

Электрохимия, 2019, T. 55, № 10, стр. 1248-1260
Квантово-химическое изучение адсорбции ионов Tl+ на Au (111)
a Институт химии твердого тела и механохимии CО РАН
630128 Новосибирск, ул. Кутателадзе, 18, Россия
b Новосибирский государственный технический университет
630073 Новосибирск, просп. К. Маркса, 20, Россия
* E-mail: rogna@mail.ru
Поступила в редакцию 10.12.2018
После доработки 10.12.2018
Принята к публикации 30.03.2019
Аннотация
На основе кластерной модели поверхности металла методом функционала плотности исследовано взаимодействие иона Tl+ с поверхностью золота. Оценены геометрические и энергетические характеристики такого взаимодействия. Ион Tl+ существует на поверхности в ад-ионной форме. Проанализировано электронное строение системы Au–${\text{Tl}}_{{{\text{ads}}}}^{ + }.$ Оценено участие адсорбированного иона таллия и ближайших к нему атомов золота в формировании молекулярных орбиталей системы. Вклад в их образование обеспечивают преимущественно s-орбитали таллия и d-орбитали золота. Установлено что в щелочных средах происходит соадсорбция ионов Tl+ и OH– с образованием TlOH на поверхности. Определена возможная геометрическая конфигурация ассоциата при низких и высоких степенях заполнения поверхности ионами OH–.
ВВЕДЕНИЕ
Взаимодействие катионов металлов с инородной металлической подложкой, начинающееся при потенциалах более анодных, чем соответствующий равновесный потенциал осаждаемого металла, получившее название “underpotential deposition (UPD)” или “осаждение при недонапряжении”, известно уже более полувека [1]. При этом имеет место субмонослойное заполнение поверхности осаждаемым металлом [2–4]. Адсорбция чужеродных металлов приводит к значительному изменению адсорбционных и каталитических свойств субстрата [5–7].
Одной из систем, для которой известен такой каталитический эффект, является поверхность золотого электрода, находящаяся в контакте в электролитом, содержащим ионы Tl+ [8–13]. Экспериментальные исследования системы золото/раствор, содержащей ионы Tl+, продемонстрировали сложную форму вольтамперограмм [8, 14–16], а также показали, что адсорбция ионов таллия на золоте начинается при потенциалах заметно более положительных, чем равновесный потенциал системы Tl+/Tl [8, 14–17]. Согласно оценкам, произведенным в [14, 15], ионы таллия в адсорбированном состоянии полностью разряжены. Однако в [16, 18, 19] допускается неполный разряд адсорбированных катионов.
Теоретические исследования взаимодействия между атомами таллия и золота показали значительную прочность связи в димере AuTl. Наибольшую величину энергии связи в ранней статье [20], дали полуэмпирические методы (–499 кДж/моль). Позднее неэмпирическими методами получены более низкие значения этого параметра в интервале от –331 до –225 кДж/моль [21–25]. Вычисленная длина связи в AuTl оказалась наибольшей (0.351 нм) при использовании полуэмпирического метода CNDO/2 [20]. Применение неэмпирических методов дало более низкие длины связи в пределах от 0.273 до 0.262 нм [21–25]. Заряд атома таллия, как элемента с меньшей электроотрицательностью [26], имел в AuTl положительное значение: 0.22 в [20] и 0.35 в [22, 23].
Экспериментальные исследования адсорбции атомарного таллия на золоте дали величины энергии адсорбции –240 [27], –285 [28] и –270 кДж/моль [29, 30]. Квантово-химическое моделирование взаимодействия таллия с гранью золота (111), выполненное на двухслойном кластере Au23 методом CNDO/2 [20], дало величину энергии адсорбции одиночного атома таллия –863 кДж/моль, значительно превышающую экспериментальную величину. Более поздние теоретические исследования адсорбции таллия на золоте, выполненные неэмпирическими методами, дали более низкие значения энергии, близкие как к экспериментальным величинам энергии адсорбции, так и к энергии связи в димере Au–Tl. Энергия взаимодействия атома таллия с двумя и тремя атомами золота составляла –240 и –253 кДж/моль соответственно [24]. Адсорбция атома таллия на грани Au (111), которая моделировалась кластерами из 16–20 атомов, была энергетически наиболее выгодна при тройной координации таллия относительно золота при энергии –256 кДж/моль [25]. Длина связи Tl–Au при такой адсорбции таллия на золоте оценивалась в 0.312 нм [25]. Она короче, чем определенная по методу CNDO/2 в [20] длина связи в димере AuTl (0.351 нм), но длиннее, чем вычисленная в [21–25] неэмпирическими методами величина этого параметра (0.262–0.273 нм). Эффективный заряд адсорбированного атома таллия определен только в [20], где он оценен в ‒0.31. Смещение электронной плотности к элементу с меньшей электроотрицательностью объяснено авторами взаимным поляризующим действием адатома таллия и кластера золота.
Исследования подобного рода, касающиеся взаимодействия катионов таллия с золотом, в литературе отсутствуют.
Цель данной работы – методами квантовой химии проанализировать взаимодействие ионов Tl+ с незаряженной поверхностью золота для изучения свойств системы Au/Tl+ на микроскопическом уровне.
МЕТОДИЧЕСКАЯ ЧАСТЬ
Квантово-химические вычисления проводились с помощью программных пакетов GAMESS-US [31] и Orca [32]. Расчеты выполнялись на основе метода функционала плотности [33] с использованием гибридного B3LYP функционала [34, 35], который довольно широко используется для вычисления в молекулярных системах, включающих в себя атомы переходных элементов. Для атомов Au и Tl вычисления проводились с использованием псевдопотенциала LANL2DZ и соответствующего ему расщепленного базисного набора для внешних и предвнешних электронных оболочек [36, 37]. Атомы кислорода и водорода описывались с помощью расширенного базисного набора 6-311+G(d, p) [38, 39], который включал в себя диффузные и поляризационные функции. Эффективные атомные заряды на адсорбированных ионах определялись на основе анализа заселенностей орбиталей по методам Левдина [40] и натуральных орбиталей (NBO) [41, 42].
Для вычисления взаимодействия ионов таллия с поверхностью золота фрагмент последней моделировался кластером. В качестве объекта исследования была выбрана грань (111). Эта грань менее склонна к изменениям вследствие ее большей термодинамической стабильности [43]. Исследования проводились на двухслойном 18-атомным кластере, содержащим 12 атомов в первом и 6 атомов во втором поверхностных слоях. На рис. 1 показан вид сверху на точки локализации адсорбированного иона для однокоординационного взаимодействия иона с кластером (1), двухкоординационной адсорбции (2) и трехкоординационных положений (3') и (3"). Два последних положения отличаются расположением адсорбированного иона относительно атомов второго слоя кластера. Адсорбированная частица находится либо над атомом второго слоя (3'), либо над точкой между тремя атомами этого слоя (3").
Рис. 1.
Модель фрагмента грани (111) золота в виде 18‑атомного кластера. Атомы второго слоя показаны более темным цветом. Точками показана локализация адсорбированного иона в вершинном однокоординационном положении (1), в мостиковом двухкоординационном положении (2) и трехкоординационном положении (3') и (3").

Положение атомов металла в кластере было принято фиксированным и соответствующим их положению в объеме металла. Межатомное расстояние для золота было принято равным экспериментальной величине 0.288 нм, соответствующей кристаллам этого металла [44]. Координаты атомов таллия в процессе вычислений варьировались.
Для расчетов, в которых рассматривалось влияние растворителя на поверхностные процессы, учет взаимодействия частиц с растворителем проводился на основе модели поляризуемого континуума C-PCM [45–47], в которой диэлектрическая полость в растворителе формируется в соответствии с молекулярной поверхностью находящейся в растворителе частицы, а учет пространственной дисперсии диэлектрической проницаемости среды производится разбиением системы на области, в которых изменение диэлектрической постоянной от низких значений до объемной величины моделируется посредством введения подборочных эффективных атомных радиусов и подгоночных коэффициентов. Описание методики определения таких параметров приведено в данных публикациях [45, 46].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Взаимодействие ионов таллия с поверхностью кластера
Энергия газофазной адсорбции ионов Tl+ ΔEads на незаряженной поверхности, характеризующая свойства связи металл-ион таллия, вычислялась как
(1)
$\Delta {{E}_{{{\text{ads}}}}} = {{E}_{{{\text{M}}{{{\text{e}}}_{{{\text{18}}}}} - {\text{T}}{{{\text{l}}}^{ + }}}}} - {{E}_{{{\text{M}}{{{\text{e}}}_{{{\text{18}}}}}}}} - {{E}_{{{\text{T}}{{{\text{l}}}^{ + }}}}},$Результаты вычислений энергии взаимодействия иона Tl+ с кластером золота для четырех положений приведены в таблице 1. В ней представлены также геометрические и зарядовые параметры, соответствующие газофазной адсорбции ионов таллия на золоте.
Как следует из таблицы, катион таллия образует прочную связь с поверхностью золота (энергия от –143 до –151 кДж/моль). Полученные величины энергий показывают, что наиболее выгодным местом адсорбции катиона Tl+ на кластере является трехцентровое положение (3"), наименее же выгодным местом адсорбции – вершинное положение (1). Однако при этом разница энергий взаимодействия для разных мест мала.
Найденная величина энергии адсорбции иона таллия оказалась почти на 100 кДж/моль меньше, чем экспериментальная энергия адсорбции атомарного таллия на золоте из [27–30]. Вычисленная для сравнения энергия взаимодействия атома таллия с золотом составляла –265 кДж/моль. Данная величина попадала в интервал экспериментальных энергий адсорбции атома таллия от ‒240 до –285 кДж/моль [27–30] и практически совпадала с результатами вычислений этого параметра из [25] (–256 кДж/моль). Таким образом, ионный таллий образует менее прочную связь с золотом, чем атомарный. Аналогичные случаи более высокой модельной энергии адсорбции незаряженных частиц на металлах по сравнению с адсорбцией заряженных форм этих компонентов продемонстрированы, например, в [51–53], где разность энергий в ряде случаев также была близка к 100 кДж/моль и даже превышала эту величину.
При адсорбции катиона Tl+ его заряд в адсорбированном состоянии сохраняется и не зависит от места адсорбции. Проведенная двумя методами оценка величины эффективного заряда дала одинаковые значения этого параметра (в пределах 3%).
Межатомная длина связи золото–таллий RMe–Tl растет при увеличении координации иона таллия на поверхности. Расстояние по нормали между ионом таллия и поверхностью z при этом практически не изменяется. Найденные геометрические величины RMe–Tl оказались большими, чем в димере AuTl (0.262–0.273 нм) [21–25] и при адсорбции атомарного таллия на золоте в положении (3") (0.312 нм) [25]. Последний факт можно связать с тем, что межъядерное отталкивание в системе при адсорбции катионов в меньшей степени компенсируется взаимодействием между ядрами и электронами по сравнению со случаем адсорбции атомарного таллия, и катиону Tl+ энергетически более выгодно немного отодвинуться от поверхности золота.
Для анализа электронного строения системы Tl+/Au (111) были рассмотрены спектры плотности состояний. Плотность состояний D(E) используется для анализа распределения молекулярных орбиталей по энергетическим уровням при их большом количестве и определяется как количество таких уровней N, находящихся в интервале энергий между E и E + dE: D(E) = dN/dE [54]. График D(E) строился на основе данных, полученных в результате обработки результатов вычислений с помощью программы GaussSum [55] для поверхности золота с адсорбированным ионом Tl+ в положении (3"), и показан на рис. 2. При построении D(E)-графиков обычно представляют область энергий, относящуюся только к заполненной области перекрывания атомных орбиталей между адсорбентом и адсорбатом. Орбитали с более низкими значениями энергии, относящиеся к внутренним орбиталям компонентов системы и не участвующие в образовании связей, традиционно в спектре плотности состояний не демонстрируются.
Рис. 2.
Спектр плотности состояний при адсорбции иона таллия на золоте: 1 – полный спектр плотности состояний (D(E)); 2 – локальная плотность состояний адсорбированного иона таллия (D(E)Tl); 3 – локальная плотность состояний трех атомов золота, связанных с таллием ($D{{(E)}_{{{\text{A}}{{{\text{u}}}_{{\text{3}}}}}}}$).

Кривая 1 отображает полный спектр плотности состояний. Основная часть энергетических уровней системы сосредоточена в области между –15 и –10 эВ. За пределами этой области распределение имеет преимущественно дискретный характер. Наименее отрицательным энергиям соответствуют сразу две орбитали при Е = –8.25 эВ, которые имеют смысл высших занятых молекулярных орбиталей (ВЗМО).
Для оценки вклада атома или группы атомов в полную плотность состояний используется локальная плотность состояний. Кривая 2 демонстрирует локальную плотность состояний для адсорбированного иона таллия. Локальная плотность состояний для трех ближайших к таллию атомов золота, т.е. атомов Au непосредственно связанных с таллием, показана кривой 3.
Участие ионов Tl+ в формировании системы золото-таллий наиболее значительно для орбитали с Е = –13.59 эВ. Доля Tl+ составляет здесь 49%. При Е = –14.25 эВ доля таллия снижается вдвое. В остальных областях энергий вклад таллия существенно менее значителен и не превышает 9%. Область ВЗМО содержит две орбитали. Вклад таллия при этом составляет всего 2% при доле ближайших к таллию атомов золота около 8%. Следовательно, ВЗМО образованы преимущественно атомами золота, которые не имеют прямого контакта с таллием.
Участие в формировании электронной структуры таллий–золото непосредственно контактирующих с ионом Tl+ трех атомов золота наиболее значительно в области от –14.8 до –13.3 эВ. Относительная доля этих атомов золота составляет около половины. Однако при Е = –13.59 эВ, когда доля таллия составляет 49%, вклад рассматриваемых трех атомов золота падает до нескольких процентов. В случае орбитали при Е = –14.25 эВ (также со значительной долей таллия) их участие возрастает до 59%. При менее отрицательных энергиях (до –10 эВ) величина локальной плотности состояний этих атомов золота колеблется, оставаясь при пиковых значениях сопоставимой с величинами при E < –13.3 эВ. Однако относительный вклад контактирующих с таллием атомов золота в электронное состояние системы сильно уменьшается.
Рисунок 3 демонстрирует вклад s- и p-орбиталей адсорбированного иона таллия (рис. 3а) и s-, p- и d-орбиталей непосредственно связанных с таллием трех атомов золота (рис. 3б) в общую плотность состояний. d-Орбитали адсорбированного таллия (в случае свободного Tl+ они имеют энергию –27.52 эВ) располагаются в узком интервале от –24.29 до –24.24 эВ. Они принадлежат только иону Tl+, т.е. являются несвязывающими орбиталями, и на графике не показаны. Участие s- и p-орбиталей таллия в формировании электронной структуры можно считать не сопоставимым. Связывание таллия с золотом осуществляется в основном за счет его s-орбитали. В изолированном ионе Tl+ она имеет энергию –16.16 эВ. При адсорбции иона таллия эта орбиталь участвует в формировании системы в широком интервале спектра от –15.6 до –9.0 эВ. Наиболее значительное ее участие наблюдается при Е = –13.59 эВ. Здесь ее доля составляет 49%. При Е = –14.25 эВ вклад s-орбитали также велик, хотя и снижается до 22%. Небольшое участие (6%) наблюдается и в районе –15.51 эВ. Несколько размытый пик около –12.5 эВ обусловлен тем, что в интервале от ‒13.18 до –11.90 эВ существует несколько молекулярных орбиталлей с долей таллия от 2 до 6%. Пик при –11.54 эВ обусловлен 8-процентным участием s-орбитали. Небольшие пики при ‒10.74 и –9.05 эВ также появляются вследствие небольшого участия этой орбитали. p-Орбитали таллия (в свободном ионе это вакантные орбитали с Е = –7.40 эВ) при E < –9.5 эВ практически не участвуют в образовании связи таллий-золото. Только при E > –9.7 эВ вклад p-орбиталей в локальную плотность состояний таллия превосходит вклад s-орбитали, хотя и остается небольшим на фоне участия золота. При Е = –9.05 эВ доля p‑орбиталей таллия увеличивается до 5%. Вклад p-орбиталей в две ВЗМО при –8.25 эВ составляет по 2%, при полном отсутствии участия иных орбиталей таллия.
Рис. 3.
Адсорбция иона Tl+ на золоте: а – вклад в плотность состояний s- (1) и p-орбиталей (2) иона таллия; б – вклад в плотность состояний d- (3), s- (4) и p-орбиталей (5) трех ближайших к таллию атомов золота.
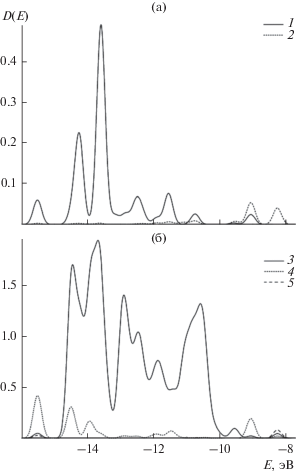
Участие непосредственно связанных с таллием трех атомов золота в образовании молекулярных орбиталей системы обеспечивается с большим преимуществом их d-орбиталями. Участие s‑орбиталей заметно менее существенно. Роль p‑орбиталей еще более незначительна. Уровень при –15.51 эВ во многом сформирован s-орбиталями (на 42%). Участие p- и d-орбиталей здесь сведено до 3 и 5%. Доля s-орбиталей заметна также и при Е = –14.50 и –13.95 эВ (16 и 7% соответственно). При менее отрицательных энергиях (до –9.0 эВ) вклад s-орбиталей колеблется от 0 до 3%. При Е = –9.05 эВ их доля увеличивается до 19%. Однако для ВЗМО она падает до 1%. Что касается p-орбиталей данных атомов золота, то их участие в формировании заполненной области ограничено орбиталью с Е = –15.51 эВ, а также ВЗМО, когда их вклад составляет всего 4%. В целом ВЗМО на 90% сформированы остальными атомами золота.
Влияние растворителя на адсорбцию ионов таллия
Рассмотрение взаимодействия адсорбированных ионов Tl+ со своим гидратным окружением было выполнено по модели поляризуемого континуума C-PCM [45–47]. Использование континуальной модели для отображения влияния растворителя на энергетику процессов, протекающих на поверхности, представляется достаточно оправданным. В качестве недостатка такой модели нередко называют пренебрежение образованием первичной сольватной оболочкой. Но, за исключением простейших континуальных моделей, их более развитые варианты учитывают образование первичной оболочки посредством разбиения области растворителя на ближайшую к иону зону и более дальнюю зону, которые различаются величиной диэлектрической постоянной растворителя [45, 47, 56, 57]. Вклад формирования первичной оболочки в процесс сольватации учитывается посредством параметризации, основанной на сопоставлении с экспериментальными данными. Важность правильного подбора количественных характеристик для отдельных атомов при выполнении такой параметризации подчеркивается, например, в публикациях [45, 46], в которых излагается и методология ее проведения. К числу достоинств континуальных моделей следует отнести учет взаимодействия с удаленными слоями растворителя. Эти слои могут оказать значительное влияние на первичную сольватную оболочку вплоть до перевода ее в разупорядоченное состояние [58]. Такое явление проявляется, например, в стоксовских радиусах, величины которых могут оказаться меньше соответствующих кристаллографических радиусов [59], или в положительных энергиях гидратации [60]. В случае сольватации многоатомных систем, когда заряд в большей или меньшей мере оказывается перераспределенным по объемистой системе, игнорирование вторичной сольватной оболочки может существенно исказить результат. Эти соображения представляются существенными при описании состояния адсорбированных ионов, когда заряд после перехода иона на поверхность принадлежит не только этому иону, а уже большому количеству атомов. Поэтому при описании электрохимической адсорбции на электродах влияние растворителя на процесс адсорбции необходимо учитывать в виде взаимодействия с растворителем как самого иона, так и металлического кластера.
При вычислениях по модели C-PCM использовалась параметризация на основе метода UAHF [45, 46]. Для атомов неметаллов параметры подбирались в соответствии с методикой, описание которой приведено в [45]. Вычисленные по такой методике энергии гидратации для используемых в дальнейших расчетах молекулы H2O и ионов H3O+ и OH– составляли соответственно –27, –441 и –438 кДж/моль и были близки к экспериментальным величинам –26, –435 и –443 кДж/моль из [45]. Для нейтральных атомов золота в вычислениях использовался ван-дер-ваальсов радиус по [61]. Что касается параметризации для ионов таллия, то в [45] подчеркивается, что, если в системе присутствуют заряженные атомы, то требуется коррекция радиусов соответствующих элементов, обусловленная величиной формального заряда. Поскольку в литературе отсутствуют сведения о параметрической поправке на заряд для ионов Tl+, для расчетов по модели С-РСМ, был проведен поиск величины радиуса, дающего величину вычисленной энергии гидратации наиболее близкой к экспериментальному значению для иона таллия –300 кДж/моль [60]. Наилучшее согласие между экспериментальной и расчетной энергиями имело место при использовании радиуса 0.192 нм. Данная величина использовалась далее в расчетах гидратационных эффектов по модели С-РСМ.
Процесс адсорбции иона из водного раствора сопровождается частичным разрушением его гидратной оболочки и вытеснением с поверхности молекул воды. Оба эти эффекта, особенно первый, требуют затрат энергии. В результате энергия адсорбции из раствора становится значительно меньше энергии адсорбции из газовой фазы. Вычисления энергии адсорбции из раствора проводились по уравнению (1), с тем отличием, что все три слагаемых этого уравнения вычислялись с использованием метода С-РСМ. Как это видно из таблицы, энергия адсорбции понижается примерно на 108–114 кДж/моль. Разность энергий адсорбции (между максимальным и минимальным значениями) на разных местах понижается с 8 до 2 кДж/моль, но вряд ли можно говорить о сильном нивелировании этой энергии растворителем.
Растворитель несколько увеличивает положительный заряд катиона таллия, но незначительно – в пределах всего 10%. Можно считать, что при адсорбции ионов Tl+ как из газовой фазы, так и из водного раствора таллий существует на незаряженной поверхности золота в ад-ионной форме.
Свободный Tl+ является катионом сильного основания и не подвергается гидролизу в водном электролите [44]. Эта устойчивость катиона таллия сохраняется и в адсорбированном состоянии. Энергия реакции
вычисленная с учетом взаимодействия с растворителем, имеет положительное значение и составляет 51 кДж/моль. Вычисленная для сравнения энергия реакции гидролиза катиона Tl+ в электролите имела величину 48 кДж/моль.Взаимодействие золото–ион таллия в щелочной среде
Катализ ионами таллия наблюдается так же и на границе между золотом и щелочными электролитами [8, 9, 12, 62]. Как уже упоминалось выше, TlOH является сильным электролитом [44]. Следовательно, и в щелочной среде формой таллия в растворе остаются катионы Tl+. Радиометрические исследования [17] продемонстрировали адсорбцию ионов таллия из сильно щелочной среды. Основное различие между процессами, протекающими на поверхности золота в нейтральных и щелочных растворах заключается в том, что в последнем случае имеет место адсорбция ионов OH– на золоте [62–64]. Адсорбируемость OH–, судя по опубликованным количественным данным [64–66], сопоставима по такому показателю с адсорбатом средней силы Cl–, но заметно уступает, например, ионам Br– или I–.
В используемых щелочных электролитах концентрация ионов OH– обычно составляет 0.01–0.1 М, а концентрация ионов таллия только 10–6–10–4 М [9, 12, 15, 17]. При таком соотношении их концентраций поток ионов OH– к поверхности на несколько порядков выше потока ионов Tl+. Следовательно, на начальном этапе контакта золота с щелочным раствором, содержащим ионы Tl+, поверхность со значительным преимуществом заполняется ионами OH–, а не ионами Tl+, и процесс адсорбции ионов таллия из щелочного электролита следует рассматривать не как взаимодействие ионов таллия со свободной поверхностью, а как переход Tl+ на поверхность золота, уже содержащую адсорбированные ионы OH–. При наличии на поверхности адсорбированных ионов противоположного знака и не очень больших степенях заполнения вследствие взаимного притяжения ионов их адсорбция усиливается [67]. При значительных заполнениях поверхности между ионами имеет место конкуренция за место на поверхности. В результате может происходить вытеснение с поверхности одних ионов другими [9, 68]. Следовательно, при установлении равновесия на поверхности золота может происходить либо вытеснение адсорбированных ионов OH– ионами таллия, либо взаимодействие ионов Tl+ с уже адсорбированными гидроксид-ионами вплоть до образования каких-либо соединений. В первом случае состояние ионов таллия на поверхности должно мало отличаться от их состояния, соответствующего кислым и нейтральным электролитам. Во втором случае состояние адсорбированных ионов таллия может быть иным. О соадсорбции ионов Tl+ и OH– говорится в [15, 16, 69–71]. Даже допускается, что в щелочной среде адсорбированный таллий может образовывать на поверхности золота соединения типа Tl2O или TlOH [69–71].
Согласно литературным данным, модельная энергия газофазной адсорбции ионов OH– на Au (111) довольно значительна и для энергетически наиболее выгодного места на поверхности, по разным данным, составляет от –189 до –440 кДж/моль [53, 72–74]. В [53] энергия адсорбции оценена только для места (2). В [72] наиболее выгодными для адсорбции названы места (3') и (2) с близкими значениями энергии. В [73] в качестве оптимального места для адсорбции называется (2). В качестве такого места в [74] предполагается точка (3''), однако выбор ограничен только местами (1) и (3") при разности энергий адсорбции для них около 50 кДж/моль.
Вследствие значительного расхождения литературных данных по адсорбции гидроксид-ионов на золоте и с целью получения результатов для условий, соответствующих данной публикации, было проведено вычисление энергии адсорбции OH– на кластере Au(111), описание которого приведено выше. Наиболее устойчивым было состояние с положением атома кислорода в положении (2) с углом между нормалью к поверхности и связью O–H 117°. Для этого состояния величина энергии газофазной адсорбции составляла –254 кДж/моль. Данная величина оказалась ближе всего к результатам из [72] (–272 кДж/моль). При учете взаимодействия с растворителем вычисленная энергия адсорбции OH– на золоте составила –24 кДж/моль.
Поскольку разность энергий адсорбции ионов OH– и Tl+ близка к 100 кДж/моль, реакция вытеснения ионов OH– катионами Tl+
(3)
${\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--O}}{{{\text{H}}}^{--}} + {\text{ T}}{{{\text{l}}}^{ + }} \to {\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--T}}{{{\text{l}}}^{ + }} + {\text{O}}{{{\text{H}}}^{--}}$В качестве альтернативы процессу вытеснения была вычислена энергия процесса соадсорбции Tl+ и OH–, который, с учетом изложенного выше, можно представить в виде реакции взаимодействия иона таллия с уже адсорбированным на поверхности ионом OH– и образования адсорбированного TlOH, что предполагается в [69].
(4)
${\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--O}}{{{\text{H}}}^{--}} + {\text{T}}{{{\text{l}}}^{ + }} \to {\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--TlOH}}.$Хотя объединение двух частиц противоположного знака является в значительной мере энергетически выгодным процессом (газофазной реакции (4) соответствует энергия –509 кДж/моль), при протекании реакции в растворе энергия реакции понижается вследствие того, что исчезает ион раствора, и на поверхности вместо заряженной частицы образуется незаряженная, т.е. происходит значительное изменение сольватного окружения. В итоге энергия реакции (4) понижается до –79 кДж/моль. Сопоставление энергий процессов (3) и (4) показывает, что соадсорбция ионов OH– и Tl+ представляется более предпочтительной по сравнению с вытеснением ионов OH–.
В [70] допускается формирование Tl2O на поверхности золота. Возможную реакцию его образования на поверхности в щелочной среде в результате взаимодействия с адсорбированными ионами OH– можно представить как
(5)
$\begin{gathered} {\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--O}}{{{\text{H}}}^{--}} + {\text{ 2T}}{{{\text{l}}}^{ + }} + {\text{ O}}{{{\text{H}}}^{--}} \to \\ \to {\text{ A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--OT}}{{{\text{l}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $При замещении иона водорода в адсорбированном OH– на два иона Tl+ и переходе H+ к свободному OH– исчезают три иона электролита и заряженная частица на поверхности превращается в незаряженную. Эти процессы сопровождаются значительными энергетическими потерями. В итоге энергия реакции (5) при ее протекании в растворе невелика и составляет всего –48 кДж/моль. Можно полагать, что образование на поверхности золота адсорбированного TlOH более вероятно, чем формирование адсорбированного Tl2O. Причиной этого является то, что процесс замещения иона водорода в адсорбированном TlOH катионом таллия, сопровождающийся исчезновением ионов электролита
(6)
${\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--TlOH }} + {\text{T}}{{{\text{l}}}^{ + }} + {\text{O}}{{{\text{H}}}^{--}} \to {\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--OT}}{{{\text{l}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}},$Также была оценена возможность замещения иона водорода ионом таллия с образованием адсорбированного аниона OTl–, который ориентирован атомом кислорода к поверхности:
(7)
${\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--O}}{{{\text{H}}}^{--}} + {\text{T}}{{{\text{l}}}^{ + }} + {\text{O}}{{{\text{H}}}^{--}} \to {\text{A}}{{{\text{u}}}_{{{\text{18}}}}}{\text{--OT}}{{{\text{l}}}^{--}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Положительное значение энергии этой реакции в электролите (13 кДж/моль) указывает на ее малую вероятность. При ориентации TlO– атомом таллия к поверхности энергия реакции замещения становилась еще более положительной (150 кДж/моль).
Сравнение энергий рассмотренных в данном разделе реакций показывает, что образование адсорбированных ассоциатов TlOH и Tl2O предпочтительнее вытеснения ионов OH– с поверхности. Судя по величине энергии реакции (4) появление TlOH на поверхности более вероятно. Данный вывод можно подтвердить сведениями о том, что Tl2O при контакте с водой образует TlOH [44].
Поскольку TlOH является наиболее предпочтительной формой при адсорбции иона таллия из щелочного раствора, представляется целесообразным подробнее рассмотреть его свойства в адсорбированном состоянии. Адсорбированный TlOH показан на рис. 4. Он имеет форму угловой молекулы, ориентированной атомом кислорода к поверхности. Атом кислорода располагается в положении, близком к (1) (смещение составляет всего 0.007 нм). В этом отношении адсорбированный TlOH напоминает адсорбированную молекулу воды, также ориентированную атомом кислорода к поверхности [52, 75, 76]. Атом таллия находится в положении (2) при z = 0.317 нм и RAu–Tl = = 0.349 нм, что несколько больше, чем при адсорбции иона Tl+ в положении (2) (см. табл. 1). Угол связи TlOH составляет 136° и несколько меньше такого угла в свободном TlOH (150°). Длина связи O–Tl в адсорбированной молекуле равна 0.233 нм и близка к длине связи в несвязанной молекуле TlOH (0.220 нм). Расстояние между атомом кислорода и поверхностью составляет 0.237 нм, что больше, чем в случае адсорбированного OH– (0.188 нм). Длина связи O–Au как для адсорбированного TlOH, так и для адсорбированного OH– одинакова (0.237 нм). Угол между нормалью к поверхности и связью O–H составляет 113°, что незначительно отличается от аналогичного параметра адсорбированного OH– (117°). Длина связи O–H одинакова для свободного и адсорбированного TlOH (0.097 нм). Такова же длина связи и в свободном и адсорбированном ионе OH–.

Таблица 1.
Энергии адсорбции иона таллия (ΔEads), геометрические параметры (RAu–Tl – расстояние от иона таллия до ближайшего поверхностного атома золота, z – расстояние по нормали от поверхности до адсорбата) и эффективные заряды таллия по Левдину (QL) и по методу натуральных орбиталей (NBO) (QNBO) для четырех мест адсорбции. В скобках приведены значения энергий адсорбции и эффективных атомных зарядов иона таллия, вычисленные с учетом влияния растворителя
| Место адсорбции | ΔEads, кДж/моль | z, нм | RAu–Tl, нм | QL | QNBO |
|---|---|---|---|---|---|
| 1 | –143 (–35) | 0.299 | 0.299 | 1.05 (1.18) | 1.08 (1.19) |
| 2 | –149 (–36) | 0.297 | 0.330 | 1.05 (1.16) | 1.08 (1.18) |
| 3′ | –148 (–36) | 0.296 | 0.340 | 1.04 (1.15) | 1.05 (1.16) |
| 3′′ | –151 (–37) | 0.297 | 0.340 | 1.05 (1.17) | 1.08 (1.19) |
В адсорбированном состоянии происходит смещение электронной плотности с молекулы TlOH на поверхность золота, и TlOH приобретает положительный заряд. Оценка заряда по Левдину дает величину 0.46. Оценка по методу NBO дает более низкое значение 0.35. Эффективный заряд атома таллия в адсорбированном TlOH при оценке двумя методами имеет близкие значения (QL = = 1.08 и QNBO = 1.14), которые мало отличаются от заряда адсорбированного иона Tl+, хотя имеют несколько более положительную величину. Заряд группы OH в адсорбированном TlOH имеет значения QL = –0.62 и QNBO = –0.79, более отрицательные, чем в адсорбированном на золоте анионе OH– (QL = –0.35 и QNBO = –0.59). Можно предположить, что при соадсорбции ионов таллия и гидроксида катион Tl+, который во многих случаях ведет себя подобно катионам щелочных металлов, способствует стабилизации отрицательного заряда расположенного рядом аниона, так как взаимодействие Tl–OH при этом усиливается. В качестве возможного подтверждения такого предположения можно привести тот факт, что реакция образования TlOH из ионов Tl+ и OH– на 220 кДж/моль энергетически выгоднее, чем реакция его образования из нейтральных Tl и OH. Другими словами, ионная составляющая связи Tl–OH весьма велика.
Взаимодействие с растворителем увеличивает заряд адсорбированного TlOH (до 0.55 по Левдину и до 0.42 по NBO). Это происходит в результате небольшого роста заряда атома таллия (QL = 1.16 и QNBO = 1.20). Заряд группы OH при этом практически не меняется (QL = –0.61 и QNBO = –0.78).
Спектр локальной плотности состояний системы TlOH/Au (111) для заполненной области взаимодействия адсорбент–адсорбат показан на рис. 5. Здесь же для сравнения приведена зависимость локальной плотности состояний для случая адсорбции ионов OH– на золоте. Поскольку энергии ВЗМО (EHOMO) для этих двух систем различаются (–5.28 эВ для адсорбированного TlOH и –2.61 эВ для адсорбированного OH–), в качестве оси абсцисс использована разность E – EHOMO (подобный прием используется при сопоставлении спектров D(E) для различающихся по какому-либо компоненту систем [77]). Кривая 1 соответствует локальной плотности состояний таллия в адсорбированном TlOH, а кривая 2 – группе ОН. D(E)-спектры по отдельности для атомов О и Н не приводятся, поскольку доля атома водорода значительно уступает вкладу атома кислорода в D(E). Как видно из графика со стороны TlOH электронное строение системы обусловлено в основном участием группы ОН. Ход графика локальной плотности состояний таллия при соадсорбции ионов Tl+ и OH– сильно изменяется по сравнению со случаем адсорбции одного лишь катиона таллия (рис. 2). Участие таллия в формировании электронного строения системы становится менее неравномерным. Как и в случае адсорбции катионов Tl+, оно обусловлено со значительным преимуществом s-орбиталью при существенно меньшей доле p-орбиталей. Со стороны группы OH система формируется со значительным преимуществом p-орбиталями. Вклад одновременно принадлежащих таллию и OH-группе орбиталей в связывание TlOH с золотом невелик и количество таких орбиталей мало. Для такого суммарного вклада TlOH не менее 5% и дополнительном условии, что доля Tl и OH по отдельности не менее 2%, этот вклад показан на графике вертикальными линиями с высотой соответствующей этому участию. Наблюдаемое в области положительнее –6 эВ совпадение положений пиков локальной плотности состояний Tl и OH (линии 1 и 2) на рис. 5 связано с тем, что орбитали, сформированные золотом и только таллием, а также золотом и только группой OH, имеют весьма близкие значения энергий. Значительный вклад общих орбиталей адсорбированного TlOH наблюдается только в области между –7 и ‒7.5 эВ. Однако здесь преобладающая роль OH очевидна. Сопоставление хода кривых 2 и 3 показывает, что свойства иона OH– на поверхности золота довольно близки, как в индивидуальной форме, так и при наличии катиона таллия. Смещение кривой 3 относительно кривой 2 к более положительным значениям энергии можно объяснить наличием отрицательного заряда, т.е. большей насыщенностью системы электронами.
Рис. 5.
Спектр плотности состояний при соадсорбции Tl+ и OH– на золоте: 1 – локальная плотность состояний атома таллия, 2 – локальная плотность состояний группы OH. Спектр плотности состояний при адсорбции ионов OH– на золоте: 3 – локальная плотность состояний иона OH–.

Реакция (4) с образованием показанного на рис. 4 TlOH представляется возможной при небольших степенях заполнения поверхности ионами OH–. В этом случае на поверхности достаточно места для того, чтобы катион таллия располагался рядом с уже адсорбированным анионом OH–. При высоких степенях заполнения на поверхности становится довольно тесно. Для того, чтобы ион таллия занял место рядом с адсорбированным OH–, необходимо вытеснение другого иона OH– с поверхности. Иначе говоря, образование ассоциата TlOH, представленного на рис. 4, должно сопровождаться десорбцией OH–. Оценка энергии такого процесса на основе энергий реакции (4) и десорбции OH– (см. выше) дала величину –55 кДж/моль.
Другим вариантом может быть образование ассоциатов, форма и положение которых на поверхности отличается от формы, показанной на рис. 4. Энергетически наиболее выгодной оказалась форма, соответствующая повороту TlOH примерно на 90° относительно поверхности (т.е., вертикальная адсорбция) со смещением атома таллия в положение (3") при связи TlOH с поверхностью через атом таллия. Геометрические параметры ассоциата при этом практически не изменяются. Энергия газофазной реакции с образованием такого ассоциата составляет –462 кДж/моль. Взаимодействие с растворителем уменьшает эту энергию до –59 кДж/моль.
Наконец, возможно обычное вытеснение ионов OH– с поверхности ионами таллия (3) (см. выше). Однако более низкая энергия этой реакции (–30 кДж/моль) по сравнению с энергиями образования ассоциатов делает ее менее вероятной. Значительная близость энергий реакций образования TlOH, связанного с поверхностью через атом кислорода при десорбции иона OH–, и вертикальной адсорбции TlOH, связанного с поверхностью через атом таллия без десорбции иона OH–, не позволяет сделать выбор между этими двумя вариантами.
ЗАКЛЮЧЕНИЕ
Исследование взаимодействия ионов таллия с поверхностью золота показало, что энергия его адсорбции очень мало зависит от места адсорбции (разность энергий находится в пределах 5%). При адсорбции катионов Tl+ они сохраняют свой заряд в адсорбированном состоянии, т.е. таллий существует на поверхности в ад-ионной форме. Проанализированы спектры плотности состояний и локальные D(E)-зависимости для адсорбированного иона таллия и непосредственно связанных с ним трех атомов золота. Участие тех и других в формировании молекулярных орбиталей системы более значительно в области более отрицательных энергий. Таллий участвует в образовании общей электронной системы благодаря в основном s-орбиталям, соседние атомы золота – за счет d-орбиталей. Растворитель не изменяет эффективный заряд таллия. В кислых и нейтральных средах ион таллия присутствует на поверхности в виде простых одноатомных ионов. Их гидролиз в таких средах не возможен.
В щелочных средах возможна соадсорбция ионов Tl+ и OH–. Образовавшийся ассоциат представляет собой угловую молекулу, связанную с золотом через атом кислорода и имеющую геометрическое строение, близкое к свободной молекуле TlOH. Адсорбированный ассоциат имеет положительный заряд. Связь TlOH с поверхностью осуществляется через атом кислорода. При этом свойства иона OH–, как при индивидуальной адсорбции, так и при соадсорбции с катионом таллия, довольно схожи.
Список литературы
Haissinsky, M., Mécanisme des dépots électrolytiques et expériences avec les radioéléments, J. Chim. Phys., 1946, vol. 43, p. 21.
Kolb, D.M., Przasnycky, M., and Gerischer, H., Underpotential deposition of metals and work function differences, J. Electroanal. Chem., 1974, vol. 54, p. 25.
Kolb, D.M., Leutloff, D., and Przasnycky, M., Optical properties of gold electrode surfaces covered with metal monolayers, Surf. Sci., 1975, vol. 47, p. 622.
Takamura, T., Watanabe, F., and Takamura, K., Electro-optical studies of submonolayers of lead formed on gold electrodes by faradaic adsorption in 1 M HClO4, Electrochim. Acta, 1974, vol. 19, p. 933.
Adžić, R.R. and Despić, A.R., Catalytic effect of metal adatoms deposited at underpotential, J. Chem. Phys., 1974, vol. 61, p. 3482.
Петрий, О.А., Лапа, А.С. Электрохимия адатомных слоев. Итоги науки, Электрохимия. Полукаров, Ю.М., Ред, М.: ВИНИТИ, 1987, Т. 24. С. 96. [Petrii, O.A. and Lapa, A.S., Electrochemistry of adatomic layers, Itogi nauki, Electrochemistry (in Russian), Polukarov, Yu.M., Ed, Moscow: VINITI, 1987, vol. 24, p. 96.]
Rhodes, A., Feliu, J.M., Aldaz, A., and Clavilier, J., The influence of polyoriented gold electrodes modified by reversibly and irreversibly adsorbed ad-atoms on the redox behaviour of the Cr(III)/Cr(II), J. Electroanal. Chem., 1989, vol. 271, p. 127.
Adžić, R., Wang, J., and Ocko. B.M., Structure of metal adlayers during the course of electrocatalytic reactions: O2 reduction on Au(111) with Tl adlayers in acid solutions, Electrochim. Acta, 1995, vol. 40, p. 83.
Pošcus, D., Agafonovas, G., and Jurgaitienė, I., Effect of thallium ions on the adsorption of cyanide-containing species from cyanide and dicyanoaurate solutions on a polycrystalline gold electrode, J. Electroanal. Chem., 1997, vol. 425, p. 107.
Gojo, M., Stankovic, V.D., and Poljacek, S.M., Electrochemical deposition of gold in citrate solution containing thallium, Acta Chim. Slov., 2008, vol. 55, p. 333.
McJntyre, J.D.E. and Peck, W.F., Electrodeposition of gold, J. Electrochem. Soc., 1976, vol. 123, p. 1800.
Бек, Р.Ю. Влияние микроколичеств нитрата таллия (1) на кинетику электроосаждения золота из щелочно-цианистых электролитов. Электрохимия. 2002. Т. 38. С. 1366. [Bek, R.Yu., Kinetics of gold electrodeposition from alkali–cyanide solutions: the effect of infinitesimal quantities of thallium(I) nitrate, Russ. J. Electrochem., 2002, vol. 38, p. 1237.]
Бек, Р.Ю., Швецова, О.Н. Влияние ионов таллия на скорость растворения золота в тиосульфатных электролитах. Электрохимия. 2012. Т. 48. С. 1148. [Bek, R.Yu. and Shevtsova, O.N., The effect of thallium ions on the gold dissolution rate in thiosulfate electrolytes, Russ. J. Electrochem., 2012, vol. 48, p. 1046.]
Wang, J.X., Adzic, R.R., Magnussen, O.M., and Ocko, B.M., Structure of electrodeposited Tl overlayers on Au (100) studied via surface X-ray scaterring, Surf. Sci., 1995, vol. 335, p. 120.
Polewska, W., Wang, J.X., Ocko, B.M., and Adzic, R.R., Scanning tunneling microscopy of electrodeposited thallium monolayers on Au(111) in alkaline solution, J. Electroanal. Chem., 1994, vol. 376, p. 41.
Niece, B.K. and Gewirth, A.A., Potential-step chronocoulometric and quartz crystal microbalance investigation of underpotentially deposited Tl on Au(111) electrodes, J. Phys. Chem. B, 1998, vol. 102, p. 818.
Pošcus, D. and Agafonovas, G., Radiotracer study of thallium underpotential deposition on a polycrystalline gold electrode in alkaline solutions, J. Electroanalyt. Chem., 2000, vol. 493, p. 50.
Shin, J.W., Bertocci, U., and Stafford, G. R., Underpotential deposition of Tl on (111)-textured Au: In situ stress and nanogravimetric measurements, J. Phys. Chem. C., 2014, vol. 114, p. 17621.
Salié, G. and Bartels, K., Partial charge transfer and adsorption at metal electrodes. The underpotential deposition of Hg(I), Tl(I), Bi(III) and Cu(II) on polycrystalline gold electrodes, Electrochim. Acta, 1994, vol. 39, p. 1057.
Кузнецов, Ан.М., Коршин, Г.В., Сайфуллин, А.Р. Квантово-химическое исследование адсорбции таллия на металлах подгруппы меди. Электрохимия. 1990. Т. 26. С. 681. [Kuznetsov, An.M., Korshin, G.V., and Saifullin, A.R., Quantum-chemical investigation of the adsorption of thallium on metals of the copper subgroup, Sov. Electrochem., 1990, vol. 26, p. 606.]
Liu, F.L., Zhao, Y.F., Li, X.Y., and Hao, F.Y., Ab initio study of the structure and stability of MnTln (M = Cu, Ag, Au; n = 1, 2) clusters, J. Mol. Struct. (Theochem), 2007, vol. 809, p. 189.
Pershina, V., Anton, J., and Jacob, T., Electronic structures and properties of MAu and MOH, where M = Tl and element 113, Chem. Phys. Lett., 2009, vol. 480, p. 157.
Pershina, V., Borschevsky, A., Anton, J., and Jacob, T., Theoretical predictions of trends in spectroscopic properties of gold containing dimers of the 6p and 7p elements and their adsorption on gold, J. Chem. Phys., 2010, vol. 133, p. 104304.
Zaitsevskii, A, Titov, A.V., Rusakov, A.A., and van Wüllen, C., Ab initio study of element 113–gold interactions, Chem. Phys. Lett., 2011, vol. 508, p. 329.
Fox-Beyer, B.S. and van Wüllen, C., Theoretical modelling of the adsorption of thallium and element 113 atoms on gold using two-component density functional methods with effective core potentials, Chem. Phys., 2012, vol. 395, p. 95.
Dean, J.A., Lange’s Handbook of Chemistry, N.Y.: McGraw-Hill, 1999. pp. 4.29, 4.7, 4.28.
König, S., Gäggeler, H.W., Eichler, R., Haenssler, F., Soverina, S., Dressler, R., Friedrich, S., Piguet, D., and Tobler, R., The production of long-lived thallium-isotopes and their thermochromatography studies on quartz and gold, PSI Annual Report 2005, Bern: Paul Scherrer Institut, 2006, p. 5.
Muther, B., Eichler, R., and Gäggeler, H.W., Thermochormatography of 212Pb and 200–202Tl on quartz and gold, PSI Annual Report 2007, Bern: Paul Scherrer Institut, 2008, p. 9.
Serov, A., Eichler, R., Türler, A., Wittwer, D., Gäggeler, H.W., Dressler, R., Piguet, D., and Vögele, A., Interaction of thallium species with quartz and gold surfaces, PSI Annual Report 2010, Bern: Paul Scherrer Institut, 2011, p. 6.
Serov, A., Eichler, R., Dressler, R., Piguet, D., Türler, A., Vögele, A., Wittwer, D., and Gäggeler, H.W., Adsorption interaction of carrier-free thallium species with gold and quartz surfaces, Radiochim. Acta, 2013, vol. 101, p. 421.
Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.H., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S.J., Windus, T.L., Dupuis, M., and Montgomery, J.A., General atomic and molecular electronic structure system, J. Comput. Chem., 1993, vol. 14, p. 1347.
Neese, F., The ORCA program system, WIREs Comput. Mol. Sci., 2012, vol. 2, p. 73.
Koch, W. and Holthausen, M.C., A chemist’s guide to density functional theory, Weinheim: Wiley-VCH and John Wiley & Sons, 2001. 293 p.
Becke, A.D., Density-functional thermochemistry. III. The role of exact exchange, J.Chem.Phys., 1993, vol. 98, p. 5648.
Stephens, P.J, Devlin, F.J., Chablowski, C.F., and Frisch, M.J., Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, vol. 98, p. 11623.
Hay, P.J. and Wadt, W.R., Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi, J. Chem. Phys., 1985, vol. 82, p. 284.
Hay, P.J. and Wadt, W.R., Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, vol. 82, p. 299.
McLean, A.D. and Chandler, G.S., Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18, J. Chem. Phys., 1980, vol. 72, p. 5639.
Krishnan, R., Binkley, J.S., Seeger, R., and Pople, J.A., Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, vol. 72, p. 650.
Löwdin, P.-O., On the nonorthogonality problem, Adv. Quantum Chem., 1970, vol. 5, p. 185.
Weinhold, F., Natural bond orbital method, in Encyclopedia of Computational Chemistry, Schleyer, P.v.R., Allinger, N.L., Clark T., Gasteiger, J., Kollman, P.A., Schaefer, H.F., and Schreiner, P.R., Eds, Chichester: John Willey & Sons, 1998. vol. 3, p. 1792.
Glendening, E.D., Landis, C.R., and Weinhold, F., Natural bond orbital methods, WIREs Comput. Mol. Sci., 2012, vol. 2, p. 1.
Titmuss, S., Wander, A., and King, D.A., Reconstruction of clean and adsorbate-covered metal surfaces, Chem. Rev., 1996, vol. 96, p. 1291.
Greenwood, N.N. and Earnshow, A., Chemistry of Elements, Oxford: Butterworth-Heinemann, 1998, p. 1176.
Barone, V., Cossi, M., and Tomasi, J., A new definition of cavities for the computation of solvation free energies by the polarizable continuum model, J. Chem. Phys., 1997, vol. 107, p. 3210.
Barone, V. and Cossi, M., Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, vol. 102, p. 1995.
Cossi, M., Rega, N., Scalmani, G., and Barone, V., Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comp. Chem., 2003, vol. 24, p. 669.
Boys, S.F. and Bernardi, F., The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, vol. 19, p. 553.
Jensen, F., Introduction to Computational Chemistry, Chichester: John Wiley & Sons, 2007, p. 227.
Pyykkö, P., Theoretical chemistry of gold, Angew. Chem. Int. Ed., 2004, vol. 43. p. 4412.
Nazmutdinov, R.R., Zinkicheva, T.T., Probst, M., Lust, K., and Lust, E., Adsorption of halide ions from aqueous solutions at a Cd(0001) electrode surface: quantum chemical modelling and experimental study, Surf. Sci., 2005, vol. 577, p. 112.
Нечаев, И.В., Введенский, А.В. Квантово-химическое моделирование адсорбции хлорид-иона и молекулы воды на металлах IB группы, Физикохим. поверхн. защ. матер., 2009, Т. 45, С. 150. [Nechaev, I.V. and Vvedenskii, A.V., Quantum chemical modeling of the adsorption of chloride ion and water molecule on group 1B metals, Prot. Met. Phys. Chem. Surf., 2009, vol. 45. p. 137.]
Liu, S., Ishimoto, T., and Koyama, M., First-principles calculation of OH−/OH adsorption on gold nanoparticles, Intern. J. Quantum Chem., 2015, vol. 115, p. 1597.
Schleyer, P.V.R., Encyclopedia of computational chemistry, Chichester: John Willey & Sons, 1998, vol. 1, p. 700.
O’Boyle, N.M., Tenderholt, A.L., and Langner, K., Software news and updates cclib: A library for package-independent computational chemistry algorithms, J. Comput. Chem., 2008, vol. 29, p. 839.
Chambers, C.C., Hawkins, G.D., Cramer, C.J., and Truhlar, D.C., Model for aqueous solvation based on class IV atomic charges and first solvation shell effects, J. Phys. Chem., 1996, vol. 100, p. 16385.
Da Silva, E.F., Svendsen, H.F., and Merz, K.M., Explicitly representing the solvation shell in continuum solvent calculations, J. Phys. Chem. A, 2009, vol. 113, p. 6404.
Desnoyers, J. E. and Jolicoeur, C., Hydratation effects and thermodynamic properties of ions, in Modern aspects of electrochemistry, Bockris, J. O’M., and Conway, B. E., Eds, N.Y.: Plenum Press, 1969, vol. 5, p. 26.
Robinson, R.A. and Stokes, R.H., Electrolyte solutions, London: Butterworth scientific publications, 1959, p. 125.
Marcus, Y., Thermodynamics of solvation of ions. Part 5. Gibbs free energy of hydration at 298.15 K, J. Chem. Soc. Trans. Farad., 1991, vol. 87, p. 2995.
Bondi, A., Van der waals volumes and radii, J. Phys. Chem., 1964, vol. 68, p. 441.
Štrbac, S. and Adžić, R.R., The influence of OH– chemisorption on the catalytic properties of gold single crystal surfaces for oxygen reduction in alkaline solutions, J. Electroanal. Chem., 1996, vol. 403, p. 169.
Бек, Р.Ю., Махнырь, Н.В., Зелинский, А.Г. Емкость двойного слоя обновляемого золотого электрода. Электрохимия. 1975. Т. 11. С. 1607. [Bek, R.Yu., Makhnyr’, N.V., and Zelinskii, A.G., Capacitance of electric double-layer at a recoverable gold electrode, Sov. Electrochem., 1975, vol. 11, p. 1503.]
Chen, A. and Lipkowski, J., Electrochemical and spectroscopic studies of hydroxide adsorption at the Au (111) electrode, J. Phys. Chem. B, 1999, vol. 103, p. 682.
Zhichao, S. and Lipkowski, J., Chloride adsorption at the Au (111) electrode surface, J. Electroanal. Chem., 1996, vol. 403, p. 225.
Zhichao, S., Lipkowski, J., Chen, A., Pettinger, B., and Bilger, C., Ionic adsorption at the Au (111) electrode, Electrochim. Acta, 1998, vol. 43, p. 2875.
Дамаскин, Б.Б., Батурина, О.А. Описание совместной специфической адсорбции катионов и анионов из бинарных растворов в рамках модели Грэма-Парсонса. Электрохимия. 1998. Т. 34. С. 403. [Damaskin, B.B. and Baturina, O.A., Specific co-adsorption of cations and anions from binary electrolytes in the framework of the Grahame-Parsons model, Russ. J. Electrochem., 1998, vol. 34, p. 366.]
Guaus, E., Sanz, F., Sluyters-Rehbach, M., and Sluyters, J.H., Competitive adsorption versus surface complexation as models for the simultaneous adsorption of metal complexes and free ligands, J. Electroanal. Chem., 1995, vol. 385, p. 121.
Amadelli, R., Marković, N., Adžić, R., and Yeager, E., Oxygen reduction on electrode surfaces modified by underpotential deposited species: Thallium on gold, J. Electroanal. Chem., 1983, vol.159, p. 391.
Wang, J.X., Adzic, R.R., O.M., and Ocko, B.M., X-ray scattering study of Tl adlayers on the Au (111) electrode in alkaline solutions: metal monolayer, OH– coadsorption, and oxide formation, J. Phys. Chem., 1994, vol. 98, p. 7182.
Stadler, R., Jusys, Z., and Baltruschat, H., Hydrogen evolution during the oxidation of formaldehyde on Au:: The influence of single crystal structure and Tl-upd, Electrochim. Acta, vol. 47, p. 4485.
Pessoa, A.M., Fajín, J.L.C., Gomes, J.R.B., and Cordeiro, M.N.D.S., Ionic and radical adsorption on the Au(hkl) surfaces: A DFT study, Surf. Sci., 2012, vol. 606, p. 69.
Shen, K., Jia, C., Cao, B., Xu, H., Wang, J., Zhang, L., Kim, K., and Wang, W., Comparison of catalytic activity between Au (110) and Au (111) for the electro-oxidation of methanol and formic acid: Experiment and density functional theory calculation, Electrochim. Acta, 2017, vol. 256, p. 129.
Нечаев, И.В., Введенский, А.В. Квантово-химическое моделирование адсорбции гидроксид-иона на металлах IB группы из водных растворов. Физикохим. поверхн. защ. матер. 2009. Т. 45. С. 358. [Nechaev, I.V. and Vvedenskii, A.V., Quantum chemical modeling of hydroxide ion adsorption on group 1B metals from aqueos solutions, Prot. Met. Phys. Chem. Surf., 2009, vol. 45, p. 391.]
Кузнецов, Ан.М., Маслий, А.Н., Шапник, М.С., Молекулярно-континуальная модель адсорбции цианид-иона на металлах подгруппы меди из водных растворов. Электрохимия. 2000. Т. 36. С. 1477. [Kuznetsov, An.M., Maslii, A.N., and Shapnik, M.S., Molecular–continuum model for the cyanide ion adsorption from aqueous solutions on copper metals, Russ. J. Electrochem., 2000, vol. 36. p. 1309.]
Liu, R., Adsorption and dissociation of H2O on Au (111) surface: A DFT study, Comput. Theor. Chem., 2013, vol. 1019, p. 141.
Bligaard, T. and Nørskov, J.K., Heterogeneous catalysis, Chemical bonding surfaces and interfaces, Nilsson, A., Petersson, L.G.M., and Nørskov, J.K., Eds. Amsterdam: Elsevier, 2008, pp. 258, 270.
Дополнительные материалы отсутствуют.