

Электрохимия, 2019, T. 55, № 10, стр. 1164-1176
Электрохимическое определение леводопы на угольно-пастовом электроде, модифицированном ДНК из молок лососевых и нанокомпозитом “восстановленный оксид графена–Fe3O4”
М. Хоссейни Галехно a, b, *, М. Мирзай a, М. Торкзаде-Махани b
a Керманский Университет Шахида Бахонара
Керман, Иран
b Университет последипломного образования в области передовых технологий
Керман, Иран
* E-mail: hmaryam703@gmail.com
Поступила в редакцию 22.11.2018
После доработки 28.01.2019
Принята к публикации 30.03.2019
Аннотация
Разработан электрохимический безмаркерный биосенсор на основе дезоксирибонуклеиновой кислоты (ДНК) для определения леводопы. В этом биосенсоре использован угольно-пастовый электрод, модифицированный восстановленным оксидом графена с магнитными наночастицами Fe3O4 и двухцепочечной ДНК. В основе определения лежит молекулярное взаимодействие между леводопой и ДНК. Вольтамперометрическое поведение леводопы на поверхности модифицированного угольно-пастового электрода исследовано методом дифференциальной импульсной вольтамперометрии, причем в качестве аналитического сигнала измеряли пиковый ток окисления леводопы. На электроде, покрытом ДНК, наблюдался существенный рост окислительного тока леводопы по сравнению с электродом, не содержавшим ДНК, что указывает на предконцентрирование леводопы в результате ее взаимодействия со слоем ДНК, локализованным на электроде. Структура синтезированных нанокомпозитов и состав электрода подтверждаются методами сканирующей электронной микроскопии, энергодисперсионного рентгеновского дифракционного микроанализа и ИК-спектроскопии с преобразованием Фурье. Электрохимические исследования показали, что модифицирование электрода существенно повышает пиковый ток окисления леводопы. В оптимальных условиях калибровочная кривая линейна в области 0.5–600 нM, а наименьшая определяемая концентрация равняется 0.11 нM. Относительное стандартное отклонение определения при концентрации, равной 200.0 нM, равняется 4.07% (n = 5). Разработанный биосенсор успешно использован при определении леводопы в образцах сыворотки человеческой крови и мочи.
ВВЕДЕНИЕ
Леводопа (биологически активная форма дофы) – это важное химическое соединение, используемое при лечении нервных расстройств, таких как болезнь Паркинсона. Это вещество – физиологический прекурсор допамина; оно участвует в метаболизме главным образом посредством ферментативной реакции (с участием дофа-декарбоксилазы), дающей допамин, компенсируя убыль последнего в мозге [1–3]. Леводопа способна устранять симптомы болезни Паркинсона и уменьшать мышечную ригидность, а также тремор и окулогирные кризы, благодаря своему превращению в допамин в ходе метаболизма в мозге [4].
Для определения леводопы в различных матрицах разработаны разнообразные аналитические методы, включая спектрофотометрию [5], жидкостную хроматографию высокого разрешения [6–8] и проточно-инъекционный анализ [9]. Однако большиноство этих методов обычно страдают такими недостатками, как дороговизна, недостаточная избирательность, использование органических растворителей, сложная процедура подготовки образцов и большая продолжительность анализов. Недавно большое внимание привлекли электрохимические биосенсоры на основе ДНК, благодаря их дешевизне, высокой чувствительности, быстрому отклику и хорошей избирательности, а также их способности к микроминиатюризации прибора. Электрохимические биосенсоры на основе ДНК содержат слой, распознающий нуклеиновую кислоту, иммобилизованный на электрохимическом преобразователе [10–12]. До настоящего времени различные типы электрохимических биосенсоров на основе ДНК имели в своей основе широкий набор наноматериалов, таких как наночастицы Au [13], углеродные нанотрубки [14] и графен [15], которые использовались для повышения чувствительности и стабильности биосенсоров на основе ДНК. В качестве одного из важнейших оксидов переходных металлов, оксид железа Fe3O4 является перспективным материалом для электродов суперконденсаторов, благодаря своей дешевизне, высокой распространенности, биосовместимости, низкой токсичности и хорошим электрохимическим свойствам [16–18]. Нанокомпозиты на основе Fe3O4 позволили приготовить проводящие материалы [19]. Например, композиты Fe3O4–графен, разработанные в качестве электродных материалов, вызвали большой интерес из-за положительного синергического эффекта между Fe3O4 и графеном [20].
Графен – это монослой sp2-гибридизованных атомов углерода, собранных в плотную кристаллическую структуру в форме пчелиных сот. Такая конфигурация придает этому материалу экстраординарные свойства, такие как выдающаяся термоустойчивость [21], очень высокая электронная проводимость [22, 23], заметная структурная гибкость [24, 25], высокая удельная площадь поверхности [26] и широкое поле применений [27] в науке и технологии наноматериалов. Поэтому изученные физико-химических свойства графена и его дешевизна позволили сделать вывод о том, что графен может быть идеальной матрицей для выращивания и закрепления большого числа функциональных веществ.
В настоящей работе разработан простой и недорогой метод определения леводопы, основанный на иммобилизации ДНК на поверхности угольно-пастового электрода, модифицированного нанокомпозитом “восстановленный оксид графена–Fe3O4” (схема 1). Исследованы параметры, оказывающие влияние на электродное поведение и аналитические характеристики угольно-пастового электрода, модифицированного ДНК и нанокомпозитом "восстановленный оксид графена–Fe3O4”.

Схема 1. Схематическое представление конструкции сенсора на основе ДНК для определения леводопы.
В предшествующей работе мы применили этот электрод для определения менадиона [28]. Задача настоящей работы – изучить возможность применения данного подхода к определению и других лекарственных веществ для целей клинического анализа.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приборы и реактивы
Все вольтамперометрические эксперименты проводили с помощью потенциостата/гальваностата Autolab PGSTAT 204, присоединенного к электрохимической ячейке и управляемого персональным компьютером (для сбора данных и контроля потенциала). Трехэлектродная система включала платиновую проволочку в качестве вспомогательного электрода, насыщенный Ag/AgCl-электрод сравнения и модифицированный угольно-пастовый электрод (рабочий электрод). Значения pH измеряли pH-метром Metrohm 827 (Herisau, Швейцария), укомплектованным системой “стеклянный электрод–электрод сравнения”. Измерения методами сканирующей электронной микроскопии (СЭМ) и энергодисперсионного рентгеновского дифракционного микроанализа проводили на приборе FESEM, модель Sigma VP (ZEISS, Германия), укомплектованном детектором EDS (Oxford Instruments, Великобритания). ИК-спектры с преобразованием Фурье снимали на спектрометре FTIR, модель Tensor 27 (Brucker, Германия) в области волновых чисел 4000–400 cм–1.
Порошок леводопы был приобретен у компании Sigma Aldrich, а двухцепочечная ДНК из молок лососевых – у компании Sigma (Сент-Луис, США). Восстановленный оксид графена также был приобретен у компании Sigma (Сент-Луис, США). Порошок чистого графита, FeCl3 · 6H2O, аскорбиновая кислота, гидразингидрат, первичный кислый фосфат натрия, ацетат натрия, уксусная кислота, фосфорная кислота и хлорная кислота были приобретены у компании Merck (Дармштадт, Германия). Исходный 0.01 мM раствор леводопы готовили, растворяя нужное количество порошка в дистиллированной воде; его хранили при 4°C. Исходный раствор ДНК (1.0 мг мл–1) готовили в Трис-HCl-буфере (pH 7.4) и хранили замороженным.
Синтез нанокомпозитов
Нанокомпозиты “восстановленный оксид графена–Fe3O4” были синтезированы по методике, описанной в предшествующих работах [28, 29]. Вкратце, приблизительно 0.036 г оксида графена диспергировали в 40 мл деионизованной воды. Затем в сосуд добавляли 0.270 г FeCl3 · 6H2O и 0.528 г аскорбиновой кислоты, и под действием ультразвука образовывался гомогенный раствор. Далее, к этой смеси при энергичном перемешивании добавляли 10 мл гидразингидрата. После этого полученный черный раствор переносили в автоклав из нержавеющей стали объемом 50 мл, выстланный тефлоном, и нагревали при 180°C в течение 8 ч. После охлаждения до комнатной температуры твердый осадок отделяли центрифугированием и трижды промывали деионизованной водой и этанолом, чтобы очистить композиты. Наконец, после сушки под вакуумом при 60°C в течение 12 ч получали композиты “восстановленный оксид графена–Fe3O4”.
Приготовление электродов
Несколько образцов с различным содержанием нанокомпозитов “восстановленный оксид графена–Fe3O4” были приготовлены и опробованы в определении леводопы. Угольно-пастовый электрод, модифицированный таким нанокомпозитом, готовили, смешивая композит “восстановленный оксид графена–Fe3O4”, порошок графита (в весовом отношении 1/100) и парафиновое масло в ступке; полученную смесь вмазывали в кончик стеклянной трубки (внутренний диаметр 3.4 мм). Наконец, в угольную пасту вставляли медную проволочку, чтобы создать электрический контакт. Из полученных результатов следует, что электрод, содержащий 1.0 вес. % нанокомпозита “восстановленный оксид графена–Fe3O4”, обладал прекрасной чувствительностью по отношению к леводопе. По этой же методике был приготовлен электрод с чистой поверхностью, т.е. без нанокомпозита “восстановленный оксид графена–Fe3O4”. Перед каждой серией измерений поверхность угольно-пастового электрода обновляли и подвергали тонкой полировке с помощью бумаги для взвешивания (бумаги, не обладающей адсорбирующими свойствами).
Эффективную площадь поверхности модифицированных электродов определяли методом циклических вольтамперограмм в модельном растворе 1 мM K3Fe(CN)6 в 0.1 M KCl, снятых при различных скоростях развертки потенциала. Для обратимого процесса пригодно следующее уравнение [30]:
Иммобилизация ДНК на поверхности угольно-пастового электрода, модифицированного нанокомпозитом “восстановленный оксид графена–Fe3O4”
Для иммобилизации ДНК угольно-пастовый электрод, модифицированный нанокомпозитом “восстановленный оксид графена–Fe3O4”, помещали в электрохимическую ячейку, заполненную 10 мл раствора 120.0 мкM ДНК в 0.5 M ацетатном буферном растворе (pH 4.8), и прикладывали к нему на 200 с потенциал, равный 0.5 В при помешивании [31]. Затем этот электрод промывали 0.5 M ацетатным буферным раствором (pH 4.8) и использовали в последующих измерениях.
Электрохимическое определение
Электрод, модифицированный нанокомпозитом “восстановленный оксид графена–Fe3O4”, погружали в 10 мл фосфатного буферного раствора (0.4 M, pH 8.0), содержащего различные концентрации леводопы, и раствор перемешивали при потенциале разомкнутой цепи в течение различного времени (стадия накопления). Затем электрод промывали дистиллированной водой и помещали в 10 мл фосфатного буферного раствора (0.3 M, pH 6.0). Вольтамперограммы записывали при развертке потенциала к положительным значениям в области от 0.0 до +400 мВ в дифференциальном импульсном режиме: ступень потенциала 9 мВ, модуляция амплитуды 50 мВ, $v$ = = 100 мВ с–1 (стадия обратного растворения). Все аналитические результаты – среднее из трех параллельных измерений. Концепция проведения анализа показана на схеме.
Образцы сыворотки человеческой крови и мочи перед анализом хранили в холодильнике при температуре 4°C. Их готовили в соответствии с литературным описанием [31], лишь с небольшими изменениями. Аликвоту образца мочи (1.0 мл) добавляли к заданному количеству леводопы и затем обрабатывали метанолом (1 мл) для осаждения белков мочи. После перемешивания образца в течение 30 с осажденные белки отделяли центрифугированием при 6000 об/мин в течение 5 мин. Прозрачный, не содержащий белков, слой надосадочной жидкости отфильтровывали сквозь фильтр Millipore (0.45 мкм) и разбавляли до 10 мл 0.40 M фосфатным буферным раствором (pH 8.0). Далее, по одному миллилитру образца сыворотки крови переносили в каждую из пробирок на центрифуге, содержащих различные известные количества леводопы, и тщательно смешивали с 1 мл метанола для осаждения белков крови. После центрифугирования при 6000 об/мин в течение 15 мин и отделения осажденных белков слой надосадочной жидкости отфильтровывали сквозь фильтр Millipore (0.45 мкм) и разбавляли до 10 мл 0.40 M фосфатным буферным раствором (pH 8.0).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В настоящей работе для модифицирования угольно-пастового электрода применяли ДНК и нанокомпозит “восстановленный оксид графена–Fe3O4”. Этот нанокомпозит увеличивал площадь поверхности и электронную проводимость. Более того, модифицирование восстановленным оксидом графена и наночастицами Fe3O4 повышало величину сигнала. Причина этого – взаимодействие между оксидом графена и наночастицами Fe3O4. ДНК использовали для предварительного концентрирования леводопы на поверхности электрода.
Характеристики нанокомпозита “восстановленный оксид графена–Fe3O4”
Для определения морфологии поверхности, размера частиц и элементного состава композита мы использовали методы сканирующей электронной микроскопии и энергодисперсионного рентгеновского дифракционного микроанализа. Однородность и морфологию поверхности нанокомпозита “восстановленный оксид графена–Fe3O4” мы исследовали методом сканирующей электронной микроскопии (рис. 1). СЭМ-микрофотографии (рис. 1a и 1б) ясно показывают относительно равномерное распределение наночастиц Fe3O4 по поверхности нанокомпозита на расстояниях 1–500 нм.
Рис. 1.
СЭМ-микрофотографии поверхности (a) угольно-пастового электрода с оксидом графена и (б) электрода, модифицированного нанокомпозитом “восстановленный оксид графена–Fe3O4”.

Спектр энергодисперсионного рентгеновского микроанализа выявил сигнал Fe(III) на поверхности нанокомпозита “восстановленный оксид графена–Fe3O4” (рис. 2). Присутствие в этом спектре только пиков углерода, кислорода и железа свидетельствует о том, что синтез проведен чисто.
Рис. 2.
Диаграмма энергодисперсионного рентгеновского дифракционного микроанализа нанокомпозита “восстановленный оксид графена–Fe3O4”.

Химическая структура приготовленного композита была также охарактеризована ИК-спектроскопией с преобразованием Фурье. Как видно из рис. 3a, в ИК-спектре с преобразованием Фурье восстановленного оксида графена присутствуют валентные колебания эпоксидной связи C–O (1163 cм–1), группы трет-C–OH (3416.20 cм–1) и связи C=O в карбонильной и карбоксильной группах (1726 cм–1). Синтезированные частицы Fe3O4 (рис. 3б) можно наблюдать как сильную полосу поглощения в спектре, которая охватывает характерное волновое число 625 cм–1. Эта конфигурация соответствует связям Fe–O, которые, как сообщалось [32], отвечают объему магнетита. Этот ИК-спектр с преобразованием Фурье нанокомпозита “восстановленный оксид графена–Fe3O4” говорит о том, что пики при 625 и 560 cм–1 отвечают валентным колебаниям связей Fe–O–Fe, что можно истолковать как присутствие оксида железа. По сравнению с оксидом графена, интенсивность полос поглощения уменьшилась благодаря алкокси-, карбокси- и карбонил/карбокси-связям. Полученные результаты показывают, что декорирование восстановленного оксида графена наночастицами Fe3O4 выполнено успешно.

Взаимодействие между леводопой и ДНК в растворе
На рис. 4 даны циклические вольтамперограммы леводопы в фосфатном буферном растворе (0.1 M, pH 7.0), снятые до и после ее взаимодействия с 50.0 нM ДНК. Видно, что после добавления ДНК потенциал пика тока окисления леводопы сдвигается в анодную область на 72 мВ. Этот результат можно объяснить одним из следующих двух факторов: (1) непроводящая ДНК может блокировать перенос электрона между леводопой и поверхностью электрода или (2) образующийся комплекс ДНК–леводопа электрохимически неактивен. Если, перенос электрона блокирован, то ток должен уменьшиться (по сравнению с электродом с чистой поверхностью), но не следует ожидать сдвига его пика. В присутствии ДНК ток должен определяться несвязанными частицами, поскольку скорость диффузии частиц, связанных с ДНК, мала [33]. Таким образом, наблюдаемое уменьшение анодного тока леводопы следует приписать уменьшению концентрации несвязанной леводопы, вызванному образованием комплекса леводопа–ДНК, как следствие добавления ДНК в раствор. Далее, после взаимодействия с ДНК потенциал пика тока сдвинулся к более положительным значениям. В литературе упоминалось [34], что отрицательный сдвиг потенциала пика тока характерен для электростатического взаимодействия между ДНК и другими веществами, в то время, как положительный сдвиг указывает на взаимодействие типа интеркаляции. Соответственно, положительный сдвиг потенциала пика тока окисления леводопы можно объяснить интеркаляцией леводопы в двойную спираль ДНК. В соответствии с этими наблюдениями, получается, что уменьшение пикового тока леводопы после добавления двухцепочечной ДНК вызвано интеркаляцией леводопы в объемистую, медленно диффундирующую молекулу ДНК, что и приводит к уменьшению кажущегося коэффициента диффузии.

Исследование иммобилизации ДНК на угольно-пастовом электроде, модифицированном нанокомпозитом “восстановленный оксид графена–Fe3O4” методом циклической вольтамперометрии
Поверхности модифицированных электродов были исследованы путем снятия циклических вольтамперограмм в 0.05 M растворе окислительно-восстановительной пары ${{{\text{Fe}}\left( {{\text{CN}}} \right)_{6}^{{3 - }}} \mathord{\left/ {\vphantom {{{\text{Fe}}\left( {{\text{CN}}} \right)_{6}^{{3 - }}} {{\text{Fe}}\left( {{\text{CN}}} \right)_{6}^{{4 - }}}}} \right. \kern-0em} {{\text{Fe}}\left( {{\text{CN}}} \right)_{6}^{{4 - }}}}$ в качестве индикатора в 0.01 M фосфатном буферном растворе и 0.1 M NaCl. Как видно из рис. 5б, пиковые токи как катодной, так и анодной волн на угольно-пастовом электроде, модифицированном нанокомпозитом “восстановленный оксид графена–Fe3O4”, повышаются, что вызвано увеличением эффективной площади поверхности электрода. После иммобилизации ДНК на поверхности этого электрода оба пика тока резко снизились (рис. 5в) из-за электростатического отталкивания между отрицательно заряженным фосфатным скелетом ДНК и окислительно-восстановительной парой (${{{\text{Fe}}\left( {{\text{CN}}} \right)_{6}^{{3 - }}} \mathord{\left/ {\vphantom {{{\text{Fe}}\left( {{\text{CN}}} \right)_{6}^{{3 - }}} {{\text{Fe}}\left( {{\text{CN}}} \right)_{6}^{{4 - }}}}} \right. \kern-0em} {{\text{Fe}}\left( {{\text{CN}}} \right)_{6}^{{4 - }}}}$). Соответственно, разница между видом циклических вольтамперограмм этих электродов позволяет оценить модифицирование угольно-пастового электрода.
Рис. 5.
Циклические вольтамперограммы 0.05 M смеси K3Fe(CN)6 и K4Fe(CN)6 в 0.1 M растворе NaCl на угольно-пастовом электроде с чистой поверхностью (a), на электроде модифицированном нанокомпозитом “восстановленный оксид графена–Fe3O4” (б) и на том же электроде с ДНК (в). Скорость развертки потенциала 100 мВ с–1.

ОПРЕДЕЛЕНИЕ ЛЕВОДОПЫ
Вольтамперометрическое поведение леводопы на поверхности угольно-пастового электрода, модифицированного ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4”
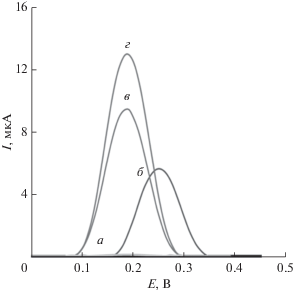
На рис. 6 даны дифференциальные импульсные вольтамперограммы, снятые в 200 нM растворе леводопы на угольно-пастовом электроде с чистой поверхностью, на электроде модифицированном нанокомпозитом “восстановленный оксид графена–Fe3O4” и на том же электроде с ДНК, снятые в 200 нM растворе леводопы в фосфатном буферном растворе: накопление леводопы в 0.40 M фосфатном буфере (pH 8.0), а окисление ее – в 0.3 M фосфатном буфере (pH 6.0). На дифференциальных импульсных вольтамперограммах, снятых на угольно-пастовом электроде, наблюдаются лишь слабые пики тока окисления. Это указывает на то, что скорость переноса электрона в процессах окисления леводопы на электроде с чистой поверхностью невелика. Рисунок 6в демонстрирует, что на угольно-пастовом электроде, модифицированном нанокомпозитом “восстановленный оксид графена–Fe3O4” скорость электроокисления леводопы эффективно повышена, и форма пика тока намного лучше. Это можно объяснить главным образом увеличением площади поверхности и низким поверхностным сопротивлением. Рисунок 6г показывает, что на угольно-пастовом электроде, модифицированном ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4”, сигнал окисления леводопы еще лучше. Это существенное увеличение пикового тока окисления говорит о том, что имеет место предварительное концентрирование благодаря адсорбции леводопы на поверхности модифицированного электрода из-за сильного ее взаимодействия со слоем иммобилизованной ДНК. Более того, интеркалятивное связывание леводопы с локализованной на поверхности ДНК способствует образованию слоя адсорбированной и предконцентрированной леводопы на поверхности покрытого ДНК угольно-пастового электрода, модифицированного нанокомпозитом “восстановленный оксид графена–Fe3O4”, что выливается в его повышенную чувствительность.
Рис. 6.
Дифференциальные импульсные вольтамперограммы, снятые в отсутствие леводопы (a) и после ее накопления на угольно-пастовом электроде с чистой поверхностью (б), на электроде модифицированном нанокомпозитом “восстановленный оксид графена–Fe3O4” (в) и на том же электроде с ДНК (г). Условия экспериментаs: накопление леводопы в 0.40 M фосфатном буферном растворе (pH 8.0), время накопления 8 мин, окисление леводопы в 0.3 M фосфатном буферном растворе (pH 6.0). Ступенька потенциала 9 мВ, амплитуда модуляции 50 мВ при скорости развертки потенциала 100 мВ с–1.
Влияние скорости развертки потенциала
Влияние скорости развертки потенциала (v) на электрохимические свойства угольно-пастового электрода, модифицированного ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4” было исследовано методом циклической вольтамперометрии (рис. S1 ). Скорость развертки потенциала от “внутренней” кривой к “внешней” равнялась 0.02, 0.04, 0.06, 0.08 и 0.10 В с–1. Показано, что анодный пиковый ток меняется с v по линейному закону в пределах от 0.02 до 0.10 В с–1. Уравнения линейной регрессии для окислительно-восстановительного процесса можно записать так:
График зависимости натурального логарифма пикового тока от натурального логарифма скорости развертки потенциала представляет собой прямую линию с наклоном, равным 0.2462 для анодного пикового тока, что отличается от теоретического значения 1.0. Это отражает смешанную природу – адсорбционную и диффузионную – контроля электродного процесса окисления леводопы в пределах 0.02–0.1 В с–1 [35].
Полученные уравнения таковы:
и
Оптимизация экспериментальных условий
Влияние времени иммобилизации ДНК на величину Ipa для накопленной леводопы в пределах от 100 до 500 с исследовали методом дифференциальной импульсной вольтамперометрии. В литературе упоминалось [36], что для наилучшего концентрирования потенциал следует поддерживать равным 0.50 В (Ag/AgCl). Иммобилизацию ДНК проводили из 120.0 мкM раствора ДНК. Показано, что с ростом времени иммобилизации количество адсорбированной ДНК увеличивается, а при приблизительно 200 с достигает насыщения. Соответственно, в последующем исследовании было выбрано время иммобилизации, равное 200 с.
Было исследовано влияние концентрации, pH и природы индифферентного электролита (буфер Бриттона–Робинсона, фосфатный буфер, Трис-HCl буфер) на стадию аккумулирования и на окисление леводопы на угольно-пастовом электроде, модифицированном ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4”. Самый высокий пик тока окисления леводопы был получен в фосфатном буферном растворе. Оценка фосфатного буферного раствора для накопления была проведена в области значений рН от 5.0 до 10.0. Как показано на рис. S2a , при pH 8.0 получен самый высокий пик тока окисления. Дополнительно была проведена оценка концентрации фосфатного буферного раствора для накопления в пределах от 0.1 до 0.6 M. Как видно из рис. S2 б, максимальный сигнал наблюдался в 0.4 M фосфатном буферном раствоpе. Именно такая концентрация буферного раствора была выбрана в качестве оптимальной.
Была исследована зависимость анодного пикового тока окисления леводопы от времени перемешивания в области 4–15 мин. Время перемешивания, равное 8 мин, было выбрано как оптимальное время накопления. Влияние pH раствора для окисления накопленной леводопы на отклик биосенсора было исследовано в буферных растворах различного типа (Трис-HCl буфер, фосфатный буфер, буфер Бриттона–Робинсона) с различными значениями pH (от 3.00 до 8.00) и концентрации (от 0.1 до 0.5 M). Полученные результаты показали, что величина Ipa была максимальна в фосфатном буферном раствоpе с pH 6.0 и концентрацией 0.3 M (рис. S2 в, г). Поэтому в последующих экспериментах стадию окисления накопленной леводопы проводили в 0.3 M фосфатном буфере с pH 6.0.
Процедура анализа
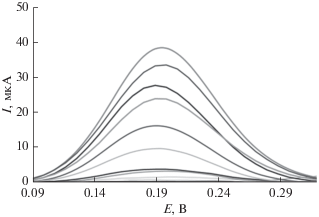
Анализы проводили методом дифференциальной импульсной вольтамперометрии, приняв в качестве детектируемого сигнала пик волны окисления леводопы при потенциале 200 мВ. На рис. 7 показан отклик дифференциальных импульсных вольтамперограмм, снятых на угольно-пастовом электроде, модифицированном ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4” при различных концентрациях леводопы. Построенная калибровочная кривая для определения леводопы разработанным биосенсором при оптимальных условиях эксперимента демонстрирует линейную связь между пиковым током и концентрацией леводопы cLD в области концентраций 0.5–600 нM. Уравнение линейной регрессии таково: I (мкA) = (0.07 ± 0.01)cLD (нM) + + (11.1 ± 0.3) с R2 = 0.9960; наименьшая определяемая концентрация составляет 0.11 нM.
Рис. 7.
Дифференциальные импульсные вольтамперограммы, снятые на угольно-пастовом электроде, модифицированном ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4” при различных концентрациях леводопы: 0.05, 2.5, 10.0, 20.0, 60.0, 80.0, 100.0, 200.0, 300.0, 400.0, 500.0 и 600.0 нM. Ступенька потенциала 9 мВ, амплитуда модуляции 50 мВ при скорости развертки потенциала 100 мВ с–1.
Стабильность и воспроизводимость биосенсора для определения леводопы
Стабильность биосенсора на основе ДНК исследовали в течение двух недель. После выдерживания в холодильнике (при 4°C) в течение двух недель биосенсор был использован для определения леводопы в той же концентрации (200.0 нM). Снижение пикового тока составило всего 5.07% от его первоначального значения. Это говорит о том, что этот биосенсор на основе ДНК весьма стабилен.
Воспроизводимость биосенсора на основе ДНК тестировали, определяя 200.0 нM леводопы с помощью пяти различных биосенсоров на основе ДНК, приготовленных независимо друг от друга. Для этих пяти независимых определений относительное стандартное отклонение составило 4.20%. Эти экспериментальные результаты показывают хорошую воспроизводимость разработанного протокола изготовления сенсоров. По окончании каждого цикла определений не требуется удалять интеркалированный аналит, потому что выбранная методика иммобилизации на угольно-пастовом электроде позволяет легко и быстро обновлять его поверхность.
Повторяемость биосенсора характеризовали, проводя пять одинаковых измерений после накопления леводопы из ее 200.0 нM раствора на угольно-пастовом электроде, модифицированном ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4”; соответствующее относительное стандартное отклонение равняется 4.06%.
Исследование возможных помех определению
Влияние различных посторонних веществ исследовали в процессе определения 200 нM леводопы. Предельно допустимое вредное воздействие мы определили как максимальную концентрацию постороннего вещества, которая вызывает приблизительно ±5%-ную относительную ошибку в определении леводопы. Это предельно допустимое вредное воздействие составило 50.0 мM для Na+, Cl−, F−, ${\text{HCO}}_{3}^{ - },$ ${\text{NO}}_{3}^{ - }$ и K+; 10.0 мM для Mg2+ и Ca2+; 3.0 мM для фолевой кислоты, мочевой кислоты, глюкозы, лактозы, фруктозы, сахарозы и ацетаминофена.
Анализ реальных образцов
Точность разработанного электрохимического биосенсора мы определяли двумя способами: (1) анализируя процент обратного извлечения известного количества аналита, намеренно добавленного в образцы сыворотки крови и мочи, и (2) сравнивая результаты предложенного метода и жидкостной хроматографии высокого разрешения (данные жидкостной хроматографии высокого разрешения были получены из аналитической лаборатории). Как показано в табл. 1, процент обратного извлечения леводопы из образцов удовлетворительный, так что разработанный биосенсор вполне применим для определения леводопы в реальных образцах. В табл. 2 даны результаты определения леводопы в образцах сыворотки крови и мочи с помощью разработанного биосенсора и жидкостной хроматографии высокого разрешения. Альтернативный F-тест (p = = 0.05) подтвердил отсутствие существенного различия между результатами, полученными этими двумя методами. Таким образом, разработанный угольно-пастовый электрод, модифицированный ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4” демонстрирует хорошую точность при анализе реальных образцов.
Таблица 1.
Определение леводопы в образцах сыворотки крови и мочи человека с использованием угольно-пастового электрода, модифицированного ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4”
| Образец | Добавлено нM | Найденоa, нM | Процент обратного извлечения, % |
|---|---|---|---|
| Сыворотка крови человека | 0.0 | 0.0 | – |
| 100 | 101 ± 1 | 101.0 | |
| 300 | 300 ± 2 | 100.0 | |
| 500 | 499 ± 1 | 99.8 | |
| Моча | 0.0 | 0.0 | – |
| 100 | 99.0 ± 0.0 | 99.0 | |
| 300 | 302 ± 1 | 100.7 | |
| 500 | 501 ± 1 | 100.2 |
Таблица 2.
Количественное определение леводопы в сыворотке крови и моче человека с использованием разработанного биосенсора и жидкостной хроматографии высокого разрешения (n = 3; P = 0.95)
| Образец | Найдено вольтамперометрическим методом, нM | Найдено методом жидкостной хроматографии высокого разрешения, нM | FRa | |
|---|---|---|---|---|
| Сыворотка крови человека | 1 | 51 ± 1 | 51 ± 2 | 1.1 |
| 2 | 71.8 ± 0.2 | 72.5 ± 0.6 | 1.7 | |
| 3 | 30 ± 2 | 29.4 ± 0.4 | 3.0 | |
| Моча | 1 | 17 ± 1 | 16.7 ± 0.6 | 2.7 |
| 2 | 34 ± 1 | 35.2 ± 0.3 | 2.3 | |
| 3 | 22 ± 1 | 21.4 ± 0.4 | 2.3 | |
Сравнение с другими методами
В табл. 3 проведено сравнение характеристик отклика разработанного биосенсора и аналитических характеристик ранее разработанных вольтамперометрических сенсоров для определения леводопы. Видно, что наш биосенсор не только демонстрирует весьма низкую наименьшую определяемую концентрацию (0.11 нM) и сравнимую или даже более широкую область линейности по концентрации, чем другие сенсоры, но он также простой, относительно недорогой и гибкий в использовании.
Таблица 3.
Сравнение разработанного биосенсора с некоторыми методами, предложенными в настоящее время для определения леводопы
| Электрод | Метод | Область линейности, M | Наименьшая определяемая концентрация, M | Ссылка |
|---|---|---|---|---|
| Угольно-пастовый, модифицированный нанопроволоками диспрозия | Квадратно-волновая вольтамперометрия с быстрым преобразованием Фурье | 1.0 × 10–9– 0.01 × 10–9 | 4.0 × 10–9 | [37] |
| Стеклоуглеродный | Дифференциальная импульсная вольтамперометрия | 2.0 × 10–7– 7.5 × 10–4 | 4.2 × 10–8 | [38] |
| Стеклоуглеродный | Дифференциальная импульсная вольтамперометрия | 2.0 × 10–6– 2.2 × 10–4 | 6.0 × 10–7 | [39] |
| Золотой, полученный методом сеткографии | Дифференциальная импульсная вольтамперометрия | 9.9 × 10–5– 1.2 × 10–3 | 6.8 × 10–5 | [40] |
| Угольно-пастовый | Дифференциальная импульсная вольтамперометрия | 1.2 × 10–4– 1.0 × 10–2 | 8.5 × 10–5 | [41] |
| Тонкопленочный оксованадий-саленовыйа | Амперометрия | 1.0 × 10–6– 1.0 × 10–4 | 8.0 × 10–7 | [1] |
| Золотые наноэлектродные ансамбли | Амперометрия | 2.5 × 10–8– 1.5 × 10–6 | 1.5 × 10–8 | [42] |
| Угольно-пастовый, модифицированный нанокомпозитом “восстановленный оксид графена–Fe3O4” | Дифференциальная импульсная вольтамперометрия | 0.5 × 10–9– 600 × 10–9 | 0.11 × 10 –9 | Настоящая работа |
ЗАКЛЮЧЕНИЕ
Нанокомпозит восстановленного оксида графена с Fe3O4 синтезирован по простой методике. Он использован для модифицирования угольно-пастового электрода с целью сконструировать биосенсор на основе ДНК для определения леводопы.
Взаимодействие между леводопой и ДНК в растворе исследовано методом дифференциальной импульсной вольтамперометрии. Мы также исследовали окисление леводопы на угольно-пастовом электроде, модифицированном ДНК и нанокомпозитом “восстановленный оксид графена–Fe3O4”. Полученные экспериментальные результаты показывают, что биосенсор на основе ДНК и нанокомпозита “восстановленный оксид графена–Fe3O4” достаточно чувствителен для определения леводопы с хорошей повторяемостью и точностью. Далее, после оптимизации основных экспериментальных параметров эксплуатационные характеристики сенсора оценивались в отношении его пригодности в качестве аналитического метода. Биосенсор на леводопу показал низкую наименьшую определяемую концентрацию, быстрый отклик, хорошую стабильность и воспроизводимость в работе. Поскольку этот биосенсор простой и недорогой, возможное применение этого подхода в клиническом анализе заслуживает дальнейшего изучения.
Список литературы
Teixeira, M.F., Marcolino-Junior, L., Fatibello-Filho, O., Dockal, E., and Bergamini, M.F., An electrochemical sensor for l-dopa based on oxovanadium-salen thin film electrode applied flow injection system, Sens. Actuators B Chem., 2007, vol. 122, p. 549.
Di Giulio, I., St George, R.J., Kalliolia, E., Peters, A.L., Limousin, P., and Day, B.L., Maintaining balance against force perturbations: impaired mechanisms unresponsive to levodopa in Parkinson9s disease, J. Neurophysiol., 2016, vol. 11, p. 493.
Gardoni, F., Morari, M., Kulisevsky, J., Brugnoli, A., Caccia, C., Mellone, M., Melloni, E., Padoani, G., Sosti, M., and Vailati, S., Safinamide modulates levodopa induced striatal glutamatergic overactivity in a rat model of Parkinson’s disease, J. Neurol. Sci., 2017, vol. 381, p. 361.
Hasan, B.A., Khalaf, K.D., and De La Guardia, M., Flow analysis-spectrophotometric determination of L‑dopa in pharmaceutical formulations by reaction with p-aminophenol, Talanta., 1995, vol. 42, p. 627.
Coello, J., Maspoch, S., and Villegas, N., Simultaneous kinetic-spectrophotometric determination of levodopa and benserazide by bi-and three-way partial least squares calibration, Talanta., 2000, vol. 53, p. 627.
Sagar, K.A. and Smyth, M.R., Simultaneous determination of levodopa, carbidopa and their metabolites in human plasma and urine samples using LC-EC, J. Pharm. Biomed., Anal. 2000, vol. 22, p. 613.
Saxer, C., Niina, M., Nakashima, A., Nagae, Y., and Masuda, N., Simultaneous determination of levodopa and 3-O-methyldopa in human plasma by liquid chromatography with electrochemical detection, J. Chromatogr. B., 2004, vol. 802, p. 299.
Tolokan, A., Klebovich, I., Balogh-Nemes, K., and Horvai, G., Automated determination of levodopa and carbidopa in plasma by high-performance liquid chromatography-electrochemical detection using an on-line flow injection analysis sample pretreatment unit, J. Chromatogr. B., 1997, vol. 698(1), p. 201.
Marcolino-Junior, L.H., Teixeira, M.F., Pereira, A.V., and Fatibello-Filho, O., Flow injection determination of levodopa in tablets using a solid-phase reactor containing lead(IV) dioxide immobilized, J. Pharm. Biomed. Anal., 2001, vol. 25, p. 393.
Ding, Y., Wang, Q., Gao, F., and Gao, F., Highly sensitive and selective DNA biosensor using a dumbbell-shaped bis-groove binder of bi-acetylferrocene ethylenediamine complex as electrochemical indicator, Electrochim. Acta, 2013, vol. 106, p. 35.
Huang, K.-J., Liu, Y.-J., Wang, H.-B., and Wang, Y.-Y., A sensitive electrochemical DNA biosensor based on silver nanoparticles-polydopamine@ graphene composite, Electrochim. Acta, 2014, vol. 118, p. 130.
Ghalehno, M.H., Mirzaei, M., and Torkzadeh-Mahani, M., Aptamer-based determination of tumor necrosis factor α using a screen-printed graphite electrode modified with gold hexacyanoferrate, Microchim. Acta, 2018, vol. 185, p. 165.
Li, L., Ma, C., Kong, Q., Li, W., Zhang, Y., Ge, S., Yan, M., and Yu, J., A 3D origami electrochemical immunodevice based on a Au@Pd alloy nanoparticle-paper electrode for the detection of carcinoembryonic antigen, J. Mater. Chem., 2014, vol. 2, p. 6669.
Peng, H.-P., Hu, Y., Liu, P., Deng, Y.-N., Wang, P., Chen, W., Liu, A.-L., Chen, Y.-Z., and Lin, X.-H., Label-free electrochemical DNA biosensor for rapid detection of mutidrug resistance gene based on Au nanoparticles/toluidine blue–graphene oxide nanocomposites, Sens. Actuators B Chem., 2015, vol. 207, p. 269.
Jiang, C., Yang, T., Jiao, K., and Gao, H., A DNA electrochemical sensor with poly-l-lysine/single-walled carbon nanotubes films and its application for the highly sensitive EIS detection of PAT gene fragment and PCR amplification of NOS gene, Electrochim. Acta, 2008, vol. 53, p. 2917.
Kumar, P.R., Jung, Y.H., Bharathi, K.K., Lim, C.H., and Kim, D.K., High capacity and low cost spinel Fe3O4 for the Na-ion battery negative electrode materials, Electrochim. Acta, 2014, vol. 146, p. 503.
Sargazi, G., Afzali, D., and Mostafavi, A., An efficient and controllable ultrasonic-assisted microwave route for flower-like Ta(V)–MOF nanostructures: preparation, fractional factorial design, DFT calculations, and high-performance N 2 adsorption, J. Porous. Mat., 2018, p. 1.
Sargazi, G., Afzali, D., and Mostafavi, A., A novel synthesis of a new thorium(IV) metal organic framework nanostructure with well controllable procedure through ultrasound assisted reverse micelle method, Ultrason. Sonochem., 2018, vol. 41, p. 234.
Vinothkannan, M., Karthikeyan, C., Kim, A.R., and Yoo, D.J., One-pot green synthesis of reduced graphene oxide (RGO)/Fe3O4 nanocomposites and its catalytic activity toward methylene blue dye degradation, Spectrochim. Acta, 2015, vol. 136, p. 256.
Zhang, W., Zheng, J., Shi, J., Lin, Z., Huang, Q., Zhang, H., Wei, C., Chen, J., Hu, S., and Hao, A., Nafion covered core–shell structured Fe3O4@ graphene nanospheres modified electrode for highly selective detection of dopamine, Anal. Chim. Acta, 2015, vol. 853, p. 285.
Balandin, A.A., Ghosh, S., Bao, W., Calizo, I., Teweldebrhan, D., Miao, F., and Lau, C.N., Superior thermal conductivity of single-layer graphene, Nano Letters., 2008, vol. 8, p. 902.
Novoselov, K.S., Jiang, Z., Zhang, Y., Morozov, S., Stormer, H.L., Zeitler, U., Maan, J., Boebinger, G., Kim, P., and Geim, A.K., Room-temperature quantum Hall effect in graphene, Science, 2007, vol. 315, p. 1379.
Novoselov, K.S., Geim, A.K., Morozov, S., Jiang, D., Katsnelson, M., Grigorieva, I., Dubonos, S., Firsov, and AA. Two-dimensional gas of massless Dirac fermions in graphene, Nature, 2005, vol. 438, p. 197.
Lee, C., Wei, X., Kysar, J.W., and Hone, J., Measurement of the elastic properties and intrinsic strength of monolayer graphene, Science, 2008, vol. 321, p. 385.
Dikin, D.A., Stankovich, S., Zimney, E.J., Piner, R.D., Dommett, G.H., Evmenenko, G., Nguyen, S.T., and Ruoff, R.S., Preparation and characterization of graphene oxide paper, Nature, 2007, vol. 448, p. 457.
Peigney, A., Laurent, C., Flahaut, E., Bacsa, R., and Rousset, A., Specific surface area of carbon nanotubes and bundles of carbon nanotubes, Carbon., 2001, vol. 39, p. 507.
Rao, C., Biswas, K., Subrahmanyam, K., and Govindaraj, A., Graphene, the new nanocarbon, J. Mater. Chem., 2009, vol. 19, p. 2457.
Ghalehno, M.H., Mirzaei, M., and Torkzadeh-Mahani, M., Double strand DNA-based determination of menadione using a Fe3O4 nanoparticle decorated reduced graphene oxide modified carbon paste electrode, Bioelectrochem., 2018, vol. 124, p. 165.
Su, J., Cao, M., Ren, L., and Hu, C., Fe3O4–graphene nanocomposites with improved lithium storage and magnetism properties, J. Phys. Chem. C, 2011, vol. 115, p. 14469.
Bard, A.J., Faulkner, L.R., Leddy, J., and Zoski, C.G., Electrochemical Methods: Fundamentals and Applications, vol. 2, New York: Wiley, 1980.
Wang, Y., Ni, Y., and Kokot, S., Voltammetric behavior of complexation of salbutamol with calf thymus DNA and its analytical application, Anal. Biochem., 2011, vol. 419, p. 76.
Bagheri, H., Afkhami, A., Saber-Tehrani, M., and Khoshsafar, H., Preparation and characterization of magnetic nanocomposite of Schiff base/silica/magnetite as a preconcentration phase for the trace determination of heavy metal ions in water, food and biological samples using atomic absorption spectrometry, Talanta, 2012, vol. 97, p. 87.
Welch, T.W. and Thorp, H.H., Distribution of metal complexes bound to DNA determined by normal pulse voltammetry, J. Phys. Chem., 1996, vol. 100, p. 13829.
Lu, X., Zhang, M., Kang, J., Wang, X., Zhuo, L., and Liu, H., Electrochemical studies of kanamycin immobilization on self-assembled monolayer and interaction with DNA, J. Inorg. Biochem., 2004, vol. 98, p. 582.
Laviron, E., Roullier, L., and Degrand, C., A multilayer model for the study of space distributed redox modified electrodes: Part II. Theory and application of linear potential sweep voltammetry for a simple reaction, Electroanalysis, 1980, vol. 112, p. 11.
Kara, P., Kerman, K., Ozkan, D., Meric, B., Erdem, A., Nielsen, P.E., and Ozsoz, M., Label-Free and Label Based Electrochemical Detection of Hybridization by Using Methylene Blue and Peptide Nucleic Acid Probes at Chitosan Modified Carbon Paste Electrodes, Electroanalysis, 2002, vol. 14, p. 1685.
Daneshgar, P., Norouzi, P., Ganjali, M.R., Ordikhani-Seyedlar, A., and Eshraghi, H., A dysprosium nanowire modified carbon paste electrode for determination of levodopa using fast Fourier transformation square-wave voltammetry method, Colloids Surf. B., 2009, vol. 68, p. 27.
Quintino, M.S.M., Yamashita, M., and Angnes, L., Voltammetric studies and determination of levodopa and carbidopa in pharmaceutical products, Electroanalysis., 2006, vol. 18, p. 655.
Babaei, A. and Babazadeh, M., A selective simultaneous determination of levodopa and serotonin using a glassy carbon electrode modified with multiwalled carbon nanotube/chitosan composite, Electroanalysis., 2011, vol. 23, p. 1726.
Bergamini, M.F., Santos, A.L., Stradiotto, N.R., and Zanoni, M.V.B., A disposable electrochemical sensor for the rapid determination of levodopa, J. Pharm. Biomed. Anal., 2005, vol. 39, p. 54.
Teixeira, M.F., Bergamini, M.F., Marques, C.M., and Bocchi, N., Voltammetric determination of L-dopa using an electrode modified with trinuclear ruthenium ammine complex (Ru-red) supported on Y-type zeolite, Talanta., 2004, vol. 63, p. 1083.
Viswanathan, S., Liao, W.-C., Huang, C.-C., Hsu, W.-L., and Ho, J.-a.A., Rapid analysis of L-dopa in urine samples using gold nanoelectrode ensembles, Talanta., 2007, vol. 74, p. 229.
Дополнительные материалы отсутствуют.