для расщепления связи С–Н вблизи одиночных атомов родия.
В плане выяснения роли электронных эффектов в электрокатализе представляют интерес сплавы платины и золота. Сунтивич и коллеги [281] представили прямые доказательства корреляции между каталитической активностью и поверхностным составом Pt–Au-наночастиц. Температурная обработка была использована для селективного обогащения платиной или золотом катализатора Au0.5Pt0.5/C и тьюнинга электрокаталитической активности сплава. Максимальный синэргетический эффект получен при 70 ат. % Au на поверхности. В случае золота эффект не может быть вызван бифункциональным механизмом, так как золото не активирует процесс образования ОНадс. Таким образом, здесь имеет место эффект снижения энергии связи Pt–COадс, вызванный электронным влиянием атомов золота на соседние атомы платины или на богатые платиной кластеры.
В случае электроокисления муравьиной кислоты природа эффекта промотирования процесса на бинарных поверхностях при низких потенциалах отлична от наблюдаемой при электроокислении метанола. Это связано, прежде всего, с тем, что СО-маршрут в этом случае представляет собой процесс дегидратации молекул НСООН. Реакция дегидратации является структурно-чувствительной и требует для своего протекания наличия на поверхности определенных ансамблей из атомов платины. Поэтому стратегия промотирования может быть основана на введении посторонних атомов (“третьего тела”), которые предотвращают возможность возникновения ансамблей атомов платины, необходимых для дегидратации молекул НСООН и образования СОадс. Термин “эффект третьего тела” был предложен Конвеем [282]. Фактически это явление можно рассматривать как разновидность ансамбль-эффекта. Лейва и коллеги [283] представили простую теоретическую модель для истолкования влияния адатомов на электроокисление муравьиной кислоты на основе эффекта “третьего тела”. Однако анализ большого экспериментального материала по влиянию адатомов на электроокисление НСООН показывает, что необходимо учитывать и целый ряд других эффектов. Фелью и сотр. [284–286] среди этих факторов отмечают электронные эффекты адатомов и бифункциональные явления. Последние могут быть вызваны влиянием адатомов на адсорбцию кислородсодержащих частиц. Влияние адатомов на электроокисление муравьиной кислоты обсуждено в [287] с использованием расчетов по теории функционала плотности. Изменение эффективности адатомов в ряду Pb–Pt(111) > > Bi–Pt(111) > Sb–Pt(111) > Te–Pt(111) > As–Pt(111) > Pt(111) > Se–Pt(111) = S–Pt(111) было связано с наличием остаточного положительного заряда на адатоме, облегчающего образование и адсорбцию формиата в конфигурации, которая облегчает разрыв связи С–Н. Этот эффект можно рассматривать как разновидность бифункционального механизма. В таком случае в качестве дескрипторов активности адатомных слоев можно рассматривать различие в электроотрицательностях адатома и субстрата.
Важную роль при этом играет и кристаллографическая структура поверхности катализаторов. Влияние декорирования поверхности Pt/C-катализатора Pb и Sb на характеристики муравьинокислого топливного элемента было обсуждено в [288] на основе двухмаршрутного механизма окисления муравьиной кислоты.
Интенсивному исследованию в электроокислении муравьиной кислоты были подвергнуты Pt–Pd-катализаторы разных типов [36, 188, 289–294] и, прежде всего, модифицированные адатомами Pd грани Pt(100) и Pt(111) [289]. На Pdad + Pt(100) было найдено значительное снижение области потенциалов окисления НСООН и отсутствие самоотравления процесса, тогда как на Pdad + Pt(111) эффекты были незначительными. В [290] методом спонтанного осаждения Pd на Pt были получены катализаторы с высокой активностью в окислении муравьиной кислоты. Aктивирующее влияние декорирования Pt-кубических наночастиц палладием на окисление муравьиной кислоты показано в [291]. Псевдоморфные слои Pd на Pt(111) и (111) ориентированных Pt–Pd-поверхностях массивных сплавов были изучены [293] электрохимическими методами и FTIR и показали ускорение процесса по сравнению с платиной и значительное снижение количества СОадс вследствие торможения дегидратации НСООН. В серии PdxPt1– x наносплавов на углеродном носителе наиболее активным и стабильным оказался сплав Pd0.9Pt0.1/C [294]. Была предложена модель из сферических частиц (ball model), которая иллюстрирует “эффект третьего тела” на примере сплава Pd–Pt. Одновременно подчеркивалась возможная роль смещения середины d-полосы при сплавлении в изменении электрокаталитической активности. В [188] показан значительный активирующий эффект (по сравнению с Pd) при отсутствии гистерезиса между анодной и катодной вольтамограммами на электроде 0.08Pt@Pd.
Значительное внимание было уделено системам Pt–Au и Pd–Au [295–308]. Ускорение электроокисления муравьиной кислоты в этих случаях объясняется ансамбль-эффектом и изменением электронной структуры поверхности. Атомный ансамбль-эффект находит подтверждение в DFT-расчетах [304, 305]. In situ SERS измерения в системе PtAu [307] и FTIR и ДЭМС результаты для системы AuPd [308] показывают, что однозначные выводы, однако, пока еще невозможны.
Абруна и сотр. [309] изучили электроокисление метанола, формальдегида и муравьиной кислоты на упорядоченных интерметаллических фазах PtPb и PtBi с помощью электрохимических методов в сочетании с ДЭМС и FTIRS. Хотя эти фазы имеют подобную структуру и межатомные расстояния, они проявляют различную активность в изученных реакциях. Во всех случаях не наблюдается образования СОадс, т.е. СО-маршрут не реализуется. Наивысшую активность в реакции окисления НСООН проявляет сплав PtPb. Тафелевский наклон поляризационной кривой окисления муравьиной кислоты на этом сплаве при низких потенциалах составляет 120 мВ, что позволяет предположить медленный перенос первого электрона в качестве лимитирующей стадии.
Стратегия использования биметаллических катализаторов широко используется в электрокатализе и стимулирует разработку теоретических подходов к конструированию таких систем. Некоторые подходы рассматривались выше. В действительности, это предмет специального обзора. Укажем некоторые обзоры, посвященные этой проблеме [310–313].
XI. ЗАКЛЮЧЕНИЕ
Итак, к настоящему времени достигнут консенсус относительно двухмаршрутного механизма электроокисления метанола до СО2 на Pt: 1) прямой путь через промежуточное образование растворимых продуктов – формальдегида и муравьиной кислоты (метилформиата); 2) СО-маршрут через образование устойчивого при низких потенциалах СО-адсорбата. В случае электроокисления муравьиной кислоты вероятен трехмаршрутный механизм, причем природа активного интермедиата пути, вносящего основной вклад в процесс, пока остается под вопросом. В ходе электроокисления метанола надежно установлено с помощью электрохимической ИК-спектроскопии образование в качестве адсорбированных интермедиатов СОадс и НСООадс. Многочисленные другие возможные адсорбированные интермедиаты (см. схемы, приведенные выше) обнаружены не были либо из-за того, что имеют очень короткие времена жизни, либо из-за того, что их спектральные характеристики не соответствуют ИК-диапазону. Ряд вопросов, касающихся природы и роли интермедиатов в окислении С1-соединений, которые требуют ответов, сформулированы в [214].
Значительный прогресс в понимании механизмов адсорции и электроокисления метанола и муравьиной кислоты оказался возможным благодаря разработке и применению методов in situ cпектроскопии и ДЭМС. Отметим, что для разработки этих методов рассматриваемые процессы явились весьма удобными модельными системами. Совершенствование методов исследования остается актуальной задачей и необходимо для дальнейшего углубления в механизм процессов. Определенные надежды в этом плане связаны и с расширяющимся использованием расчетов по теории функционала плотности. Выше приведены некоторые примеры того, как подобные расчеты указывают на возможные новые маршруты реакций (например, кроссоверные маршруты), требующие экспериментального подтверждения. Детальное знание механизма процессов необходимо для разработки подходов к синтезу новых высокоэффективных катализаторов и дескрипторов электрокаталитической активности.
В настоящее время интерес к электроокислению метанола стимулируется также решением задачи эффективного получения водорода из воды [314–316]. Идея заключается в том, чтобы заменить реaкцию выделения кислорода на аноде электролизера реакцией электроокисления метанола, которая протекает при более низких потенциалах. Общий процесс в таком электролизере можно представить уравнением:
Таким образом, можно констатировать, что исследование механизмов электроокисления С1-соединений будет оставаться в числе мейнстримных направлений и в будущем.
Список литературы
Electrocatalysis. Eds. Lipkowski J., Ross P.N. Wiley-VCH. 1998.
Handbook of Fuel Cells. Fundamentals Technology and Applications. V. 2. Electrocatalysis. Eds. Vielstich, W., Lamm, A., N.A. Gasteiger. Wiley. 2003.
Petrii, O.A., Podlovchenko, B.I., Frumkin, A.N., and Hira Lal, The behaviour of platinized-platinum and platinum-ruthenium electrodes in methanol solutions, J. Electroanal. Chem., 1965, vol. 10, p. 253.
Podlovchenko, B.I., Petrii, O.A., Frumkin, A.N., and Hira Lal, The behavior of a platinized-platinum electrode in solutions of alcohols containing more than one carbon atom, aldehydes and formic acid, J. Electroanal. Chem., 1966, vol. 11, p. 12.
Bagotzky, V.S., Vassiliev, Yu.B., and Khazova, O.A., Generalized scheme of chemisorption, electrooxidation and electroreduction of simple organic compounds on platinum group metals, J. Electroanal. Chem., 1977, vol. 81, p. 229.
Bagotzky, V.S. and Vassilyev, Yu.B., Mechanism of electro-oxidation of methanol on the platinum electrode, Electrochim. Acta, 1967, vol. 12, p. 1323.
Kazarinov, V.E., Tysyachnaya, G.Ya., and Andreev, V.N., On the reasons for the discrepancies in the data on methanol adsorption on platinum, J. Electroanal.Chem., 1975, vol. 65, p. 391.
Problemy elektrokatalyza (Problems of electrocatalysis) (in Russian). Ed. Bagotzky, V.S. Izd. “Nauka,” Moscow, 1980. 271 p.
McNicol, B.D., Rand, D.A.J., and Williams, K.R., Direct methanol-air fuel cells for road transportation, J. Power Sources, 1999, vol. 83, p. 15.
Lamy, C., Lima, A., LeRhun, V., Delima, F., Countanceau, C., and Leger, J.-M., Recent advances in the development of direct alcohol fuel cell (DAFC), J. Power Sources, 2002, vol. 105, p. 283.
Yu, X.W. and Pickup, P.G., Recent advances in direct formic acid fuel cells (DFAFC), J. Power Sources, 2008, vol. 182, p. 124.
Rees, N.V. and Compton, R.G., Sustainable energy: a review of formic acid electrochemical fuel cells, J. Solid-State Electrochem., 2011, vol. 15, p. 2095.
An, L. and Chen, R., Direct formate fuel cells: A review, J. Power Sources, 2016, vol. 320, p. 127.
Hampson, N.A., Willars, M.J., and McNicol, B.D., The methanol-air fuel cell: a selective review of methanol oxidation mechanisms at platinum electrodes in acid electrolytes, J. Power Sources, 1979, vol. 4, p. 191.
Lamy, C., Leger, J.M., Clavilier, J., and Parsons, R., Structural effects in electrocatalysis. A comparative study of the oxidation of CO, HCOOH and CH3OH on single crystal Pt electrodes, J. Electroanal.Chem., 1983, vol. 150, p. 71.
Parsons, R. and VanderNoot, T., The Oxidation of Small Organic Molecules: A Survey of Recent Fuel Cell Related Research, J. Electroanal. Chem., 1988, vol. 257, p. 9.
Beden, B., Leger, J.-M., and Lamy, C., Electrocatalytical oxidation of oxygenated aliphatic organic compounds at noble metal electrodes, Modern asp. Electrochem., vol. 22, p. 97. Plenum Press. N.Y. 1992. Bockris J.O., M., Conway B.E., White R.E. Eds.
Hamnett, A., Mechanism and electrocatalysis in the direct methanol fuel cell, Catal. Today, 1997, vol. 38, p. 445.
Wasmus, S. and Kuver, A., Methanol oxidation and direct methanol fuel cell, J. Electroanal.Chem., 1999, vol. 461, p. 14.
Arico, A., Srinivasan, S., and Antonucci, V., DMFCs: From fundamental aspects to technology development, Fuel Cells, 2001, vol. 1, p. 133.
Iwasita, T., Electrocatalysis of methanol oxidation, Electrochim. Acta, 2002, vol. 47, p. 3663.
Markovic, N.M. and Ross P.N., Jr., Surface science studies of model fuel cell electrocatalysts, Surf. Sci. Reports, 2002, vol. 45, p. 117.
Mayrhofer, K.J.J., Arenz, M., Blizanak, B., Stamenkovich, V., Ross, P.N., and Markovic, N.M., CO surface electrochemistry on Pt-nanoparticles: a selective review, Electrochim. Acta, 2005, vol. 50, p. 5144.
Yu, E.H., Wang, X., Krewer, U., Li, L., and Scott, K., Direct oxidation alkaline fuel cells: from materials to systems, Energy Environ. Sci., 2012, vol. 5, p. 5668.
Bartrom, A. and Haan, J., The Direct Formate Fuel Cell with alkaline anion exchange membrane, J. Power Sources, 2012, vol. 214, p. 68.
Jiang, J. and Wieckowski, A., Prospective direct formate fuel cell, Electrochem. Commun., 2012, vol. 18, p. 41.
Tolmachev, Yu.V. and Petrii, O.A., Pt–Ru electrocatalysts for fuel cells: developments in the last decade, J. Solid State Electrochem., 2017, vol. 21, p. 613.
Cohen, J.L., Volpe, D.J., and Abruna, H.D., Electrochemical determination of activation energies for methanol oxidation on polycrystalline platinum in acidic and alkaline electrolytes, PhysChemChemPhys., 2007, vol. 7, p. 49.
Housmans, T.H.M. and Koper, M.T.M., Methanol oxidation on stepped Pt[n(111) × (110)] electrodes: a chronoamperometric study, J. Phys. Chem. B., 2003, vol. 107, p. 8557.
Grozovski, V., Climent, V., Herrero, E., and Feliu, J.M., The role of the surface structure in the oxidation mechanism of methanol, J. Electroanal. Chem., 2011, vol. 662, p. 43.
Clavilier, J., Pulsed linear sweep voltammetry with pulses of constant level in a potential scale, demanding condition in the study of platinum single crystal electrodes, J. Electroanal. Chem., 1987, vol. 236, p. 87.
Xu, W., Lu, T., Liu, C., and Xing, W., Supplement of the theory of normal puls voltammetry and its application to the kinetic study of methanol oxidation on a polycrystalline platinum electrode, J. Phys. Chem., 2005, vol. 109, p. 7872.
Murthy, A. and Manthiram, A., Electrocatalytic oxidation of methanol to soluble products on polycrystalline platinum: Application of convolutive potential sweep voltammetry in the estimation of kinetic parameters, Electrochim. Acta, 2011, vol. 56, p. 6078.
Okamoto, H., Kon, W., and Mukouyama, Y., Five current peaks in voltammograms for oxidation of formic acid, formaldehyde and methanol on platinum, J. Phys. Chem. B., 2005, vol. 109, p. 15659.
Chung, D.Y., Lee, K-J., and Sung, Y.-E., Methanol electrooxidation on the Pt surface: revisiting the cyclic voltammetry interpretation, J. Phys.Chem. C., 2016, vol. 120, p. 9028.
Lee, Y.-W., Lee, J.-J., Kwak. D.-H., Hwang, E.-T., Jang, I.S., and Park, K.-W., Pd@Pt core–shell nanostructures for improved electrocatalytic activity in methanol oxidation reaction, Appl. Catal. B: Environmental, 2015, vol. 179, p. 178.
Hofstead-Duffy, A.M., Chen, O.J., Sun, S.G., and Tong, Y.J., Origin of the current peak of negative scan in the cyclic voltammetry of methanol electro-oxidation on Pt-based electrocatalysts: A revisit to current ratio criterion, J. Mater. Chem., 2012, vol. 22, p. 5205.
Vidakovich, T., Christov, M., and Sundmacher, K., Rate expression for electrochemical oxidation of methanol on a direct methanol fuel cell anode, J. Electroanal. Chem., 2005, vol. 580, p. 105.
Petrii, O.A., Pt–Ru electrocatalysts for fuel cells: a representative review, J. Solid State Electrochem., 2008, vol. 12, p. 609.
Bao, W.-Q., He, X.-D., Wang, Y., and He, J.-B., Diffusion-restriction electrodeposition of platinum on solid carbon paste for electrocatalytic oxidation of methanol, Catal.Today, 2016, vol. 264, p. 198.
Piela, P., Fields, R., and Zelenay, P., Electrochemical Impedance Spectroscopy for direct methanol fuel cell diagnostics, J. Electrochem. Soc., 2006, vol. 153, p. A1902.
Bruckenstein, S. and Gadde, R.R., Use of a porous electrode for in situ mass spectrometric determination of volatile electrode reaction products, J. Am. Chem. Soc., 1971, vol. 93, p. 793.
Wolter, O. and Heitbaum, J., Differential electrochemical mass spectrometry (DEMS)—a new method for the study of electrode processes, Ber. Bunsenges. Phys. Chem., 1984, vol. 88, p. 2.
Willsau, J., Wolter, O., and Heitbaum, J., On the nature of the adsorbate during methanol oxidation at platinum, J. Electroanal. Chem., 1985, vol. 185, p. 163.
Hartung, T. and Baltruschat, H., Differential electrochemical mass spectrometry using smooth electrodes: adsorption and H/D-exchange reactions of benzene on Pt, Langmuir, vol. 6, p. 953.
Wang, H., Loffler, T., and Baltruschat, H., Formation of intermediates during methanol oxidation: A quantitative DEMS study, J. Appl. Electrochem., 2001, vol. 31, p. 759.
Wonders, A.H., Housmans, T.H.M., Rosca, V., and Koper, M.T.M. On-line mass spectrometry system for measurements at single-crystal electrodes in hanging meniscus configuration, J. Appl. Electrochem., 2006, vol. 36, p. 1215.
Lu, J., Hua, X., and Long, Y.-T., Recent advances in real-time and in situ analysis of an electrode-electrolyte interface by mass spectrometry, Analyst, 2017, vol. 142, p. 691.
Willsau, J. and Heitbaum, J., Analysis of adsorbed intermediates and determination of surface potential shifts by DEMS, Electrochim. Acta, 1986, vol. 31, p. 943.
Chen, Y.-X., Heinen, M., Jusys, Z., and Behm, R.J., Kinetic isotope effects in complex reaction networks: formic acid electrooxidation, Chem. Phys. Chem., 2007, vol. 8, p. 380.
Jusys, Z. and Behm, R.J., DEMS analysis of small organic molecule electrooxidation: a high-temperature high-pressure DEMS study, ECS Trans., 2008, vol. 16, p. 1243.
Baltruschat H., Differential Electrochemical Mass Spectrometry, J. Am. Soc. Mass Specrom., 2004, vol. 15, p. 1693.
Wang, H., Alden, L., DiSalvo, E.J., and Abruna, H.D., Electrocatalytic mechanism and kinetics of SOC oxidation on ordered PtPb and PtBi intermetallic compounds: DEMS and FTIRS study, PhysChemChemPhys., 2008, vol. 10, p. 3739.
Wang, H., Rus, E., and Abruna, H.D., New double-band-electrode channel flow differential electrochemical mass spectrometry cell: application for detecting product formation during methanol electrooxidation, Anal. Chem., 2010, vol. 82, p. 4319.
Zhou, W., Jusys, Z., and Behm, R.J., Complete quantitative online analysis of methanol electrooxidation products via electron impact and electrospray ionization mass spectrometry, Anal. Chem., 2012, vol. 84, p. 5479.
Cheng, S., Wu, Q., Dewald, H.D., and Chen, H., Online monitoring of methanol electrooxidation reactions by ambient mass spectrometry, J. Am. Soc. Mass Spectrom., 2017, vol. 28, p. 1005.
Bewick, A., Beden, B., Lamy, C., and Kunimatzu, K., Electrosorption of methanol on a platinum electrode. IR spectroscopic evidence for adsorbed CO species, Electroanal. Chem., 1981, vol. 121, p. 343.
Osawa, M., Dynamic processes in electrochemical reactions studied by surface-enhanced infrared absorption spectroscopy (SEIRAS), Bull. Chem. Soc. Jpn., 1997, vol. 70, p. 2861.
Chen, W., Cai, J., Yang, J., Sartin, M.M., and Chen, Y.-X., The kinetics of methanol oxidation at a Pt film electrode, a combining mass and infrared spectroscopic study, J. Electroanal. Chem., 2017, vol. 800, p. 89.
Heinen, M., Chen, Y.X., Jusys, Z., and Behm, R.J., In situ ATR-FTIRS coupled with on-line DEMS under controlled mass transfer conditions—A novel tool for electrocatalytic reaction study, Electrochim. Acta, 2007, vol. 52, p. 5634.
Pronkin, S. and Wandlowski, Th., ATR-SEIRAS approach to probe the reactivity of Pd-modified quasi-single crystal gold film electrodes, Surf. Sci., 2004, vol. 573, p. 109.
Пронькин, С., Хара, M., Вандловский, T. Электрокаталитические свойства Au(111)-Pd квазимонокристаллических пленочных электродов, изученные методом ATR-SEIRAS. Электрохимия. 2006. Т. 42. С. 1312. [Pronkin, S., Hara, M., and Wandlowski, T., Electrocatalytic Properties of Au(111)-Pd Quasi-Single Crystal Film Electrodes as Probed by ATR-SEIRAS, Russ. J. Electrochem., 2006, vol. 42, p. 1177.]
Xu, Q., Pobelov, I.V., Wandlowski, T., and Kuzume, A., ATR-SEIRAS study of formic acid adsorption and oxidation on Rh modified Au(111-25 nm) film electrodes in 0.1 M H2SO4, J. Electroanal. Chem., 2017, vol. 793, p. 70.
Wain, A.J. and O’Connell, M.A., Surface-enhanced vibrational spectroscopy at electrochemical interfaces, Advances in Physics. X, 2017, vol. 2, p. 188.
Лазоренко-Маневич, Р.М. Адатомная гипотеза как преобладающий механизм усиленного поверхностью Рамановского рассеяния: обзор экспериментальных подтверждений. Электрохимия. 2005. Т. 41. С. 899. [Lazorenko-Manevich, R.M., Adatom hypothesis as a predominant mechanism of surface enhanced Raman scattering: A review of experimental argumentation, Russ. J. Electrochem., 2005, vol. 41, p. 799.]
Wu, D.-Y., Li, J.-F., Ren, B., and Tian, Z.-Q., Electrochemical surface-enhanced Raman spectroscopy of nanostructures, Chem. Soc. Rev., 2008, vol. 37, p. 1025.
Leung, L.-W.H. and Weaver, M.J., Extending surface-enhanced Raman spectroscopy to transition-metal interfaces: carbon monoxide adsorption and electrooxidation on platinum- and palladium-coated gold electrodes, J. Am. Chem. Soc., 1987, vol. 109, p. 5113.
Zou, S. and Weaver, M.J., Surface-enhanced scattering in uniform transition-metal films: toward a versatible adsorbate vibrational strategy for solid-nonvacuum interfaces, Analyt. Chem., 1998, vol. 70, p. 2387.
Li, J.-F., Zhang, Y.-J., Ding, S.-Y., Panneerscaan, R., and Tian, Z.-Q., Core-shell nanoparticle-enhanced Raman spectroscopy, Chem. Rev., 2017, vol. 117, p. 5002.
Jeong, H. and Kim, J., Insights into the electrooxidation mechanism of formic acid on Pt layers on Au examined by electrochemical SERS, J. Phys. Chem.C., 2016, vol. 120, p. 24271.
Stamenkovich, V., Chou, K.C., Somorjai, G.A., Ross, P.N., and Markovic, N.M., Vibrational properties of CO at the Pt(111)-solution interface: The anomalous Stark-tuning slope, J. Phys. Chem. B., 2005, vol. 109, p. 678.
Zhang, P., Wei, Y., Cai, J., Chen, Y.-X., and Tian, Z.-Q., Nonlinear Stark effect observed for carbon monoxide chemisorbed on gold core/palladium shell nanoparticles film electrodes, using in situ surface-enhanced Raman spectroscopy, Chin. J. Catal., 2016, vol. 37, p. 1156.
Beltramo, G.L., Shubina, T.E., and Koper, M.T.M., Oxidation of formic acid and carbon monoxide on gold electrodes studied by surface-enhanced Raman spectroscopy and DFT, Chem. Phys. Chem., 2005, vol. 6, p. 2597.
Loupe, M., Dean, J., and Smotkin, E.S., Twenty years of operando IR, X-ray absorption and Raman spectroscopy: direct methanol and hydrogen fuel cell, Catal. Today, 2017, vol. 283, p. 11.
Behrens, R.L., Lagutchev, A., Dlott, D.D., and Wieckowski, A., Broad-band sum frequency generation study of formic acid chemisorptions on Pt(100) electrode, J. Electroanal. Chem., 2010, vol. 649, p. 32.
Tong, Y., Cai, K., Wolf, M., and Campen, R.K., Probing the electrooxidation of weakly adsorbed formic acid on Pt(100), Catal. Today, 2016, vol. 260, p. 66.
Lai, S.C.S., Lebedeva, N.P., Housmans, T.H.M., and Koper, M.T.M., Mechanism of carbon monoxide and methanol oxidation at single-crystal electrodes, Top. Catal., 2007, vol. 46, p. 320.
Kua, J. and Goddard, W.A., III, Oxidation of Methanol on 2nd and 3rd Row Group VIII Transition Metals (Pt, Ir, Os, Pd, Rh, and Ru): Application to Direct Methanol Fuel Cells, J. Am. Chem. Soc., 1999, vol. 121, p. 10928.
Ishikawa, Y., Liao, M.-S., and Cabrera, C.R., Oxidation of methanol on platinum, ruthenium and mixed Pt–M metals (M = Ru, Sn): a theoretical study, Surf. Sci., 2000, vol. 463, p. 66.
Desai, S.K., Neurock, M., and Kourtakis, K., A periodic Density Functional Theory study of the dehydrogenation of methanol over Pt(111), J. Phys. Chem. B, 2002, vol. 106, p. 2559.
Okamoto, Y., Sugino, O., Mochizuki, Y., Ikeshoji, T., and Morikawa, Y., Comparative study of dehydrogenation of methanol at Pt(111)/water and Pt(111)/vacuum interfaces, Chem. Phys. Lett., 2003, vol. 377, p. 236.
Greely, J. and Mavrikakis, M., Competitive paths for methanol decomposition on Pt(111), J. Am. Chem. Soc., 2004, vol. 126, p. 3910.
Shubina, T.E., Hartnig, C., and Koper, M.T.M., Density functional theory study of the oxidation of CO by OH on Au(110) and Pt(111) surfaces, PhysChemChemPhys., 2004, vol. 6, p. 4215.
Cao, D., Lu, G.Q., Wieckowski, A., Wasileski, S.A., and Neurock, M. Mechanisms of methanol decomposition on platinum: a combined experimental and ab initio approach, J. Phys. Chem. B, 2005, vol. 109, p. 11622.
Hartnig, C. and Spohr, E., The role of water in the initial steps of methanol oxidation on Pt(111), Chem. Phys., 2005, vol. 319, p. 185.
Kandoi, S., Greeley, J., Sanchez-Castillo, M.A., Evans, S.T., Gonhale, A.A., Dumesic, J.A., and Mavrikakis, M., Prediction of experimental methanol decomposition rates on platinum from first principles, Top. Catal., 2006, vol. 37, p. 17.
Janik, M.J., Taylor, C.D., and Neurock, M., First principles analysis of the electrooxidation of methanol and carbon monoxide, Top. Catal., 2007, vol. 46, p. 306.
Hartnig, C., Grimminger, J., and Spohr, E., The role of water in the initial steps of methanol oxidation on Pt(211), Electrochim. Acta, 2007, vol. 52, p. 2236.
Hartnig, C., Grimminger, J., and Spohr, E., Adsorption of formic acid on Pt(111) in the presence of water, J. Electroanal. Chem., 2007, vol. 607, p. 133.
Ferrin, P., Njkelar, A.U., Greeley, J., Mavrikakis, M., and Rossmeisl, J., Reactivity descriptors for direct methanol fuel cell anode catalysts, Surf. Sci., 2008, vol. 602, p. 3424.
Neurock, M., Janik, M., and Wieckowski, A., A first principles comparison of the mechanism and site requirements for the electrocatalytic oxidation of methanol and formic acid over Pt, Faraday Discuss., 2008, vol. 140, p. 363.
Wang, H.-F. and Liu, Z.-P., Formic acid oxidation at Pt/H2O interface from Periodic DFT calculations integrated with a continuum solution model, J. Phys. Chem. C, 2009, vol. 113, p. 17502.
Ferrin, P. and Mavrikakis, M., Structure sensitivity of methanol electrooxidation on transition metals, J. Am. Chem. Soc., 2009, vol. 131, p. 14381.
Schnur, S. and Gross, A., Challenges in the first-principles description of reactions in electrocatalysis, Catal. Today, 2011, vol. 165, p. 129.
Rossmeisl, J., Ferrin, P., Tritsaris, G.A., Nilekar, A.U., Koh, S., Bai, S.E., Brankovic, S.R., Strasser, P., and Mavrikakis, M., Bifunctional anode catalysts for direct methanol fuel cell, Energy Environ. Sci., 2012, vol. 5, p. 8335.
Zhong, W., Wang, R., Zhang, D., and Liu, C., Theoretical study of the oxidation of formic acid on the PtAu(111) surface in the continuum water solution phase, J. Phys. Chem. C, 2012, vol. 116, p. 24143.
Luo, Q., Beller, M., and Jiao, H., Formic acid dehydrogenation on surfaces—A review of computational aspect, J. Theor. Comput. Chem., 2013, vol. 12, p. 1330001.
Miller, A.V., Kaichev, V.V. Prosvirin, I.P., and Bukhtiyarov, V.I., Mechanistic study of methanol decomposition and oxidation on Pt(111), J. Phys. Chem. C, 2013, vol. 117, p. 8189.
Braunchweig, B., Hibbitts, D., Neurock, M., and Wieckowski, A., Electrocatalysis: a direct alcohol fuel cell and surface science perspective, Catal. Today, 2013, vol. 202, p. 197.
Anderson, A.B. and Asiri, H.A., Reversible potentials for steps in methanol and formic acid oxidation and CO2 reduction, PhysChemChemPhys., 2014, vol. 16, p. 10587.
Scaranto, J. and Mavrikakis, M., Density functional theory studies of HCOOH decomposition on Pd(111), Surf. Sci., 2015, vol. 650, p. 111.
Wang, X., Chen, L., and Li, B., A density functional theory study of methanol dehydrogenation on the PtPd3(111) surface, Intern. J. Hydrogen Energy, 2015, vol. 40, p. 9656.
Zhong, W., Qi, Y., and Dong. M., The ensemble effect of formic acid oxidation on platinum-gold electrode studied by first-principles calculations, J. Power Sources, 2015, vol. 278, p. 203.
Fang. Y.-H. and Liu. Z.-P., Tafel kinetics of electrocatalytic reactions: from experiment to first principles, ACS Catal., 2014, vol. 4, p. 4364.
Ding, Q., Xu, W., Sang, P., Xu, J., Zhao, L., Xe, X., and Guo, W., Insight into reaction mechanisms of methanol on PtRu/Pt(111): a Density Functional study, Appl. Surf. Sci., 2016, vol. 369, p. 257.
Gasper, R.J. and Ramasubramamian, A., Density functional theory studies of methanol decomposition reaction on grapheme-supported Pt13 nanoclusters, J. Phys. Chem. C, 2016, vol. 120, p. 17498.
Fang, Y.-H. and Liu, Z.-P., First principles Tafel kinetics of methanol oxidation on Pt(111), Surf. Sci., 2016, vol. 631, p. 42.
Scaranto, J. and Mavrikakis, M., HCOOH decomposition on Pt(111): A DFT study, Surf. Sci., 2016, vol. 648, p. 201.
Sakong, S. and Gross, A., The importance of the electrochemical environmental in the electrooxidation of methanol on Pt(111), ACS Catal., 2016, vol. 6, p. 5574.
Wang, Q.-Y. and Ding, Y.-H., Mechanism of methanol oxidation on graphene-supported Pt: Defect is better or not?, Electrochim. Acta, 2016, vol. 216, p. 140.
Sakong, S. and Gross, A., Methanol oxidation on Pt(111) from first-principles in heterogeneous and electrocatalysis, Electrocatalysis, 2017, vol. 8, p. 577.
Ou, L. and Huang, J., DFT-based study in the optimal CH3OH decomposition pathways in aqueous-phase : Homolysis versus heterolysis, Chem. Phys. Lett., 2017, vol. 679, p. 66.
Du, P., Wu, P., and Cai, C., Mechanism of methanol decomposition of the Pt3Ni surface: DFT study, J. Phys. Chem. C, 2017, vol. 127, p. 9348.
Park, S., Xie, Y., and Weaver, M., Electrocatalytic pathways on carbon-supported platinum nanoparticles: comparison of particle-size-dependent rates of methanol, formic acid and formaldehyde electrooxidation, Langmuir, 2002, vol. 18, p. 5792.
Breiter, M.W., A study of intermediates adsorbed on platinized platinum during the steady-state oxidation of methanol, formic acid and formaldehyde, J. Electroanal. Chem., 1967, vol. 14, p. 407.
Breiter, M.W., Role of adsorbed species for the anodic methanol oxidation on platinum in acidic electrolytes, Discuss. Faraday Soc., 1968, vol. 45, p. 79.
Housmans, T.H.M., Wanders, A.H., and Koper, M.T.M., Structure sensitivity of methanol electrooxidation pathways on platinum: an on-line electrochemical mass spectrometry study, J. Phys. Chem. B, 2006, vol. 110, p. 10021.
Chen, Y.X., Miki, A., Ye, S., Sakai, H., and Osawa, M., Formate, an active intermediate for direct oxidation of methanol on Pt-electrode, J. Am. Chem. Soc., 2003, vol. 125, p. 3680.
Cuesta, A., At least three contiguous atoms are necessary for CO formation during methanol electrooxidation on platinum, J. Am. Chem. Soc., 2006, vol. 128, p. 13332.
Cuesta, A., Escudero, M., Lanova, B., and Baltruschat, H., Cyclic Voltammetry, FTIRS, and DEMS Study of the Electrooxidation of Carbon Monoxide, Formic Acid, and Methanol on Cyanide-Modified Pt(111) Electrodes, Langmuir, 2009, vol. 25, p. 6500.
Kunimatsu, K., Hanawa, N., Uchida, H., and Watanabe, M., Role of adsorbed species in methanol oxidation on Pt studied by ATR-FTIRAS combined with linear potential sweep voltammetry, J. Electroanal. Chem., 2009, vol. 632, p. 109.
Franaszsczuk, K., Herrero, E., Zelenay, P., Wieckowski, A., Wang, J., and Masel, R.I., A comparison of electrochemical and gas-phase decomposition of methanol on platinum surfaces, J. Phys. Chem. B, 1992, vol. 96, p. 8509.
Lebedeva, N.P., Koper, M.T.M., Feliu, J.M., and van Santen, R.A., Mechanism and kinetic of the electrochemical CO adlayer oxidation on Pt(111), .Electroanal. Chem., 2002, vol. 524–525, p. 242.
Sriramulu, S., Jarvi, T.D., and Stuve, E.M., Reaction mechanism and dynamics of methanol electrooxidation on platinum(111), J. Electroanal. Chem., 1999, vol. 467, p. 132.
Batista, E.A., Malpass, G.R.P., Motheo, A.J., and Iwasita, T., New mechanistic aspects of methanol oxidation, J. Electroanal. Chem., 2004, vol. 571, p. 273.
Wang, H. and Baltruschat, H., DEMS study on methanol oxidation at poly- and monocrystalline platinum electrodes: the effects of anion, temperature, surface structure, Ru adatoms and potential, J. Phys. Chem. C, 2007, vol. 111, p. 7038.
Mostafa, E., Abd-El-Latif, A. A., and Baltruschat, H., Electrocatalytic Oxidation and Adsorption Rate of Methanol at Pt Stepped Single-Crystal Electrodes and Effect of Ru Step Decoration: A DEMS Study, Chem. Phys. Chem., 2014, vol. 15, p. 2029.
Liao, L.W., Liu, S.X., Tao, Q., Geng, B., Zhang, P., Wang, C.M., Chen, Y.X., and Ye, S., A method for kinetic study of methanol oxidation on Pt-electrode by electrochemical in situ infrared spectroscopy, J. Electroanal. Chem., 2010, vol. 650, p. 233.
Liu, S.X., Liao, L.W., Tao, Q., Chen, Y.X., and Ye, S., The kinetics of CO pathway in methanol oxidation at Pt electrode, a quantitative study by ATR-FTIR spectroscopy, PhysChemChemPhys., 2011, vol. 13, p. 9725.
Tao, Q., Chen, W., Yao, Y., Yousaf, A.B., and Chen, Y.-X., Study of methanol oxidation at Pt and PtRu electrodes by combining in situ infrared spectroscopy and differential electrochemical mass spectrometry, Chin. J. Chem. Phys., 2014, doi 10.1063/1674-oo68/27/05/541-547
Reichert, R., Schnaidt, J., Jusys, Z., and Behm, R.J., The influence of reactive side products in electrocatalytic reactions: methanol oxidation as case study, Chem. Phys. Chem., 2013, vol. 14, p. 3678.
Reichert, R., Schnaidt, J., Jusys, Z., and Behm, R.J., The influence of reactive side products on the electrooxidation of methanol—a combined in situ infrared spectroscopy and on line mass spectrometry study, PhysChemChemPhys., 2014, vol. 16, p. 13780.
Seidel, Y.E., Schneider, A., Jusys, Z., Wiesman, B., Kasemo, B., and Behm, R.J., Mesoscopic mass transport effects in electrocatalytic processes, Faraday Discuss., 2008, vol. 140, p. 167.
Majidi, P., Altarawnef, R.M., Ryan, N.D.W., and Pickup, P.G., Determination of the efficiency of methanol oxidation in a direct methanol fuel cell, Electrochim. Acta, 2016, vol. 199, p. 210.
Iwasita, T. and Vielstich, W., On-line mass spectroscopy of volatile products during methanol oxidation at platinum in acid solutions, J. Electroanal. Chem., 1986, vol. 201, p. 403.
Jusys, Z., Kaiser, J., and Behm, R.J., Methanol electrooxidation over Pt/C fuel cell catalysts: dependence of products yelds on catalyst loading, Langmuir, 2003, vol. 19, p. 6759.
Abd-El-Latif, A.A. and Baltruschat, H., Formation of methylformate during methanol oxidation revisited: The mechanism, J. Electroanal. Chem., 2011, vol. 662, p. 204.
Wang, H., Alden, L.R., DiSalvo, F.J., and Abruna, H.D., Methanol Electrooxidation on PtRu Bulk Alloys and Carbon-Supported PtRu Nanoparticle Catalysts: A Quantitative DEMS Study, Langmuir, 2009, vol. 25, p. 7725.
Nakagawa, N., Sekimoto, K., Masdar, M.S., and Noda, R., Reaction analysis of a direct methanol fuel cell employing a porous carbon plate at high methanol concentrations, J. Power Sources, 2009, vol. 186, p. 45.
Capon, A. and Parsons, R., The oxidation of formic acid at noble metal electrodes. Part III. Intermediates and mechanism on platinum electrodes, J. Electroanal. Chem., 1973, vol. 45, p. 205.
Sun, S.G., Clavilier, J., and Bewick, A., The mechanism of electrocatalytic oxidation of formic acid on Pt(100) and Pt(111) in sulphuric acid solution: an EMIRS study, J. Electroanal. Chem., 1988, vol. 249, p. 147.
Miki, A., Ye, S., and Osawa, M., Surface-enhanced IR absorption on platinum nanoparticles: an application to real-time monitoring of electrocatalytic reactions, Chem. Commun., 2002, p. 1500.
Miyake, H., Okada, T., Samjeske, G., and Osawa, M., Formic acid electrooxidation on Pd in acidic solutions studied by surface-enhanced infrared absorption spectroscopy, PhysChemChemPhys., 2008, vol. 10, p. 3662.
Columbia, M.R. and Thiel, P.A., The interaction of formic acid with transition metal surfaces, studied in ultrahigh vacuum, J. Electroanal. Chem., 1994, vol. 369, p. 1.
Samjeske, G. and Osawa, M., Current oscillations during formic acid oxidation on Pt-electrode: insight into the mechanism by time-resolved IR spectroscopy, Angew. Chem., 2005, vol. 117, p. 5840.
Samjeske, G., Miki, A., Ye, S., and Osawa, M., Mechanistic study of electrocatalytic oxidation of formic acid at platinum in acidic solution by time-resolved surface-enhanced infrared spectroscopy, J. Phys. Chem. B, 2006, vol. 110, p. 16559.
Osawa, M., Komatsu, K., Samjeski, G., Uchida, T., Ikeshoji, T., Cuesta, A., and Gutierrez, C., The role of bridge-bonded adsorbed formate in the electrocatalytic oxidation of formic acid on platinum, Angew. Chem. Int. Ed., 2011, vol. 50, p. 1159.
Cuesta, A., Cabello, G. Gutierrez, C., and Osawa, M., Adsorbed formate: the key intermediate the oxidation of formic acid on platinum electrodes, PhysChemChemPhys., 2011, vol. 13, p. 20091.
Cuesta, A., Cabello, G., Osawa, M., and Gutierrez, C., Mechanism of the electrocatalytic oxidation of formic acid on metals, ACS Catal., 2012, vol. 2, p. 728.
Grozovski, V., Vidal-Iglesias, F.J., Herrero, E., and Feliu, J.M., Adsorption of Formate and Its Role as Intermediate in Formic Acid Oxidation on Platinum Electrodes, Chem. Phys. Chem., 2011, vol. 12, p. 1641.
Wieckowski, A. and Sobkowski, J., Comparative of adsorption and oxidation of formic acid and methanol on platinized electrodes in acidic solution, J. Electroanal. Chem., 1975, vol. 63, p. 365.
Chen, Y., Heinen, M., Jusys, Z., and Behm, R., Kinetics and mechanism of the electrooxidation of formic acid—Spectroscopical study in a flow cell, Angew. Chem. Int. Ed., 2006, vol. 45, p. 981.
Chen, Y.-X., Heinen, M., Jusys, Z., and Behm, R.J., Bridge-Bonded Formate: Active Intermediate or Spectator Species in Formic Acid Oxidation on a Pt Film Electrode, Langmuir, 2006, vol. 22, p. 10399.
Chen, Y.X., Ye, S., Heinen, M., Jusys, Z., Osawa, M., and Behm, R.J., Application of in situ Attenuated Total Reflection-Fouries Transform Infrared Spectroscopy for the understanding of complex reaction mechanism and kinetics: formic acid oxidation on a Pt film electrode at elevated temperatures, J. Phys. Chem. B, 2006, vol. 110, p. 9534.
Chen, Y.-X., Heinen, M., Jusys, Z., and Behm, R.J., Kinetic isotope effects in complex reaction networks: formic acid electrooxidation, Chem. Phys. Chem., 2007, vol. 8, p. 380.
Xu, J., Yuan, D.F., Yang, F., Mei, D., Zhang, Z., and Chen, Y.-X., On the mechanism of the direct path way formic acid oxidation at Pt(111) electrodes, PhysChemChemPhys., 2013, vol. 15, p. 4367.
Okamoto, H., Numata, Y., Gojuki, T., and Mukouyama, Y., Different behavior of adsorbed bridge-bonded formate from that of current in the oxidation of formic acid on platinum, Electrochim. Acta, 2014, vol. 116, p. 263.
Gao, W., Keith, J.A., Anton, J., and Jacob, T., Theoretical Elucidation of the Competitive Electro-oxidation Mechanisms of Formic Acid on Pt(111), J. Am. Chem. Soc., 2010, vol. 132, p. 18377.
Gao, W., Mueller, J.E., Jiang, Q., and Jacob, T., The role of co-adsorbed CO and OH in the electrooxidation of formic acid on Pt(111), Angew. Chem. Int. Ed., 2012, vol. 51, p. 9448.
Joo, J., Uchida, T., Cuesta, A., Koper, M.T.M., and Osawa, M., Importance of acid-base equilibrium in electrocatalytic oxidation of formic acid on platinum, J. Am. Chem. Soc., 2012, vol. 135, p. 9991.
Joo, J., Uchida, T., Cuesta, A., Koper, M.T.M., and Osawa, M., The effect of pH on the electrocatalytic oxidation of formic acid/formate on platinum: A mechanistic study by surface-enhanced infrared spectroscopy coupled with cyclic voltammetry, Electrochim. Acta, 2014, vol. 129, p. 127.
Koper, M.T.M., Theory of multiple proton-electron transfer reactions and its implication to electrocatalysis, Chem. Sci., 2013, vol. 4, p. 2719.
Mei, D., He, Z.D., Jiang, D.C., Cai, J., and Chen, Y.X., Modeling of potential oscillation during galvanostatic electrooxidation of formic acid at platinum electrode, J. Phys. Chem. C, 2014, vol. 118, p. 6335.
Perales-Rondon, J.V., Herrero, E., and Feliu, J.M., Effects of the anion adsorption and pH on the formic acid oxidation reaction on Pt(111) electrodes, Electrochim. Acta, 2014, vol. 140, p. 511.
Brimaud, S., Solla-Gullon, J., Weber, I., Feliu, J.M., and Behm, R.J., Formic acid electrooxidation on noble-metal electrodes: role and mechanistic implication of pH, surface structure, and anion adsorption, Chem. ElectroChem., 2014, vol. 1, p. 1075.
Perales-Rondon, J.V., Brimaud, S., Solla-Gullon, J., Herrero, E., Behm, R.J., and Feliu, J.M., Futher Insights into the Formic Acid Oxidation Mechanism on Platinum: pH and Anion Adsorption Effects, Electrochim. Acta, 2015, vol. 180, p. 479.
Jiang, K., Zhang, H.-X., Zou, S., and Cai, W.-B., Electrocatalysis of formic acid on palladium and platinum surfaces: from fundamental mechanisms to fuel cell applications, PhysChemChemPhys., 2014, vol. 16, p. 20360.
Gao, W., Song, E.H., Jiang, Q., and Jacob, T., Revealing the active intermediate in the oxidation of formic acid on Au and Pt(111), Chem. Eur. J., 2014, vol. 20, p. 11005.
Qi, Y.Y., Li, J.J., Zhang, D.J., and Liu,C.B., Reexamination of formic acid decomposition on the Pt(111) surface in the absence and in the presence of water, from periodic DFT calculations, Catal. Sci. Technology, 2015, vol. 5, p. 3322.
Schwarz, K.A., Sundararaman, R., Moffat, T.P., and Allison, T.C., Formic acid oxidation on platinum: a simple mechanistic study, PhysChemChemPhys., 2015, vol. 17, p. 20805.
Cuesta, A., Cabello, G., Hartl, F.W., Escudero-Escriberro, M., Vaz-Dominguez, C., Kibler, L.A., Osawa, M., and Guttierrez, C., Electrooxidation of formic acid on gold: An ATR-SEIRAS study of the role of adsorbed formate, Catal. Today, 2013, vol. 202, p. 79.
McPherson, I.J., Ash, P.A., Jacobs, R.M.J., and Vincent, K.A., Formate adsorption on Pt nanoparticles during formic acid electrooxidation: Insights from in situ infrared spectroscopy, Chem. Commun., 2016, vol. 52, p. 12665.
Christensen, P.A., Hamnett, A., and Linares-Moya, D.J., The electro-oxidation of formate ions at a polycrystalline Pt electrode in alkaline solution: an in situ FTIR study, PhysChemChemPhys., 2011, vol. 13, p. 11739.
John, J., Wang, H., Rus, E.D., and Abruna, H.D., Mechanistic studies of formate oxidation on platinum in alkaline medium, J. Phys. Chem. C, 2012, vol. 116, p. 5810.
Jiang, J., Scott, J., and Wieckowski, A., Direct evidence of triple-path mechanism of formate electrooxidation on Pt black in alkaline media at varying temperature. Part I: The electrochemical studies, Electrochim. Acta, 2013, vol. 104, p. 124.
Jusys, Z. and Behm, R.J., Dynamics of the interaction of formic acid with a polycrystalline Pt film electrode a time-resolved ATR-FTIR spectroscopy study at low potentials and temperatures, Electrocatalysis, 2017, vol. 8, p. 616.
Uhm, S., Lee, H.J., and Lee, J., Understanding underlying processes in formic acid fuel cells, PhysChemChemPhys., 2009, vol. 11, p. 9326.
Yu, X. and Pickup, P.G., Mechanistic study of the deactivation of carbon supported Pd during formic acid oxidation, Electrochem. Commun., 2009, vol. 11, p. 2012.
Wang, J.-Y., Zhang, H.-X., Jiang, K., and Cai, W.-B., From HCOOH to CO on Pd electrodes: A surface-enhanced infrared spectroscopic study, J. Am. Chem. Soc., 2011, vol. 133, p. 14876.
Zhang, R.G., Liu, H.Y., Wang, B.J., and Ling, I.X., Insight into the preference of CO2 formation from HCOOH decomposition on Pd surface, J. Phys. Chem. C, 2012, vol. 116, p. 22266.
Jeon, H., Jeong, B., Loo, J., and Lee, J., Electrocatalytic oxidation of formic acid: closing gap between fundamental study and technical applications, Electrocatalysis, 2015, vol. 6, p. 20.
Wang, Y., Qi, Y., Zhang, D., and Liu, C., New insight into decomposition of formic acid on Pd(111): Competing formation of CO and CO2,J. Phys. Chem. C, 2014, vol. 118, p. 2067.
Capon, A. and Parsons, R., The oxidation of formic acid on noble metal electrodes. II. A comparison of the behavior of pure electrodes, J. Electroanal. Chem., 1973, vol. 44, p. 239.
Arenz, M., Stamenkovic, V., Schmidt, T.J., Wandelt, K., Ross, P.N., and Markovic, N.M., The electro-oxidation of formic acid on Pt-Pd single crystal bimetallic surfaces, Phys. Chem. Phys. Chem., 2003, vol. 5, p. 4242.
Zhang, H.X., Wang, S.H., Jiang, K., Andre, T., and Cai, W.B., In situ spectroscopic investigation of CO accumulation and poisoning on Pt black surfaces in concentrated HCOOH, J. Power Sources, 2012, vol. 199, p. 163.
Obradovic, M.D. and Gojkovic, S.Lj., HCOOH oxidation on thin Pd adlayers on Au: Self-poisoning by the subsequent reaction of the reaction product, Electrochim. Acta, 2013, vol. 88, p. 384
Vidal-Iglesias, F.J., Aran-Aris, R.M., Solla-Gullon, J., Garnier, E., Herrero, E., Aldaz, A., and Feliu, J.M., Shape-dependent electrocatalysis: formic acid electrooxidation on cubic Pd nanoparticles, PhysChemChemPhys., 2012, vol. 14, p. 10258.
Obradovic, M.D. and Gojkovic, S.L., Pd black decorated by Pt sub-monolayers as an electrocatalyst for the HCOOH oxidation, J. Solid State Electrochem., 2014, vol. 18, p. 2599.
Chen, X. and Koper, M.T.M., Mass-transport-limited oxidation of formic acid on Pd ML Pt(100) electrode in perchloric acid, Electrochem. Commun., 2017, vol. 82, p. 155.
Haan, J.L. and Masel, R.T., The influence of solution pH on rates of an electrocatalytic reactions: Formic acid electrooxidation on platinum and palladium, Electrochim. Acta, 2009, vol. 54, p. 4073.
Joo, J., Choun, N., Jeong, J., and Lee, J., Influence of solution pH on Pt anodic catalyst in direct formic acid fuel cells, ACS Catal., 2015, vol. 5, p. 6848.
Abdelrahman, A., Hermann, J.M., and Kibler, L.A., Electrocatalytic oxidation of formate and formic acid on platinum and gold: study of pH dependence with phosphate buffers, Electrocatalysis, 2017, vol. 8, p. 509.
Jiang, K.,Wang, J.-Y., Zhao, T.-T., and Cai, W.-B., Formic acid oxidation on palladium electrode in acidic media containing chloride anions: An in situ ATR-SEIRAS investigation, J. Electroanal. Chem., 2017, vol. 800, p. 77.
Wetzel, R., Gunther, H., and Muller, L., A switch effect in the oxidation behavior of formate on Pt in alkaline solution, J. Electroanal. Chem., 1979, vol. 103, p. 271.
Gunther, H., Wetzel, R., and Muller, L., A new method for pH measurement in the immediate vicinity of the electrode surface, Electrochim. Acta, 1979, vol. 24, p. 237.
Liao, L.W., Li, M.F., Kang, J., Chen, D., Chen, Y.-X., and Ye, S., Electrode reaction induced pH change at the Pt electrode/electrolyte interface and its impact on electrode processes, J. Electroanal. Chem., 2013, vol. 688, p. 207.
Wei, Y., Zuo, K.Q., He, Z.D., Chen, W., Lin, C.H., Cai, J., Sartin, M., and Chen, Y.-X., The mechanisms of HCOOH/HCOO– oxidation on Pt electrodes: Implication from pH effect and H/D kinetic isotope effect, Electrochem. Commun., 2017, vol. 81, p. 1.
Subbaraman, R., Danilovic, N., Lopes, P., Tripkovic, D., Strmcnik, D., Stamenkovic, V., and Markovic, N., Origin of anomalous activities for electrocatalysts in alkaline electrolytes, J. Phys. Chem. C, 2012, vol. 16, p. 22231.
Strmcnik, D., Kodama, K., van der Vliet, D., Greely, J., Stamenkovic, V.B., and Markovic, N.M., The role of non-covalent interactions in electrocatalytic fuel-cell reactions on platinum, Nature Chem., 2009, vol. 1, p. 466.
Dunwell, M., Wang, J., Yan, Y., and Xu, B., Surface enhanced spectroscopic investigation of adsorption of cations on electrochemical surfaces, PhysChemChemPhys., 2017, vol. 19, p. 971.
Previdello, B.A., Machado, E.G., and Varela, H., The effect of the alkali metal cation on the electrocatalytic oxidation of formate on platinum, RSC Adv., 2014, vol. 4, p. 15271.
McCrum, I.T. and Janik, M.J., pH and alkali cation effects in the Pt cyclic voltammogram explained using Density Functional Theory, J. Phys. Chem. C, 2016, vol. 120, p. 457.
Nagao, R., Epstein, I.R., Gonzalez, E.R., and Varela, H., Temperature (over)compensation in an oscillatory surface reaction, J. Phys. Chem. A, 2008, vol. 112, p. 4617.
Angelucci, C.A., Varela, H., Herrero, E., and Feliu, J.M., Activation energies of the electrooxidation of formic acid on Pt(100), J. Phys. Chem. C, 2009, vol. 113, p. 18835.
Gilman, S., The mechanism of electrochemical oxidation of carbon monoxide on platinum. II. The “reactant pair” mechanism for electrochemical oxidation of carbon monoxide and methanol, J. Phys. Chem., 1964, vol. 68, p. 70.
Garcia, G. and Koper, M.T.M., Carbon monoxide oxidation on Pt single crystal electrodes: understanding the catalysis for low temperature fuel cell, Chem. Phys. Chem., 2011, vol. 12, p. 2064.
Herrero, E., Feliu, J.M., Bluis, S., Radovic-Hrapovic, Z., and Jerkiewicz, G., Temperature dependence of CO chemisorptions and its oxidative desorption on Pt(111) electrode, Langmuir, 2000, vol. 16, p. 4779.
Garcia, G. and Koper, M.T.M., Mechanism of electrooxidation of carbon monoxide on stepped platinum electrodes in alkaline media: A chronoamperometric and kinetic modeling study, PhysChemChemPhys., 2009, vol. 11, p. 11437.
Kunimatsu, K., Sato, T., Uchida, H., and Watanabe, M., Adsorption/oxidation of CO on highly dispersed Pt catalyst studied by combined electrochemical and ATR-SEIRAS methods: Oxidation of CO adsorbed on carbon supported Pt catalyst and unsupported Pt black, Langmuir, 2008, vol. 24, p. 3590.
Samjeske, G., Komatsu, K., and Osawa, M., Dynamics of CO oxidation on a polycrystalline platinum electrode: a time-resolved infrared study, J. Phys. Chem. C, 2009, vol. 113, p. 10222.
Zhu, Y., Uchida, H., and Watanabe, M., Oxidation of carbon monoxide at a platinum film electrode studied by Fourier Transform Infrared Spectroscopy with attenuated total reflection technique, Langmuir, 1999, vol. 15, p. 8757.
Breiter, M.W., Adsorption and oxidation of carbon monoxide on platinized platinum, J. Phys. Chem., 1968, vol. 72, p. 1305,
Cuesta, A., The oxidation of adsorbed CO on Pt(100) electrodes in the pre-peak region, Electroanalysis, 2010, vol. 1, p. 7.
Cuesta, A., Electrooxidation of C1 organic molecules on Pt-electrodes, Cur. Opin. Electrochem., 2017, vol. 4, p. 32.
Strmcnik, D.S., Tripcovic, D.V., van der Vliet, D., Chang, K.-C., Kominicky, V., You, H., Karapetov, G., Greely, J.P., Stamenkovic, V.R., and Markovic, N.M., Unique activity of platinum adislands in the CO electrooxidation reaction, J. Am. Chem. Soc., 2008, vol. 130, p. 15332.
Farias, M.J.S., Camara, G.A., and Feliu, J.M., Understanding the CO preoxidation and the intrinsic catalytic activity of step sites on stepped Pt surfaces in acidic medium, J. Phys. Chem. C, 2015, vol. 119, p. 20272.
Yan, Y.-G., Yang, Y.-Y., Peng, B., Malkhandi, S., Band, A., Stimming, U., and Cai, W.-B., Study of CO oxidation on polycrystalline Pt electrodes in acidic solution by ATR-SEIRAS, J. Phys. Chem. C, 2011, vol. 115, p. 16378.
Brimaud, S., Pronier, S., Coutanceau, C., and Leger, J.M., New insights on CO electrooxidation at Pt nanoparticle surfaces, Electrochem. Commun., 2008, vol. 10, p. 1703.
Urchaga, P., Baranton, S., and Coutanceau, C., Jerkiewiez, G., Electrooxidation of COchem on Pt nanosurfaces: Solution of the peak multiplicity puzzle, Langmuir, 2012, vol. 28, p. 3658.
Wang, H., Jusys, Z., Behm, R.J., and Abruna, H.D., New insights into the mechanism and kitetics of adsorbed CO electrooxidation on platinum: online mass spectroscopy and kinetic Monte Carlo simulation studies, J. Phys. Chem. C, 2012, vol. 116, p. 11040.
Wang, H. and Abruna, H.D., Origin of multiple peaks in potentiodynamic oxidation of CO adlayers on Pt- and Ru-modified Pt-electrodes, J. Phys. Chem. Lett., 2015, vol. 6, p. 1899.
Farias, M.J.S., Buso-Rogero, C., Vidal-Iglesias, F.J., Solla-Gullon, J., Camara, G.A., and Feliu, J.M., Mobility and oxidation of adsorbed CO on shape-controlled Pt nanoparticles in acidic medium, Langmuir, 2017, vol. 33, p. 865.
Lebedeva, N.P., Koper, M.T.M., Feliu, J.M., and van Santen, R.A., Role of crystalline defects in electrocatalysis: Mechanism and kinetics of CO adlayer oxidation on stepped platinum electrodes, J. Phys. Chem. B, 2002, vol. 106, p. 12938.
McCallum, C. and Pletcher, D., An investigation of the mechanism of the oxidation of carbon monoxide adsorbed onto a smooth Pt electrode in aqueous acid, J. Electroanal. Chem., 1976, vol. 70, p. 277.
Chang, S.C. and Weaver, M.J., In situ infrared spectroscopy of CO adsorbed at ordered Pt(110)-aqueous interfaces, Surf. Sci., 1990, vol. 230, p. 222.
McPherson, I.J., Ash, P.A., Jones, L., Varambhia, A., Jacobs, R.M.J., and Vincent, K.A., Electrochemical CO oxidation at platinum on carbon studied through analysis of anomalous in situ IR spectra, J. Phys. Chem. C, 2017, vol. 121, p. 17176.
Maillard, F., Savinova, E.R., and Stimming, U., CO monolayer oxidation on Pt nanoparticles: Further insights into the particle size effects, J. Electroanal. Chem., 2007, vol. 599, p. 221.
Podlovchenko, B.I., Manzhos, R.A., and Maksimov, Yu.M., Interaction of HCO-substances with adsorbed oxygen on platinum electrodes: open-circuit transient reactions of HCOOH and CO, Electrochim Acta, 2005, vol. 50, p. 4807.
Manzhos, R.A., Maksimov, Yu.M., and Podlovchenko, B.I., Transients of the open-circuit potential observed during the interaction of formic acid with preliminarily adsorbed oxygen on a platinized platinum electrode, Russ. J. Electrochem., 2005, vol. 41, p. 832.
Manzhos, R.A., Podlovchenko, B.I., and Maksimov, Yu.M., Specific features of interaction between formic acid and oxygen adsorbed on smooth polycrystalline platinum: transients of the open-circuit potential, Russ. J. Electrochem., 2006, vol. 42, p. 658.
Podlovchenko, B.I., Manzhos, R.A., and Maksimov, Yu.M., Kinetics and mechanism of interaction between methanol and adsorbed oxygen on a smooth polycrystalline platinum electrodes: transients of the open-circuit potential, Russ. J. Electrochem., 2006, vol. 42, p. 1061.
Manzhos, R.A., Podlovchenko, B.I., and Maksimov, Yu.M., Specific features of methanol interaction with adsorbed oxygen at platinized platinum electrode: transients of open-circuits potential, Russ. J. Electrochem., 2007, vol. 43, p. 1268.
Sitta, E. and Varela, H., On the open-circuit interaction between methanol and oxidized platinum, J. Solid State Electrochem., 2008, vol. 12, p. 554.
Batista, B.C., Sitta, E., Eiswirth, M., and Varela, H., Autocatalysis in the open circuit interaction of alcohol molecules with oxidized Pt surfaces, PhysChemChemPhys., 2008, vol. 10, p. 6686.
Batista, B.C. and Varela, H., Open circuit interaction of formic acid with oxidized Pt surfaces: experiment, modeling and simulations, J. Phys. Chem. C, 2010, vol. 114, p. 18494.
Tao, Q., Zheng, Y.-L., Jiang, D.-C., Chen, Y.-X., Jusys, Z., and Behm, R.J., Interaction of C1 molecules with Pt electrode at open circuit potential: a combined infrared and mass spectroscopic study, J. Phys. Chem. C, 2014, vol. 118, p. 6799.
Jurzinsky, N., Kurzhals, P., and Crevers, C., A novel differential electrochemical mass spectrometry method to determine the product distribution from parasitic Methanol oxidation reaction on oxygen reduction catalysts, J. Power Sources, 2018, vol. 389, p. 61.
Climent, V. and Feliu, J.M., Surface electrochemistry with Pt single-crystal electrodes, In: Nanopatterned and nanoparticle-modified electrodes. Eds. Alkire, R.C., Bartlett, N., and Lipkowski, J., 2017, Wiley-VCH. Ch. 1. P. 1–57
Motoo, S. and Furuya, N., Effect of terraces and steps in the electrocatalysis for formic acid oxidation on platinum, Ber. Bunsen-Ges. Phys. Chem., 1987, vol. 91, p. 457.
Grozovski, V.,Climent, V., Herrero, E., and Feliu, J.M., Intrinsic activity and poisoning rate for HCOOH oxidation at Pt(100) and vicinal surfaces containing monoatomic (111) steps, Chem. Phys. Chem., 2009, vol. 10, p. 1922.
Grozovski, V.,Climent, V., Herrero, E., and Feliu, J.M., Intrinsic activity and poisoning rate for HCOOH oxidation on platinum stepped surfaces, PhysChemChemPhys., 2010, vol. 12, p. 8822.
Koper, M.T.M., Structure sensitivity and nanoscale effects in electrocatalysis, Nanoscale, 2011, vol. 3, p. 2054.
Cuesta, A., Atomic ensemble effects in electrocatalysis: The site-knockout strategy, Chem. Phys. Chem., 2011, vol. 12, p. 2375.
Park, S., Xie, Y., and Weaver, M., Electrocatalytic pathways on carbon-supported platinum nanoparticles: comparison of particle-size-dependent rates of methanol, formic acid and formaldehyde electrooxidation, Langmuir., 2002, vol. 18, p. 5792.
Scheijen, F.J.E., Beltramo, G.L., Hoeppener, S., Housmans, T.H.M., and Koper, M.T.M., The electrooxidation of small organic molecules on platinum nanoparticles supported on gold: influence of platinum deposition procedure, J. Solid-State Electrochem., 2008, vol. 12, p. 483.
Solla-Gullon, J., Vidal-Iglesias, F.J., Lopez-Cudero, A., Garnier, E., Feliu, J.M., and Aldaz, A., Shape-dependent electroocatalysis: methanol and formic acid electrooxidation on preferently oriented Pt nanoparticles, PhysChemChemPhys., 2008, vol. 10, p. 3689.
Lee, S.W., Chen, S., Sheng, W., Yabuuchi, N., Kim, Y.-T., Mitani, T., Vescovo, E., and Shao-Horn, Y., Roles of surface steps on Pt nanoparticles in electro-oxidation of carbon monoxide and methanol, J. Am. Chem. Soc., 2009, vol. 131, p. 15669.
Chumillas, S., Buso-Rogero, C., Solla-Gullon, J., Vidal-Iglesias, F.J., Herrero, E., and Feliu, J.M., Size and diffusion effects on the oxidation of formic acid and ethanol on platinum nanoparticles, Electrochem. Commun., 2011, vol. 13, p. 1194.
Hoshi, N., Kida, K., Nakamura, M., Nakada, M., and Osada, K., Structural effects of electrochemical oxidation of formic acid on single crystal electrodes of palladium, J. Phys. Chem. B, 2006, vol. 110, p. 12480.
Hoshi, N., Nakamura, M., and Kida, K., Structural effects on the oxidation of formic acid on the high index planes of palladium, Electrochem. Commun., 2007, vol. 9, p. 279.
Zhou, W.P., Lewera, R., Larsen, R., Masel, R.I., Bagus, P.S., and Wieckowski, A., Size effects in electronic and catalytic properties of unsupported palladium nanoparticles in electrooxidation of formic acid, J. Phys. Chem. B, 2006, vol. 110, p. 13393.
Zhou, W. and Lee, J.Y., Particle size effects in Pd-catalyzed electrooxidation of formic acid, J. Phys. Chem. C, 2008, vol. 112, p. 3789.
Suo, Y. and Hsing, I.M., Size-controled synthesis and impedance-based mechanistic understanding of Pd/C nanoparticles for formic acid oxidation, Electrochim. Acta, 2009, vol. 55, p. 210.
Choi, S.-L., Harron, J.A., Scaranto, J., Huang, H., Wang, Y., Xia, X., Tian, Lv., Park, J., Peng, H.-C., Mavrikakis, M., and Xia, Y., A comprehensive study of formic acid on palladium nanocrystals with different types of facets and twin defects, ChemCatChem., 2011, vol. 7, p. 2077.
Zheng, W., Qu, J., Hong, X., Tedsree, K., and Tsang, S.C.E., Probing the size and shape effects of cubic- and spherical-shaped palladium nanoparticles in the electrooxidation of formic acid, ChemCatChem., 2015, vol. 7, p. 3826.
Ju, W., Valiollahi, R., Ojami, R., Schneider, O., and Stimming, U., The electrooxidation of formic acid on Pd nanoparticles: an investigation of size-dependent performance, Electrocatalysis, 2016, vol. 7, p. 149.
Watanabe, M. and Motoo, S., Electrocatalysis by ad-atoms. Part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms, J. Electroanalyt. Chem., 1975, vol. 60, p. 267.
Gasteiger, H.A., Markovic, N., Ross, P.N., and Cairns, E.J., Methanol electrooxidation on well-characterized Pt–Ru alloys, J. Phys. Chem., 1993, vol. 97, p. 12029.
Markovic, N., Gasteiger, H.A., Ross, P.N., Jiang, X., Villegas, I., and Weaver, M.J., Electro-oxidation mechanisms of methanol and formic acid on Pt–Ru alloy surfaces, Electrochim. Acta, 1995, vol. 40, p. 91.
Gojkovic, S.Lj., Vidakovic, T.R., and Durovic, D.R., Kinetic study of methanol oxidation on carbon-supported PtRu electrocatalyst, Electrochim. Acta, 2003, vol. 48, p. 3607.
Tong, Y.Y., Kim, H.S., Babu, P.K., Waszczuk, P., Wieckowski, A., and Oldfield, E., An NMR investigation of CO tolerance in a Pt/Ru fuel cell catalyst, J. Am. Chem. Soc., 2002, vol. 124, p. 468.
Pinheiro, A.L.N., Zei, M.S., and Ertl, G., Electro-oxidation of carbon-monoxide and methanol on bare and Pt-modified Ru(1010) electrodes, PhysChemChemPhys., 2005, vol. 7, p. 1300.
Sawy, E.N., El-Sayed, H.A., and Birss, V., Clarifying the role of Ru in methanol oxidation at Rucore@Ptshell nanoparticles, PhysChemChemPhys., 2015, vol. 17, p. 27509.
Du, B., Rabb, S.A., Zangmeister, C., and Tong, Y.Y., A volcano curve: optimizing methanol electro-oxidation on Pt-decorated Ru nanoparticles, PhysChemChemPhys., 2009, vol. 11, p. 8231.
Kuznetsov, A.N., Simonov, P.A., Zaikovskii, V.I., Parmon, V.N., and Savinova, E.R., Temperature effects in carbon monoxide and methanol electrooxidation on platinum-ruthenium: influence of grain boundaries, J. Solid State Electrochem., 2013, vol. 17, p. 1903.
Rigsby, M.A., Zhou, W.P., Lewera, A., Duong, H.T., Bagus, P.S., Jaegermann, W., Hynger, R., and Wieckowski, A., Experiment and theory of fuel cell electrocatalysis: methanol and formic acid decomposition on nanoparticle Pt/Ru, J. Phys. Chem. C, 2008, vol. 112, p. 15595.
Islam, M., Basnayake, R., and Korzeniewski, C., A study of formaldehyde formation during methanol oxidation over PtRu bulk alloys and nanometer scale catalyst, J. Electroanal. Chem., 2007, vol. 599, p. 31.
Liu, B.J., Jin, J.M., Lin, X., Hardacre, C., Hu, P., Ma, C.A., and Lin, W.F., The effects of stepped sites and ruthenium adatom decoration on methanol dehydrogenation over platinum based catalyst surfaces, Catal. Today, 2015, vol. 242, p. 230.
Ding, Q., Xu, W., Sang, P., Xu, J., Zhao, L., Xe, X., and Guo, W., Insight into reaction mechanisms of methanol on PtRu/Pt(111): a Density Functional study, Appl. Surf. Sci., 2016, vol. 369, p. 257.
Wang, K., Gasteiger, H.A., Markovic, N.M., and Ross, P.N., Jr., On the reaction pathway for methanol and carbon monoxide electrooxidationn on Pt–Sn alloy versus Pt–Ru alloy surfaces, Electrochim. Acta, 1996, vol. 41, p. 2583.
Colmati, F., Antolini, E., and Gonzalez, E.R., Electrocatalysts for methanol oxidation synthesized by reduction with formic acid, Electrochim. Acta, 2005, vol. 50, p. 5496.
Wei, Z.D., Li, L.L., Luo, Y.H., Yan, C., Sun, C.X., Yin, G.Z., and Shen, P.K., Electrooxidation methanol on upd-Ru and upd-Sn modified Pt-electrodes, J. Phys. Chem. B, 2006, vol. 110, p. 26055.
Liu, P., Logadottir, A., and Norskov, J.K., Modeling the electro-oxidation of CO and H2/CO on Pt, Ru, Pt‒mRu and Pt3Sn, Electrochim. Acta, 2005, vol. 48, p. 3734.
Stevanovic, S., Tripkovic, D., Tripkovic, V., Minic, D., Gavrilovic, A., Tripkovic, A., and Jovanovic, V.M., Insight into the effect of Sn on CO and formic acid oxidation at PtSn catalysts, J. Phys. Chem. C, 2014, vol. 118, p. 278.
Farias, M.J.S., Cheuquepan, W., Tanaka, A.A., and Feliu, J.M., Non-uniform synergistic effect of Sn and Ru in site-specific catalytic activity of Pt at bimetallic surfaces toward CO electrooxidation, ACS Catal., 2017, doi 10.1021/acscatal.7b00257
Rizo, R., Pastor, E., and Koper, M.T.M., CO electrooxidation on Sn-modified Pt single crystals in acid media, J. Electroanal. Chem., 2017, vol. 80, p. 32.
Lu, X., Ding, Z., Guo, C., Wang, W., Wei, S., Ng, S.-P., Chen, X., Ding, N., Guo, W., and Wu, C.-M.L., Methanol oxidation on Pt3Sn(111) for direct methanol fuel cells: methanol decomposition, ACS Appl. Mater. Interfaces, 2016, vol. 8, p. 12194.
Tritsaris, G.A. and Rossmeisl, J., Methanol excitation on model elemental and bimetallic transition metal surfaces, J. Phys. Chem. C, 2012, vol. 116, p. 11980.
Choi, J.H., Park, K.W., Park, I.S., Nam, W.H., and Sung, Y.E., Methanol electrooxidation and direct methanol fuel cell using Pt/Rh and Pt/Ru/Rh alloy catalysts, Electrochim. Acta, 2004, vol. 50, p. 787.
Sheng, T. and Sun, S.-G., Insight into the promoting role of Rh doped on Pt(111) in methanol electro-oxidation, J. Electroanal. Chem., 2016, vol. 781, p. 24.
Suntivich, J., Xu, Z., Carlton, C.E., Kim, J., Han, B., Lee, S.W., Bonnet, N., Mazzari, N., Allard, L.F., Hasteiger, H.A., Hamad-Schifferli, K., and Shao-Horn, Y., Surface composition tunning of Au–Pt bimetallic nanoparticles for enhanced carbon monoxide and methanol electro-oxidation, J. Am. Chem. Soc., 2013, vol. 135, p. 7985.
Conway, B.E., Angerstein-Kozlowska, H., and Czartoryska, G., “Third body” effect in the auto-inhibition of formic acid oxidation on electrodes, Z. Physik. Chem. N.F., 1978, vol. 112, p. 195.
Leiva, E., Iwasita, T., Herrero, E., and Feliu, J.M., Effect of adatoms in the electrocatalysis of HCOOH oxidation. A theoretical model, Langmuir, 1997, vol. 13, p. 6287.
Vidal-Iglesias, F.J., Lopez-Cudero, A., Sulla-Gullon, J., and Feliu, J.M., Towards more active and stable electrocatalysts for formic acid electrooxidation: antimony decorated octahedral platinum nanoparticles, Angew. Chem. Int. Ed., 2013, vol. 52, p. 964.
Buso-Rogero, C., Peralles-Rondon, J.V., Farias, F.J., Vidal-Iglesias, F.J., Solla-Gullon, J., Herrero, E., and Feliu, J.M., Formic acid electrooxidation on thallium-decorated shape-controlled platinum nanoparticles : an improvement in electrocatalytic activity, PhysChemChemPhys., 2014, vol. 16, p. 13616.
Ferre-Vilaplana, A., Perales-Rondon, J.V., Feliu, J.M., and Herrero, E., Understanding the effect of the adatoms in formic acid oxidation mechanism on Pt(111) electrodes, ACS Catal., 2015, vol. 5, p. 645.
Perales-Rondon, J.V., Solla-Gullon, J., Herrero, E., and Sanchez-Sanchez, C.M., Enhanced catalytic activity and stability for the electrooxidation of formic acid on lead modified shape controlled platinum nanoparticles, Appl. Catal. B: Enviromental, 2017, vol. 201, p. 2095.
Yu, X. and Pickup, P.G., Pb and Sb modified Pt/C catalysts for direct formic acid fuel cell, Electrochim. Acta, 2010, vol. 55, p. 7354.
Llorca, M.J., Feliu, J.M., Aldaz, A., and Clavilier, J., Formic acid oxidation on Pdad + Pt(100) and Pdad + Pt(111) electrodes, J. Electroanal. Chem., 1994, vol. 376, p. 151.
Waszczuk, P., Barnard, T.M., Race, C., Masel, R.L., and Wieckowski, A., A nanoparticle catalyst with superior activity for electrooxidation of formic acid, Electrochem. Commun., 2002, vol. 4, p. 599.
Lee, H., Habas, S.E., Somorjai, G.A., and Yang, P., Localized Pd overgrowth on cubic Pt nanocrystals for enhanced electrooxidation of formic acid, J. Am. Chem. Soc., 2008, vol. 130, p. 5406.
Liu, B., Li, H.Y., Die, L., Zhang, X.-H., Fan, Z., and Chen, Y.H., Carbon nanotubes supported PtPd hollow nanospheres for formic acid electrooxidation, J. Power Sources, 2009, vol. 186, p. 62.
Vidal-Iglesias, F.J., Solla-Gullon, J., Herrero, E., Aldaz, A., and Feliu, J.M., Pd decorated (100) preferentially oriented Pt nanoparticles for formic acid electrooxidation, Angew. Chem. Int. Ed., 2010, vol. 49, p. 6998.
Zhang, H.X., Wang, C., Wang, J.Y., Zhai, J.J., and Cai, W.B., Carbon-Supported Pd–Pt Nanoalloy with Low Pt Content and Superior Catalysis for Formic Acid Electrooxidation, J. Phys. Chem. C, 2010, vol. 114, p. 6446.
Kim, J., Jung, C., Rhee, C.K., and Lim, T., Electrocatalytic oxidation of formic acid and methanol on Pt deposits on Au(111), Langmuir, 2007, vol. 23, p. 10831.
Park, I.S., Lee, K.S., Choi, J.H., Park, H.Y., and Sung, Y.E., Surface structure of Pt-modified Au nanoparticles and electrocatalytic activity in formic acid electrooxidation, J. Phys. Chem. C, 2007, vol. 111, p. 19126.
Xu, J.B., Zhao, T.S., and Liang, Z.X., Carbon supported platinum-gold alloy catalyst for direct formic acid fuel cell, J. Power Sources, 2008, vol. 185, p. 857.
Kristian, N. and Yu, Y.L., Ptshell-Aucore/C electrocatalyst with a controlled shell thickness and improved Pt utilization for fuel cell reactions, Electrochem. Commun., 2008, vol. 10, p. 12.
Wang, S.Y., Kristian, N., Jiang, S.P., and Wang, X., Controlled deposition of Pt on Au nanorodes and their catalytic activity towards formic acid oxidation, Electrochem. Commun., 2008, vol. 10, p. 961.
Kristian, N., Yu, Y.L., Gunawan, P., Xu, R., Deng, W., Liu, X., and Wang, X., Controlled synthesis of Pt-decorated Au nanostructure and its promoted activity toward formic acid electro-oxidation, Electrochim. Acta, 2009, vol. 54, p. 4916.
Obradovich, M.D., Tripkovic, A.V., and Gojkovic, S.L., The origin of high activity of Pt–Au surfaces in the formic acid oxidation, Electrochim. Acta, 2009, vol. 55, p. 204.
Liu, Y., Wang, L.W., Wang, G., Dong, C., Wu, B., and Gao, Y., High active carbon supported PdAu catalyst for formic acid electrooxidation and study of the kinetics, J. Phys. Chem. C, 2010, vol. 114, p. 21417.
Obradovic, M.D., Rogan, J.R., Babic, B.M., Tripkovic, A.V., Guntam, A.R.S., Radmilovic, V.R., and Gojkovic, S.L., Formic acid oxidation on Pt-Au nanoparticles: relation between the catalyst activity and poisoning rate, J. Power Sources, 2012, vol. 197, p. 72.
Yuan, D.W. and Liu, Z.R., Atomic ensemble effects on formic acid oxidation on PdAu electrode studied by first-principles calculations, J. Power Sources, 2013, vol. 224, p. 241.
Zhong, W., Qi, Y., and Dong, M., The ensemble effect of formic acid oxidation on platinum-gold electrode studied by first-principles calculations, J. Power Sources, 2015, vol. 278, p. 203.
Duan, T., Zhang, R., Ling, I., and Wang, B., Insights into the effect of Pt atomic ensemble on HCOOH oxidation over Pt-decorated Au bimetallic catalyst to optimize Pt utilization, J. Phys. Chem. C, 2016, vol. 120, p. 2234,
Jeong, H. and Kim, J., Insights into the electrooxidation mechanism of formic acid on Pt layers on Au examined by Electrochemical SERS, J. Phys. Chem. C, 2016, vol. 120, p. 24271.
Cellorio, V., Quaino, P.U., Santos, E., Florez-Montano, J., Humphray, J.J.L., Quillon-Villafuerte, O., Plana, D., Lazaro, M.J.,Pastor, E., and Fermin, D.J., Strain effects in the oxidation of CO and HCOOH on Au–Pd core–shell nanoparticles, ACS Catal., 2017, vol. 7, p. 3826.
Wang, H., Alden, L., DiSalvo, E.J., and Abruna, H.D., Electrocatalytic mechanism and kinetics of SOC oxidation on ordered PtPb and PtBi intermetallic compound: DEMS and FTIRS study, PhysChemChemPhys., 2008, vol. 10, p. 3739.
Emirci, U.B., Theoretical means for searching bimetallic alloys as anode electrocatalysts for direct liquid-feed fuel cells, J. Power Sources, 2007, vol. 173, p. 11.
Norskov, J.K., Bligaard, T., Rossmeisl, J., and Christensen, C.H., Towards the computational design of solid catarysts, Nat. Chem., 2009, vol. 1, p. 34.
Calle-Vallejo, F., Koper, M.T.M, and Bandarenka, A.S., Tailoring the catalytic activity of electrodes with monolayer amounts of foreign metals, Chem. Soc. Rev., 2013, vol. 42, p. 5210.
Koper, M.T.M., Volcano activity relationships for proton-coupled electron transfer reactions in electrocatalysis, Top. Catal., 2015, vol. 58, p. 1153.
Sasikumar, G., Mathumeenal, A., Pethaiah, S.S., Nachiappen, N., and Balaji, R., Aqueous methanol electrolysis using proton conducting membrane for hydrogen production, Intern. J. Hydrogen Energy, 2008, vol. 33, p. 5905.
Cloutier, P.R. and Wilkinson, D.P., Electrolytic production of hydrogen from aqueous acidic methanol solutions, Intern. J. Hydrogen Energy, 2010, vol. 35, p. 3967.
Lamy, C., Guenot, B., Cretin, M., and Pourcelly, G., Kinetics analysis of the electrocatalytic oxidation of methanol inside DMFC working as a PEM electrolytic cell (PEMEC) to generate clean hydrogen, Electrochim. Acta, 2015, vol. 177, p. 352.
Дополнительные материалы отсутствуют.
Инструменты
Электрохимия