

Электрохимия, 2019, T. 55, № 12, стр. 1435-1451
Влияние рH раствора на окисление сульфит-ионов и образование оксидов на золотом электроде
А. Г. Зелинский a, *, О. Н. Новгородцева a, b, **
a Институт химии твердого тела и механохимии СО РАН
630128 Новосибирск, ул. Кутателадзе, 18, Россия
b Новосибирский государственный технический университет, кафедра ХХТ
630073 Новосибирск, просп. К. Маркса, 20, корп. 1, Россия
* E-mail: azelinsky@solid.nsc.ru
** E-mail: shevtsova@solid.nsc.ru
Поступила в редакцию 20.12.2018
После доработки 04.04.2019
Принята к публикации 10.05.2019
Аннотация
В работе представлены результаты изучения влияния pH раствора на электродный процесс, который протекает на золотом электроде в растворах сульфита натрия, серной кислоты, щелочи и в растворах универсальной буферной смеси Бриттона–Робинсона (pH 2–14). В растворах сульфита натрия электродный процесс представляет собой комбинацию процессов окисления сульфитных частиц и образования оксидов на поверхности золота, которые протекают одновременно и в одной области потенциалов. В работе показано, что pH раствора и процесс окисления сульфитных частиц не влияют на количество образованного α-оксида золота. В то же время pH раствора оказывает сильное влияние на процесс окисления сульфитных частиц. Так, в растворах с величинами pH в области примерно от 2 до 11 измеренные вольтамперограммы одинаковы, т.е. скорость процесса не зависит от кислотности и парциального состава раствора. Эти результаты указывают на то, что в этой области pH окисление сульфит-ионов можно интерпретировать брутто-реакцией: $2{\text{SO}}_{3}^{{2 - }} \to {{{\text{S}}}_{{\text{2}}}}{\text{O}}_{6}^{{2 - }} + 2{\text{e}}$ с образованием дитионат-ионов. В сильнощелочных растворах (pH 12.5–14) потенциал окисления смещается в сторону более отрицательных потенциалов, а величина тока уменьшается при увеличении pH. Эти результаты указывают на то, что в сильнощелочных растворах окисление сульфит-ионов может протекать на частично блокированной поверхности электрода по реакции: ${\text{SO}}_{3}^{{2 - }} + 2{\text{O}}{{{\text{H}}}^{ - }} \to {\text{SO}}_{4}^{{2 - }} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + 2{\text{e}}$ с образованием сульфат-ионов. Изменение механизма окисления сульфит-ионов происходит в узкой области потенциалов в растворах с pH в диапазоне от 11 до 12.5 и сопровождается аномально резкими изменениями измеряемого тока. Эти аномалии связываются с особым характером динамики процесса пассивации/депассивации поверхности электрода оксидами золота.
ВВЕДЕНИЕ
Электрохимическое окисление сульфитных частиц, а именно сульфит-ионов (${\text{SO}}_{3}^{{2 - }}$), ионов гидросульфита (${\text{HSO}}_{3}^{ - }$) и диоксида серы (${\text{S}}{{{\text{O}}}_{2}}(ж)$), интенсивно изучается в связи с индустриальными возможностями этих процессов [1]. Особый интерес вызывает окисление этих частиц на электродах из благородных металлов, так как эти металлы проявляют высокую каталитическую активность. Исследования на золотых электродах выполнялись в работах [2–6] и рассматривались в обзоре [7]. Результаты исследований показывают, что для этой системы характерна сложная комбинация нескольких процессов, которые протекают одновременно в одной той же области потенциалов. Так, известно, что фарадеевский ток окисления указанных соединений S(IV) наблюдается в анодной области потенциалов, где на поверхности электрода протекают фарадеевские процессы образования оксидных пленок золота. Оксидные слои на поверхности металла активны в гетерогенных электрокаталитических процессах и разными путями могут оказывать влияние на скорость окисления диоксида серы. Оксиды золота могут химически окислять сульфитные частицы [2]. Оксидная пленка может резко уменьшать скорость окисления диоксида серы путем блокировки активных мест на поверхности электрода [3, 4, 8] или путем вытеснения адсорбированных сульфитных частиц с поверхности электрода [9]. Поэтому можно ожидать, что изменение условий формирования оксидов золота будет приводить к изменениям в процессе окисления сульфитных частиц.
Одним из факторов, которые влияют как на процессы образования и восстановления оксидов золота [10, 11], так и на процессы окисления сульфитных частиц, является кислотность раствора. В растворах с разными значениями pH не одинаковы как область потенциалов образования оксидов, так и их состав. Плотный (негидратированный) $\alpha $-оксид, а также рыхлый (гидратированный) $\beta $-оксид могут формироваться на поверхности золота при протекании анодного тока и могут быть восстановлены катодным током [12]. Процессам образования и восстановления оксидов на золотом электроде посвящено большое число работ. Чаще всего эти исследования выполнялись в сильнокислой среде серной и хлорной кислот. Отдельные исследования были выполнены также в нейтральных [13] и щелочных растворах [14]. В данной работе будут представлены результаты систематического изучения этих процессов в буферных растворах Бриттона–Робинсона и небуферных водных растворах сульфата натрия. Отметим, что систематические исследования процессов образования и восстановления оксидов золота в широком диапазоне pH для растворов с буферными свойствами ранее не проводились. Сравнивая результаты, полученные в буферных и небуферных растворах, можно оценить степень влияния pH в приэлектродном слое раствора на процессы образования и восстановления оксидов золота.
Кислотность раствора может оказывать влияние не только на процессы образования оксидов на поверхности золотого электрода, но также и на кинетику процесса окисления сульфитных частиц и на количественный и качественный состав электроактивных частиц в объеме раствора. Действительно, при растворении в воде газообразного диоксида серы ${\text{S}}{{{\text{O}}}_{2}}(г)$ или соли из щелочного металла и аниона ${\text{SO}}_{3}^{{2 - }}$ равновесный состав частиц в объеме раствора определяется константами реакций диссоциации этих соединений в водной среде. Расчеты [15] показывают, что в растворах сульфита натрия с величиной pH ≥ 9.0 доминируют частицы ${\text{SO}}_{3}^{{2 - }}.$ В области pH ≤ 1.0 доминирует водный раствор сернистой кислоты ${{{\text{H}}}_{{\text{2}}}}{\text{SO}}_{{\text{3}}}^{{}}$ (${\text{S}}{{{\text{O}}}_{2}}(ж)$), который быстро распадается до сернистого газа. В области 1.0 < pH < 9 присутствуют также анионы ${\text{HSO}}_{{\text{3}}}^{ - }.$ В данной работе будут представлены результаты измерения скорости окисления сульфитных частиц в зависимости от кислотности раствора в области от pH 2 до pH 14. Так как в указанном диапазоне pH в объеме раствора присутствуют разные электроактивные частицы, то такие измерения дают возможность оценить, как изменяется скорость окисления сульфитных частиц в растворах разного состава. Ранее систематические исследования скорости изучаемого процесса в широкой области pH не проводились.
Основная цель данной работы в том, чтобы выяснить особенности окисления сульфитных частиц на золотом электроде при изменении pH раствора. Сопутствующей и необходимой частью такого исследования является детальное изучение влияния pH на процесс формирования и восстановления оксидов на поверхности золота. Это следует из существования тесной связи между этими двумя процессами. Предпосылками такого исследования являются результаты нашей предыдущей работы [16], в которой наблюдался факт отсутствия явной зависимости скорости окисления сульфит-ионов от pH раствора. Проверка и подтверждение этого феномена, как и попытка дать ему приемлемое объяснение, явились стимулом к выполнению этой работы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для приготовления растворов использовались дважды дистиллированная вода, соли Na2SO3, Na2SO4 (“ч. д. а.”), а также щелочь NaOH и кислота H2SO4 (“ос. ч.”). Водный раствор Na2SO4 и универсальная буферная смесь Бриттона–Робинсона с добавками требуемых количеств NaOH использовались в качестве фонового электролита. Величина pH растворов сульфата натрия обеспечивалась добавлением соответствующего количества щелочи или кислоты и измерялась с помощью стеклянного электрода. Растворы, содержащие сульфит натрия, приготавливались непосредственно перед измерением. Зависимости плотности тока (j) от потенциала (E) в изучаемой системе измерялись на золотом электроде с помощью потенциостата IPC-compact (ИФХЭ РАН). Измерения проводились в 3-электродной ячейке с вспомогательным Pt-электродом и Ag/AgCl-электродом сравнения. В тексте статьи потенциалы приведены в шкале стандартного водородного электрода (НВЭ). Для расчетов величин плотности тока использовалась площадь геометрической поверхности электрода.
Вольтамперограммы в растворах фонового электролита и в растворах с добавками сульфита натрия были измерены на рабочем электроде, который представлял собой торец золотой проволоки диаметром 0.5 мм. Боковая поверхность проволоки была изолирована композитом на основе эпоксидной смолы. Подготовка рабочего электрода перед измерением заключалась в механическом обновлении его поверхности путем среза тонкого (≈5 мкм) слоя металла резцом из твердого сплава. Схема установки для обновления поверхности электрода, которая использовалась в настоящей работе, была представлена в [17]. Срез осуществляется непосредственно в исследуемом растворе. Такой метод подготовки поверхности электрода обеспечивает быстрое получение “свежей” и воспроизводимой поверхности металла. Поэтому на результаты измерений предыдущая история “жизни” поверхности электрода оказывает незначительное влияние.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Окисление золота в 0.01 М растворах H2SO4 и NaOH
На рис. 1 (кривая 1) показана вольтамперограмма, которая была измерена в 0.01 М (pH 2) растворе H2SO4. Перед измерением поверхность золотого электрода обновлялась путем среза, после чего потенциал электрода изменялся со скоростью 50 мВ/с в следующей последовательности: от E = 0.5 до 1.8 В, затем в отрицательном направлении до –0.3 В и назад до 0.5 В. Вольтамперограмма имеет вид, который является типичным для процессов формирования и восстановления оксидов золота [18–23]. Классический механизм этих процессов сформулирован в работах [12, 24–26]. Согласно этим представлениям, при изменении потенциала электрода от стационарного в положительную сторону вначале имеет место “предокисление” поверхности золота. На рис. 1 (кривая 1) это область потенциалов между примерно 0.5 и 1.26 В. При этих потенциалах на поверхности золотого электрода формируется субмонослой хемосорбированного кислорода (${\text{Au--O}}$) [27]. Преобразование субмонослоя этих частиц в тонкий и компактный $\alpha $-оксид начинается при потенциале, близком к 1.26 В, и сопровождается резким увеличением фарадеевского тока. Механизм преобразований связан с реакцией обмена местами атомов кислорода и золота в адсорбционном слое (${\text{Au--O}} \leftrightarrow {\text{O--Au}}$) и фарадеевскими реакциями образования AuO и ${\text{A}}{{{\text{u}}}_{{\text{2}}}}{{{\text{O}}}_{3}}.$ Брутто-реакции этого процесса обычно записываются в виде:
(1)
${\text{Au}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{AuO}} + 2{{{\text{H}}}^{ + }} + 2{\text{e}},$(2)
${\text{2Au}} + 3{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to {\text{A}}{{{\text{u}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}} + 6{{{\text{H}}}^{ + }} + 6{\text{e}}.$Рис. 1.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.01 М H2SO4 (кривая 1) и 0.01 М NaOH (кривая 2).

Плато тока положительнее 1.26 В на рис. 1 (кривая 1), по-видимому, является результатом кинетических затруднений указанных реакций. Плато прерывается экспоненциальным увеличением тока примерно при потенциале 1.70 В. Вблизи этого потенциала заканчивается образование, по меньшей мере, монослойного покрытия поверхности золотого электрода оксидной пленкой, в составе которой преобладают негидратированные $\alpha $-оксиды типа ${\text{AuO}}$ [29], но содержится и некоторое количество ${\text{A}}{{{\text{u}}}_{{\text{2}}}}{{{\text{O}}}_{3}}.$ После образования компактной пленки из α-оксидов на золотом аноде начинается выделение газообразного кислорода O2(г). Одновременно с выделением кислорода на золоте имеет место образование осадка $\beta $-оксидов, которые имеют состав типа ${\text{A}}{{{\text{u}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{3}}}} \cdot n{{{\text{H}}}_{{\text{2}}}}{\text{O}}{\text{.}}$ Рыхлая пленка этих оксидов растет поверх подслоя из $\alpha $-оксидов при потенциалах близких к 2.0 В и положительнее этого потенциала. Образованные в анодном процессе $\alpha $-оксиды золота восстанавливаются при развертке потенциала в отрицательную сторону (пик катодного тока вблизи 1.0 В). Катодный пик тока характеризует количество оксидов на поверхности электрода. В указанных на рис. 1 (кривая 1) условиях их количество Q составляет 880 мкКл (примерно 2 монослоя) на единице геометрической площади поверхности электрода и, по-видимому, относится к $\alpha $-оксидам золота. При потенциалах отрицательнее 0.5 В на j,E-кривых (рис. 1) наблюдается характерное плато катодного тока восстановления молекулярного кислорода. Процессы восстановления кислорода не оказывают влияние на плотность тока восстановления $\alpha $-оксидов золота и не обсуждаются в данной работе.
Описанное выше поведение и параметры вольтамперограммы, которая была измерена в 0.01 М растворе серной кислоты на обновляемом золотом электроде (рис. 1, кривая 1), количественно согласуются с известными результатами предыдущих исследований в этой системе [12, 18], если учесть различия в концентрациях исследуемых растворов и различия в шкалах стандартного (НВЭ) и обратимого (ОВЭ) водородного электродов.
На рис. 1 (кривая 2) также показана вольтамперограмма, которая была измерена в 0.01 М (pH 12) растворе NaOH. Перед измерением поверхность золотого электрода обновлялась путем среза, после чего потенциал электрода изменялся со скоростью 50 мВ/с в следующей последовательности: от E = 0.2 до –0.9 В, затем в положительном направлении до 1.3 В и назад до 0.2 В. Как видно из этого рисунка, вольтамперограмма, измеренная в щелочи, смещена в сторону отрицательных потенциалов по отношению к вольтамперограмме, которая была измерена в кислоте. В шкале обратимого водородного электрода величина смещения составляет примерно –0.1 В, как и в работе [12]. “Предокисление” поверхности золота в щелочи имеет место в области потенциалов примерно от 0.2 до 0.5 В. Однако в щелочи источником появления кислорода на поверхности золота, скорее всего, являются ионы ${\text{O}}{{{\text{H}}}^{ - }},$ а не молекулы воды [28]. Хемосорбция гидроксид-ионов приводит к образованию на поверхности золота дипольных частиц, подобных тем, которые формируются в кислых растворах. В то же время степень переноса заряда и степень заполнения поверхности этими частицами в кислых и щелочных растворах могут различаться. Плато тока на j–E-кривой, где происходит формирование оксидной пленки в щелочном растворе, наблюдается в области от 0.6 до 1.0 В. Пик катодного тока при восстановлении оксидной пленки в щелочи расположен при 0.37 В (НВЭ) или 1.08 В (ОВЭ). Количество оксидов, которые были образованы в щелочи, составляют 860 мкКл/см2 (рис. 1, кривая 2). Эти параметры примерно совпадают с теми, которые были получены в кислом растворе. Поэтому можно допустить, что в указанных условиях катодный пик тока в щелочи, как и в кислоте, в основном обусловлен восстановлением $\alpha $‑оксидов золота, которые были сформированы в области плато анодного тока. Из данных, которые приведены на рис. 1, видно, что для указанного диапазона потенциалов измеренная в щелочи вольтамперограмма в основном повторяет форму той, которая наблюдается в кислоте. Различия в форме вольтамперограмм в области потенциалов, отрицательнее пиков восстановления оксидов золота, по-видимому, связаны с различиями в кинетике восстановления кислорода в кислой и щелочной среде и в настоящей работе не обсуждаются. Измеренные в растворах кислоты и щелочи вольт-амперные зависимости будут в дальнейшем использоваться для сравнения с результатами измерений в растворах, содержащих сульфит натрия.
Результаты на рис. 1 показывают, что и в кислоте и в щелочи на j,E-кривых наблюдается характерное плато анодной плотности тока, которое отражает процесс формирования $\alpha $-оксидов золота и предшествует выделению кислорода. Эти плато наблюдаются в различных областях потенциалов, но величины плотности тока в них примерно одинаковы. Другими словами скорость образования $\alpha $-оксидов золота при изменении концентрации ионов ${\text{O}}{{{\text{H}}}^{ - }}$ на 10 порядков остается примерно одинаковой. С целью проверки этого результата были проведены измерения в универсальной буферной смеси Бриттона–Робинсона.
Окисление золота в буферных растворах (pH 2–12)
На рис. 2 показана серия j,E-кривых, которые были измерены в области потенциалов формирования $\alpha $-оксидов (плато анодного тока j,E-кривой) в растворах буферной смеси Бриттона–Робинсона (pH 2–12). Приведенные результаты показывают, что измеренные в области плато значения плотности тока (≈100 мкА/см2), как и количество (Q) образованных и восстановленных $\alpha $-оксидов (≈0.90 мКл/см2), не зависят от pH и практически совпадают с результатами на рис. 1. Отсюда следует, что гетерогенная реакция, которая описывает лимитирующую стадию скорости образования $\alpha $-оксидов, имеет нулевой порядок по концентрации ионов гидроксида. Этот вывод, по-видимому, подтверждает представление о том, что замедленной стадией образования оксидов золота является реакция (${\text{Au--O}} \leftrightarrow {\text{O--Au}}$) [24]. Вместе с тем результаты на рис. 2 показывают, что изменение pH раствора влияет на величину потенциала образования $\alpha $-оксидов золота. В указанном интервале изменения pH раствора наблюдается смещение потенциала анодного тока образования $\alpha $-оксидов золота на величину 74 мВ при изменении кислотности раствора в 10 раз (${{\Delta E} \mathord{\left/ {\vphantom {{\Delta E} {{\text{pH}}}}} \right. \kern-0em} {{\text{pH}}}}$ = –74 мВ) (вставка 1 на рис. 2). Вставка 2 на рис. 2 показывает, что величина ${{\Delta E} \mathord{\left/ {\vphantom {{\Delta E} {{\text{pH}}}}} \right. \kern-0em} {{\text{pH}}}}$ для максимума катодного тока, который характеризует процесс восстановления $\alpha $-оксидов золота, в указанном интервале pH составляет –67 мВ. Полученные значения ${{\Delta E} \mathord{\left/ {\vphantom {{\Delta E} {{\text{pH}}}}} \right. \kern-0em} {{\text{pH}}}}$ мало отличаются от данных, полученных в работах [10, 11, 29]. В целом, результаты измерений в буферных растворах (рис. 2) подтверждают, что скорость образования и количество образованных на поверхности электрода $\alpha $-оксидов золота остаются примерно одинаковыми при изменении концентрации ионов ${\text{O}}{{{\text{H}}}^{ - }}$ в объеме раствора в диапазоне от 10‒2 до 10–12 М. При этом скорость образования оксидов золота в буферных и небуферных растворах примерно одинакова.
Рис. 2.
Вольтамперограммы, измеренные на золотом электроде в буферных растворах Бриттона–Робинсона. Величина pH составляет: 3.06 (кривая 1), 5.10 (2), 6.54 (3), 8.82 (4), 10.5 (5), 12.0 (6). Врезка 1 – $\Delta E{\text{/pH}}$ = –74 мВ. Врезка 2 – $\Delta E{\text{/pH}}$ = –67 мВ.

Однако процесс восстановления оксидов золота в небуферных растворах имеет свои особенности. На рис. 3 приведены вольтаперограммы, которые были измерены в небуферных растворах сульфата натрия. Обращает внимание, что в отличие от поведения пиков восстановления в буферных растворах, в которых наблюдается линейное смещение потенциала пика с наклоном –67 мВ на единицу pH в интервале pH от 2 до 12, в небуферных растворах указанное значение ${{\Delta E} \mathord{\left/ {\vphantom {{\Delta E} {{\text{pH}}}}} \right. \kern-0em} {{\text{pH}}}}$ наблюдается только при значенияx pH < 4 или pH > 10 (рис. 3). В интервале pH от 4 до 10 величины смещения ${{\Delta E} \mathord{\left/ {\vphantom {{\Delta E} {{\text{pH}}}}} \right. \kern-0em} {{\text{pH}}}}$ в небуферных растворах составляют примерно 10–20 мВ (вставки 1 и 2 в рис. 3). При этом, в небуферных растворах внутри указанного интервала pH могут наблюдаться два раздельных катодных пика восстановления $\alpha $-оксидов (j,E-кривая для pH 3.9). Раздвоение пика восстановления оксидов золота наблюдалось ранее в работе [30] и объяснялось сопутствующим восстановлением некоторого количества β-оксида золота. Результаты на рис. 3 показывают, что раздвоенный пик, который наблюдается в растворах с pH 3.9, скорее всего фиксирует восстановление только $\alpha $-оксида золота. Этот вывод следует из того факта, что количество (Q) восстановленного оксида золота во всем указанном интервале pH примерно одинаково, несмотря на различие формы катодных кривых, и близко к величинам Q в буферных растворах и в растворах кислоты или щелочи (рис. 1). Поэтому, различие j,E-кривых восстановления $\alpha $-оксида золота, которое наблюдается в буферных (рис. 2) и небуферных (рис. 3) растворах, по-видимому, связано с неконтролируемым изменением кислотности приэлектродного слоя раствора. Отмеченные выше особенности восстановления оксидов золота при изменении pH раствора ранее не наблюдались и требуют более детального изучения. Тем не менее, приведенные результаты показывают, что буферная смесь Бриттона–Робинсона является пригодной для использования в качестве фонового раствора. Напротив, применение небуферных растворов усложняет интерпретацию результатов измерения скорости процессов образования/восстановления оксидов золота в интервале значений pH от 4 до 10.

Окисление золота в щелочном (pH 12) и кислом (pH 2) растворах 0.1 М Na2SO4
На рис. 4 показаны циклические вольтамперограммы, которые были измерены в кислых (pH 2) и щелочных (pH 12) 0.1 M растворах Na2SO4 в интервале потенциалов от 0.2 до 2.0 В. Видно, что в щелочном растворе (кривая 1) при сканировании потенциала от 0.2 до 2.0 В величины тока для E > > 1.0 В намного больше, чем ток, который наблюдается в кислых растворах. При последующем инверсном сканировании потенциала (от 2.0 до 0.2 В) вблизи потенциала E = 0.26 В наблюдается максимум катодного тока. Его величина и соответствующее ему количество электричества, затраченное на восстановление оксидной пленки, принципиально не отличаются от тех, которые наблюдаются в кислом растворе (рис. 4, кривая 2). Нет также заметных различий между величинами анодного тока, который расходуется на образование оксидов золота в кислых и щелочных растворах (вставка в рис. 4). Поэтому есть основания считать, что и в щелочном, и в кислом растворах образуется и восстанавливается примерно одинаковое количество $\alpha $-оксидов золота, несмотря на значительную разницу в величинах анодного тока при E > 1.0 В. Отсюда следует, что в щелочном растворе только очень малая часть наблюдаемого при E > 1.0 В фарадеевского тока расходуется на процесс образования оксидов золота. Так как золото в растворах сульфата натрия не растворяется, то наблюдаемая при E > 1.0 В плотность тока практически полностью обусловлена выделением кислорода на поверхности, покрытой оксидами золота. При этом кинетика анодного выделения кислорода на золотом электроде в щелочном растворе отличается от кинетики этого процесса в кислоте. Так, на j,E-кривой в щелочном растворе наблюдаются две волны предельного тока в области 1.2–1.4 В и в области 1.6–1.9 В, которых нет в кислом растворе. Отметим, что скорость (величина тока в области E > 1.0 В) выделения кислорода увеличивается линейно при увеличении концентрации ионов гидроксида [OH–]. Это обстоятельство указывает, что в реакции выделения кислорода, которая на золотом электроде протекает в щелочных растворах при потенциалах положительнее 1.0 В, участвуют не только молекулы воды, но и гидроксид-ионы. Более подробное обсуждение вопросов об особенностях кинетики и механизма выделения кислорода на золотом электроде не представляется целесообразным, так как они выходят за рамки темы данной работы.

Окисление сульфит-ионов в щелочных (pH ≈ 12) растворах 0.1 M Na2SO4
На рис. 5 (кривая 1) показана вольтамперограмма, которая была измерена в щелочном (pH 12) растворе 0.1 M Na2SO4 c добавлением малого количества Na2SO3 (при этих значениях pH в растворе присутствуют исключительно ионы ${\text{SO}}_{3}^{{2 - }}$). Потенциал электрода изменялся со скоростью 50 мВ/с от 0.0 до 2.0 В, затем в отрицательном направлении до –0.9 В и назад до 0.0 В. На этом же рисунке (кривая 2) для сравнения показана j,E-кривая в растворе без добавки Na2SO3. Из сравнения этих кривых следует, что окисление сульфит-ионов в указанном растворе начинается при потенциале примерно 0.5 В, имеет максимум анодного тока вблизи E = 0.6 В и прослеживается вплоть до потенциала примерно 1.4 В. Этот факт показывает, что окисление сульфит-ионов имеет место в области потенциалов, где в растворах щелочного фона на поверхности золота происходит формирование пленки $\alpha $-оксидов. Окисление сульфит-ионов наблюдается также и в области потенциалов E = 1.0–1.4 В, где в растворах фонового электролита фиксируется плато тока выделения кислорода (рис. 4). При более положительных потенциалах (E > 1.4 В) j,E-кривые в растворах фонового электролита и в растворах с добавками сульфита натрия не имеют заметного различия.
Рис. 5.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.1 M Na2SO4 + xМ Na2SO3 (рН 12): x = 4 × 10–3 (кривая 1), 0.0 (2).
Известно, что электрохимическое окисление сульфит-ионов на золотом электроде можно описать следующими уравнениями электродных реакций [31]:
(3)
${\text{SO}}_{3}^{{2 - }} + 2{\text{O}}{{{\text{H}}}^{ - }} \to {\text{SO}}_{4}^{{2 - }} + {{{\text{H}}}_{{\text{2}}}}{\text{O}} + 2{\text{e}},$(4)
$2{\text{SO}}_{3}^{{2 - }} \to {{{\text{S}}}_{{\text{2}}}}{\text{O}}_{6}^{{2 - }} + 2{\text{e}}.$При этом на золотом электроде количество ${\text{SO}}_{3}^{{2 - }},$ превращенное в ${{{\text{S}}}_{{\text{2}}}}{\text{O}}_{6}^{{2 - }},$ составляет примерно 30% [32] или 45% [33], а суммарный выход по току этих реакций близок к 100%. Другими словами электрохимическое окисление сульфит-ионов на золотом электроде приводит к образованию в растворе смеси ионов сульфата и дитионата. Эти продукты реакций окисления сульфит-ионов в изучаемой области потенциалов являются электрохимически неактивными [31, 34]. Поэтому их образование не может изменить величину плотности тока, который протекает в системе при инверсной развертке потенциала.
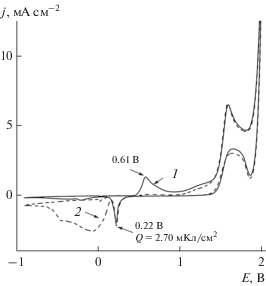
Рисунок 5 показывает, что на инверсной ветви вольтамперограммы величина тока близка к нулю вплоть до потенциала равного примерно 0.22 В, при котором наблюдается максимум катодного тока. Так как ионы сульфата и дитионата неактивны, то можно ожидать, что этот максимум обусловлен процессом восстановления $\alpha $-оксидов золота, которые были образованы во время сканирования потенциала электрода в прямом направлении. Это предположение подтверждается тем фактом, что количество оксида на электроде (2.7 мКл/см2) и значение потенциала катодного максимума практически совпадают с параметрами аналогичного максимума в растворе фонового электролита (рис. 4, 5). Приведенные выше результаты показывают, что в области потенциалов отрицательнее примерно 1.4 В процессы образования/восстановления $\alpha $-оксидов протекают на поверхности золотого электрода одновременно с процессом электрохимического окисления сульфит-ионов. При этом процесс окисления ${\text{SO}}_{3}^{{2 - }}$ не оказывает влияние на количество образованных $\alpha $-оксидов золота. Характерно, что величина тока на инверсном участке j,E-кривой близка к нулю до тех пор, пока потенциал электрода не достигнет потенциала восстановления оксидов. Отсюда следует, что образование компактной оксидной пленки на поверхности золота препятствует окислению ${\text{SO}}_{3}^{{2 - }}.$ Тот факт, что оксидная пленка блокирует доступную для окисления сульфит-ионов поверхность золотого электрода, был показан в работах [4, 8] и обсуждался в [1].
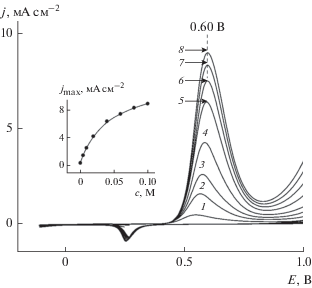
На рис. 6 приведены результаты более подробного изучения зависимости скорости окисления ${\text{SO}}_{3}^{{2 - }}$ от его концентрации в щелочном (pH 12) растворе 0.1 M Na2SO4. При увеличении концентрации (c) сульфита натрия до 0.01 M на измеряемой j,E-зависимости наблюдается единственный максимум анодного тока (jпик) при потенциале примерно 0.6 В. Плотность тока в этом максимуме линейно зависит от концентрации ${\text{SO}}_{3}^{{2 - }}$ в растворе (вставка на рис. 6). Линейный характер зависимости j от c, согласно известному уравнению нестационарной диффузии jпик = = 272n3/2D1/2v1/2c [35], может указывать на диффузионные ограничения скорости окисления сульфит-ионов в этих растворах. Величина коэффициента диффузии (D), рассчитанная по указанному уравнению из результатов на рис. 6, равна примерно 0.41 × 10–5 см2/с. Это значение практически совпадает с величиной коэффициента диффузии D = = 0.43 × 10–5 см2/с, которая была найдена ранее в работе [16] при изменении скорости развертки потенциала. При потенциалах положительнее этого максимума нестационарная диффузия разряжающихся ионов ${\text{SO}}_{3}^{{2 - }}$ приводит к снижению плотности тока, однако при достижении потенциала примерно 1.0 В плотность тока начинает увеличиваться. Увеличение анодной плотности тока при потенциалах положительнее 1.0 В, обусловлено тем, что наряду с окислением сульфит-ионов в щелочном растворе начинается выделение кислорода (см. рис. 4).
Рис. 6.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.1 M Na2SO4 + x М Na2SO3 (рН 12): x = 2 × 10–4 (кривая 1), 6 × 10–4 (2), 1 × 10–3 (3), 2 × 10–3 (4), 4 × 10–3 (5), 6 × 10–3 (6), 8 × 10–3 (7), 1 × × 10–2 (8). На врезке: зависимость плотности тока в максимуме от концентрации сульфит-ионов.

Приведенные на рис. 6 результаты показывают, что в указанных растворах зависимости плотности тока от потенциала при инверсном сканировании потенциала одинаковы. Плотность тока на инверсной ветви j,E-кривой близка к нулю вплоть до потенциала, при котором наблюдается катодный максимум скорости восстановления оксидной пленки. Величина и положение на оси потенциалов этого максимума не зависят от концентрации ${\text{SO}}_{3}^{{2 - }}$ в растворе и совпадают с результатами измерения скорости восстановления оксидов золота в чистой щелочи (рис. 1). Отсюда следует, что количество восстановленных, а следовательно, и образованных на поверхности золотого электрода $\alpha $-оксидов не зависит от концентрации сульфит-ионов в растворе. Рисунок 6 показывает также, что на инверсном участке j,E-кривой поверхность электрода остается заблокированной оксидами золота при различных концентрациях сульфита натрия в растворе, вплоть до их восстановления при потенциале 0.36 B.
Приведенные выше в этом разделе результаты характерны для растворов с относительно малой (≤0.01 М) концентрацией ${\text{SO}}_{3}^{{2 - }},$ однако при более высоких концентрациях сульфита натрия в растворах с pH 12 наблюдается аномальный вид зависимости плотности тока от потенциала электрода. На рис. 7 приведены j,E-зависимости, которые были измерены в щелочных (pH 12.1) растворах с концентрациями Na2SO3 в диапазоне от 0.01 до 0.1 M. Видно, что при концентрациях ${\text{SO}}_{3}^{{2 - }}$ выше 0.02 M на вольт-амперных зависимостях наблюдаются резкие скачки тока. Они наблюдаются в узкой области потенциалов и представляют собой один или несколько затухающих по амплитуде импульсов фарадеевского тока (см. вставки на рис. 7). Аналогичные скачки плотности тока имеют место также при измерениях в буферных растворах (pH 11.8) с концентрациями ${\text{SO}}_{3}^{{2 - }}$ выше 0.02 M (рис. 8). Поэтому причина появления этих скачков тока, по-видимому, не связана с резкими изменениями величин pH приэлектродного слоя в небуферных растворах.
Рис. 7.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.1 M Na2SO4 + x М Na2SO3 (рН 12.1): x = 1 × 10–2 (кривая 1), 2 × 10–2 (2), 3 × 10–2 (3), 4 × 10–2 (4), 5 × 10–2 (5), 6 × 10–2 (6), 8 × 10–2 (7). На врезке 1: x = 0.05, скорость развертки потенциала 2 мВ/с, на врезке 2: x = 0.05, скорость развертки потенциала 50 мВ/с.

Рис. 8.
Вольтамперограммы, измеренные на золотом электроде в буферных растворах Бриттона–Робинсона) + x M Na2SO3 (pH 11.8): x = 2 × 10–2 (кривая 1), 4 × 10–2 (2), 6 × 10–2 (3), 8 × 10–2 (4), 1 × 10–1 (5). На врезке: x = 6 × 10–2, скорость развертки потенциала 2 мВ/с (кривая 1), 50 мВ/с (2).

Представленный на рис. 7 и 8 аномальный характер изменения величины плотности тока при изменении потенциала электрода отличается от наблюдавшихся ранее на Pt-электроде гармонических осцилляций в кислых растворах диоксида серы [7, 36]. Такие гармонические осцилляции можно объяснить участием дитионат- [36] или иодид-ионов [37] в промежуточных стадиях реакции окисления диоксида серы. Однако, наблюдаемые на рис. 7 и 8 резкие изменения величины фарадеевского тока более характерны для процессов пассивации/депассивации и, вероятно, связаны с изменением свойств поверхности золотого электрода.
Следует обратить внимание, что аномально резкое изменение скорости окисления сульфит-ионов наблюдается в узкой области потенциалов между 0.62 и 0.82 В (рис. 7). Первое из этих значений характеризует положение максимума плотности тока окисления ${\text{SO}}_{3}^{{2 - }}$ в щелочных (pH 12) растворах с концентрациями сульфита натрия 0.01 M и меньше (см. рис. 6). Второе значение (вблизи 0.8 В) характерно для максимума тока в растворах с концентрациями ${\text{SO}}_{3}^{{2 - }}$ выше 0.02 М. Как будет видно из материала следующего раздела, потенциал 0.8 В характеризует положение единственного максимума плотности тока окисления сульфитных частиц в более кислых растворах. Можно поэтому предположить, что скачкообразные изменения плотности тока в указанном интервале потенциалов связаны с резким изменением свойств поверхности золотого электрода в растворах с относительно высоким содержанием сульфита натрия при изменении кислотности раствора. Результаты более подробного изучения отмеченного выше аномального поведения j,E-кривых будут представлены в разделе 6.
Рисунок 7 показывает, что на j,E-зависимостях, которые измерены в щелочных (pH ≈ 12) растворах ${\text{SO}}_{3}^{{2 - }}$ с концентрациями больше 0.02 M, наблюдается максимум тока, расположенный вблизи 0.8 В. Этот максимум и последующее плато тока имеют признаки, характерные для электродных процессов, протекающих в режиме нестационарной диффузии электроактивных компонентов раствора. Действительно, при уменьшении скорости развертки потенциала величина плотности тока уменьшается, а форма зависимости тока от потенциала принимает вид плато без максимума (вставки на рис. 7 и 8). Кроме того, величина плотности тока в максимуме кривой вблизи 0.8 В линейно зависит от концентрации сульфита натрия. При этом, величина коэффициента диффузии, которая была рассчитана аналогично тому, как это было сделано для данных рис. 6, составляет 4.05 × 10–6 см2/с. Это значение практически совпадает с величиной D которая была найдена в растворах с концентрациями меньше 0.01 M (рис. 6). Можно, поэтому, допустить, что в растворах с величиной pH ≈ 12 как при малой (c < < 0.01 M), так и при высокой концентрации ${\text{SO}}_{3}^{{2 - }}$ (c > 0.02 M) окисление сульфит-ионов в области потенциалов 0.8–1.3 В имеет диффузионные ограничения скорости процесса, а в диффузии участвует один и тот же электроактивный компонент раствора.
Рисунки 7 и 8 показывают также, что плато диффузионного тока, которое наблюдается в растворах с относительно высоким содержанием сульфит-ионов, прерывается резким снижением плотности тока окисления ${\text{SO}}_{3}^{{2 - }}.$ Спад тока начинается при потенциале 1.3 В и заканчивается вблизи потенциала 1.45 В. Есть основания считать, что в этом интервале потенциалов на поверхности электрода происходит окончательное формирование компактной пленки $\alpha $-оксидов золота, которое приводит к практически полной пассивации электрода по отношению к реакции окисления сульфит-ионов [8].
Окисление сульфитных частиц в кислых (pH 2) растворах 0.1 M Na2SO4
На рис. 9 (кривая 1) показана вольтамперограмма, которая была измерена в кислом (pH 2) растворе 0.1 M Na2SO4 c добавлением малого количества Na2SO3 (при этих значениях pH в растворе присутствуют сернистая кислота H2SO3 в виде растворенного SO2(газ) и ионы ${\text{HSO}}_{3}^{ - },$ обозначенные дальше как “сульфитные частицы”). На этом же рисунке (кривая 2) для сравнения показана j,E-кривая в растворе без добавки Na2SO3. Из сравнения этих кривых следует, что окисление сульфитных частиц в указанном растворе начинается при потенциале, равном примерно 0.5 В, и имеет максимум анодного тока вблизи E = 0.6 В. При дальнейшем увеличении анодного потенциала наблюдается плато анодного тока, а положительнее потенциала, равного примерно 1.26 В, анодный ток окисления сульфитных частиц суммируется с током, который расходуется на процесс образования $\alpha $-оксидов золота. Эти результаты показывают, что окисление сульфитных частиц в кислом растворе, как и в щелочной среде (см. рис. 5), протекает в области потенциалов, где в растворах фона происходит “предокисление” и формирование оксидной пленки на поверхности золота.
Рис. 9.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.1 M Na2SO4 + x М Na2SO3 (рН 2): x = 1 × 10–3 (кривая 1), 0.0 (2).

Известно, что продукты окисления сульфитных частиц не восстанавливаются в изучаемой области потенциалов. Поэтому инверсный ток суммируется из величины парциального катодного тока восстановления оксидов золота, которые были образованы на поверхности электрода при сканировании потенциала в прямом направлении, и величины парциального анодного тока окисления сульфитных частиц. Рисунок 9 показывает, что при сканировании потенциала от предельного анодного значения примерно до 1.0 В инверсный ток имеет малую величину. В этой области потенциалов на поверхности электрода присутствует плотная пленка $\alpha $-оксидов золота. Ее восстановление в кислом (pH 2) растворе сульфита натрия наблюдается в виде катодного пика вблизи 1.0 В (кривая 1). Это значение примерно совпадает с катодным пиком восстановления $\alpha $‑оксидов золота, который наблюдается в растворе фонового электролита (кривая 2). Приведенные результаты показывают, что образование оксидов золота на поверхности электрода в кислых растворах происходит одновременно с процессом окисления сульфитных частиц аналогично тому, как это наблюдается в щелочной среде. Кроме того, полученные результаты указывают, что в кислых растворах, как и в щелочной среде, пленка $\alpha $-оксидов золота препятствует протеканию реакции окисления сульфитных частиц вплоть до момента ее восстановления.
После восстановления оксидной пленки инверсным током вблизи E = 1.0 В поверхность золотого электрода становится доступной для процесса окисления сульфитных частиц. Поэтому отрицательнее этого потенциала инверсный ток увеличивается и принимает значение, характерное для скорости анодного окисления сульфитных частиц. При потенциалах отрицательнее 0.5 В процесс окисления сульфитных частиц не протекает, и поэтому инверсный ток уменьшается до значений, близких к нулю. При дальнейшем сканировании потенциала в отрицательном направлении наблюдаются изменения величины тока, которые связаны с фарадеевскими процессами восстановления сульфитных частиц в кислой среде. Эти процессы обсуждались в работах [1, 9], где отмечалось, что они могут оказывать влияние на вид циклических j,E-зависимостей в изучаемой системе за счет адсорбции серосодержащих продуктов восстановления этих частиц. В настоящей работе эти процессы рассматриваться не будут, а их влияние исключено выбором соответствующего алгоритма сканирования потенциала и применением методики обновления поверхности электрода.
Вольтамперограммы, которые были измерены в кислых (pH 2) растворах 0.1 M Na2SO4 с различными концентрациями добавки сульфита натрия, приведены на рис. 10 и 11. Измерения проводились в диапазоне концентраций сульфита натрия от 0.0 до 0.1 M. Потенциал электрода сканировался от 0.2 до 1.8 В и обратно. Увеличение концентрации сульфита натрия в растворе приводит к увеличению плотности тока его окисления в области потенциалов от 0.5 В примерно до 1.3 В. Положительнее этого потенциала наблюдается резкий спад скорости окисления сульфитных частиц в результате пассивации поверхности электрода компактной пленкой $\alpha $-оксидов золота. Вставка на рис. 10 показывает, что величины плотности тока в максимуме j,E-кривой линейно зависят от концентрации сульфита натрия в растворе. Линейный характер зависимости j от c может указывать на диффузионные ограничения скорости окисления сульфитных частиц в этих растворах. Более аргументировано диффузионная кинетика анодного процесса в этой системе была установлена в работах [4, 38]. Коэффициент диффузии, который был рассчитан по данным рис. 10 и 11, имеет величину 0.9 × 10–5 см2/с. Это значение отличается от соответствующего коэффициента в щелочном растворе, который равен 0.43 × 10–5 см2/с (рис. 5). Разница этих значений указывает на различия в диффузионных характеристиках частиц, которые обеспечивают транспорт в щелочном и в кислом растворах сульфита натрия, и может быть связана с изменением состава сульфитных частиц в растворе при переходе от щелочной к кислой среде.
Рис. 10.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.1 M Na2SO4 + x М Na2SO3 (рН 2): x = 1 × 10–3 (кривая 1), 2 × 10–3 (2), 4 × 10–3 (3), 6 × 10–3 (4), 8 × 10–3 (5), 1 × 10–2 (6). На врезке: зависимость плотности тока в максимуме от концентрации сульфита натрия, $\Delta j{\text{/}}\Delta c \approx 0.5.$

Рис. 11.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.1 M Na2SO4 + x М Na2SO3 (рН 2): x = 1 × 10–2 (кривая 1), 2 × 10–2 (2), 4 × 10–2 (3), 6 × 10–2 (4), 1 × 10–1 (5).

Результаты на рис. 10 показывают, что при увеличении концентрации сульфита натрия в растворе изменяется форма инверсного участка вольтамперограмм. Эти изменения наглядно демонстрируют, что катодный пик восстановления оксидов золота, который наблюдается при малых концентрациях сульфитных частиц вблизи 1.0 В (рис. 9, кривая 1) маскируется анодным током окисления этих частиц. При этом в растворах с относительно большой концентрацией сульфита натрия катодный ток восстановления оксидной пленки составляет малую величину от суммарного тока и не фиксируется на инверсном участке измеряемой вольт-амперной кривой (рис. 11). Поэтому в кислом растворе с относительно большими концентрациями сульфита натрия процесс восстановления оксидов золота наблюдается не в виде катодного максимума, а в виде анодного скачка измеряемого инверсного тока (рис. 10 и 11). Характерно, что восстановление оксидной пленки в кислом растворе сульфита натрия протекает с высокой скоростью, что приводит к резкому нестационарному пику инверсного тока. Потенциал этого пика (1.14 В) практически не зависит от концентрации сульфитных частиц в растворе и расположен близко к потенциалу восстановления $\alpha $-оксидов в растворе фона. Это обстоятельство указывает, что в кислых растворах с различной концентрацией сульфита натрия количество образованного на поверхности электрода оксида золота примерно одинаково.
Приведенные выше результаты показывают, что процессы электрохимического окисления сульфитных частиц в кислом (pH 2) и щелочном (pH 12) растворах протекают на золотом электроде во многом одинаково. Однако имеется одно принципиально важное отличие. Оно заключается в том, что при окислении сульфитных частиц в кислом (pH 2) растворе (рис. 11) на j,E-кривых не наблюдаются те аномальные и резкие скачки тока, которые имеют место в щелочном (pH 12) растворе (рис. 7) при относительно высоких концентрациях сульфита натрия. Чтобы выяснить условия и возможные причины указанного отличия j,E-кривых, были проведены более подробные измерения зависимости скорости окисления сульфитных частиц от их концентрации и величины pH раствора.
Окисление сульфитных частиц в зависимости от концентрации и pH раствора
На рис. 12 показана эволюция формы вольтамперограмм, которые были измерены в щелочных растворах 0.05 М сульфата натрия при изменении pH в области от 11 до 14 единиц. Очевидный результат состоит в том, что в указанной области pH могут наблюдаться два различных типа зависимости плотности тока от потенциала электрода. При pH 11 зависимость j от E имеет единственный максимум, расположенный вблизи 0.8 В. Величина тока в этом максимуме составляет примерно 22 мА/см2. Однако в сильнощелочных растворах уже при pH 12.4 на j,E-кривой наблюдается единственный максимум, который расположен при потенциале 0.63 В, а величина плотности тока не превышает 8.5 мА/см2. Значительное изменение этих характеристик может свидетельствовать о смене механизма окисления сульфитных частиц при сравнительно малом изменении pH раствора. В указанных растворах смена механизма окисления сульфитных частиц происходит в области потенциалов, расположенной между обозначенными пиками j,E-кривых и сопровождается резким скачком тока. Аналогичное поведение скорости окисления сульфитных частиц наблюдается и в буферных растворах (рис. 13). Поэтому наблюдаемые аномалии тока не являются случайными и не связаны с неконтролируемым изменением pH приэлектродного слоя раствора.
Рис. 12.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.1 M Na2SO4 + 0.05 М Na2SO3. Величины pH 11 (кривая 1), 12.05 (2), 12.4 (3), 12.8 (4), 13.5 (5), 14 (6).

Рис. 13.
Вольтамперограммы, измеренные на золотом электроде в буферных растворах Бриттона–Робинсона + 0.05 М Na2SO3. Величины pH 10 (кривая 1), 11 (2), 12 (3), 12.5 (4), 13.5 (5).

Из результатов на рис. 12 и 13 следует, что в сильнощелочных (pH > 12.5) растворах сульфита натрия скорость окисления сульфит-ионов зависит от концентрации ионов ${\text{O}}{{{\text{H}}}^{ - }}.$ Прежде всего отметим, что наблюдаемый на j,E-кривой потенциал максимума тока смещается в отрицательную сторону при увеличении концентрации гидроксид-ионов. Сам по себе этот факт указывает, что в реакции электрохимического окисления ${\text{SO}}_{3}^{{2 - }}$ в таких растворах участвуют ионы ${\text{O}}{{{\text{H}}}^{ - }}.$ При этом величина смещения потенциала максимума равна примерно 75–80 мВ на единицу изменения pH при постоянной концентрации ${\text{SO}}_{3}^{{2 - }}$ в растворе. Эти значения $\Delta E{\text{/pH}}$ близки к значениям, которые характерны для термодинамики электрохимической реакции (3). Поэтому можно, по-видимому, считать, что в сильнощелочных растворах окисление сульфит-ионов протекает в соответствии с реакцией (3) и продуктом этой реакции являются ионы ${\text{SO}}_{4}^{{2 - }}.$ Механизм этой реакции, известный как прямой перенос кислорода от адсорбированных на поверхности металла гидроксил- радикалов на сульфит-ионы, рассматривался в [39, 40].
Результаты измерений (рис. 12 и 13) показывают также, что увеличение pH раствора приводит к уменьшению плотности тока. Объяснить этот эффект можно, если предположить, что увеличение концентрации гидроксид-ионов приводит к уменьшению доступной для окисления сульфит-ионов площади поверхности электрода. Иначе говоря допустить, что в сильнощелочных (pH > 12.5) растворах сульфита натрия имеет место частичная блокировка поверхности золотого электрода. Причина частичной блокировки, по-видимому, связана с окислением поверхности золота, так как приведенные выше значения $\Delta E{\text{/pH}}$ для величин максимума тока окисления ${\text{SO}}_{3}^{{2 - }}$ в сильнощелочных растворах практически совпадают с величинами, которые наблюдаются при образовании оксидов золота (рис. 2). Поэтому нельзя исключить, что в сильнощелочных растворах окисление сульфит-ионов происходит на золотом электроде, поверхность которого частично блокирована оксидами золота. Возможность блокировки поверхности электрода оксидами золота рассматривалась в работе [8].
Результаты, которые были получены в сильнощелочных растворах с постоянной величиной pH и разными значениями концентрации сульфита натрия, показаны на рис. 14 (сульфатный фон) и рис. 15 (буферный раствор Бриттона–Робинсона). Увеличение концентрации сульфита натрия в растворах с pH > 12.5 приводит к увеличению плотности тока окисления ${\text{SO}}_{3}^{{2 - }}.$ Однако зависимость плотности тока от концентрации сульфита натрия в указанной области pH не является линейной (см. вставки на рис. 14 и 15). При увеличении концентрации сульфита натрия скорость процесса стремится к пределу. Этот факт указывает, что в растворах с pH > 12.5 скорость окисления сульфит-ионов обусловлена не только диффузионным переносом реагента в зону реакции, но и, по-видимому, скоростью гетерогенных химических реакций, которые протекают на поверхности золотого электрода. Возможность протекания гетерогенной химической реакции между ${\text{SO}}_{3}^{{2 - }}$ и гидроксил-радикалами на поверхности золотого электрода рассматривалась в [8, 39, 40] в качестве одной из стадий в механизме реакции (3).
Рис. 14.
Вольтамперограммы, измеренные на золотом электроде в растворах 0.1 M Na2SO4 + х М Na2SO3 (рН 14): x = 5 × 10–3 (кривая 1), 1 × 10–2 (2), 2 × 10–2 (3), 3 × 10–2 (4), 5 × 10–2 (5), 7 × 10–2 (6), 8 × 10–2 (7), 9 × × 10–2 (8), 1 × 10–1 (9). На врезке: зависимость плотности тока в максимуме от концентрации сульфита натрия.

Рис. 15.
Вольтамперограммы, измеренные на золотом электроде в буферных растворах Бриттона–Робинсона + х М Na2SO3 (рН 13): x = 1 × 10–3 (кривая 1), 5 × 10–3 (2), 1 × 10–2 (3), 2 × 10–2 (4), 4 × 10–2 (5), 6 × × 10–2 (6), 8 × 10–2 (7), 1 × 10–1 (8). На врезке: зависимость плотности тока в максимуме от концентрации сульфита натрия.
Иное поведение j,E-кривых наблюдается в более кислых растворах сульфита натрия. Детальные измерения, которые были проведены в буферных растворах сульфита натрия в диапазоне pH от 2.0 до 12.5 (рис. 16) показывают, что в области pH от 2.0 до примерно pH 11 ни величина плотности тока, ни потенциал максимума j,E-зависимости практически не изменяются при изменении концентрации ионов гидроксида. Отметим, что в указанном диапазоне pH в растворе могут присутствовать различные соединения четырехвалентной серы (${{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{{\text{3}}}},$ ${\text{HSO}}_{3}^{ - },$ ${\text{SO}}_{3}^{{2 - }}$). При изменении pH раствора общая концентрация частиц S(IV) остается постоянной, а парциальный состав раствора претерпевает изменения. Тем не менее, согласно результатам на рис. 16, изменения в составе раствора не приводят к изменению скорости электрохимического процесса. Этот результат может означать, что непосредственный перенос заряда через границу раздела осуществляется частицами одного сорта, несмотря на их различие в объеме раствора. Однако это предположение требует более детального анализа и дополнительных исследований.
Рис. 16.
Вольт-амперные зависимости, измеренные на золотом электроде в буферных растворах Бриттона–Робинсона + 0.05 М Na2SO3. Величина pH: 2 (кривая 1), 4 (2), 6 (3), 9 (4), 10 (5), 12.5 (6).

В то же время известно, что в растворах с pH > 9 присутствуют исключительно ионы ${\text{SO}}_{3}^{{2 - }}$ и, следовательно, по результатам рис. 16 скорость окисления этих ионов одинакова в диапазоне pH от 9 до 11. Это обстоятельство указывает, что электрохимическая реакция окисления сульфит-ионов протекает без участия протонов или ионов ${\text{O}}{{{\text{H}}}^{ - }}.$ Поэтому можно предположить, что окисление сульфит-ионов в растворах с величинами pH < 11 протекает в соответствии с реакцией (4) и продуктом этой реакции являются дитионат-ионы. Реакция образования дитионат-ионов обычно рассматривается как реакция димеризации сульфит-радикалов [34, 40]. Скорость этой реакции ограничена диффузионным переносом реагирующих частиц к поверхности электрода [4, 31, 38]. Из результатов на рис. 16 следует также, что в указанном диапазоне pH в отличие от сильнощелочных растворов отсутствует частичная блокировка поверхности электрода оксидами золота.
Приведенные выше результаты показывают, что изменение кислотности раствора может привести к изменению механизма процесса электрохимического окисления сульфита натрия. В растворах с pH > 12.5 процесс электрохимического окисления сульфит-ионов на золоте вероятно протекает по реакции (3) с образованием ионов ${\text{SO}}_{4}^{{2 - }},$ в отличие от процесса окисления сульфит-ионов в растворах с pH < 11, который протекает по реакции (4) с образованием ионов ${{{\text{S}}}_{{\text{2}}}}{\text{O}}_{{\text{6}}}^{{{\text{2}} - }}{\text{.}}$ Высокая степень покрытия поверхности электрода хемосорбированными гидроксид-ионами в сильнощелочных растворах стимулирует выбор окисления сульфитных частиц путем реакции (3), тогда как их недостаток, по-видимому, объясняет выбор реакции (4) в растворах с pH < 11. Область pH растворов между pH 11 и pH 12.5 – это область, где осуществляется трансформация способа окисления сульфитных частиц между реакциями (3) и (4). Эта трансформация сопровождается аномальными скачками фарадеевского тока.
Наблюдаемые в изучаемой системе аномально резкие изменения величин измеряемого тока, по-видимому, можно объяснить специфическим характером динамики формирования оксидов на поверхности золотого электрода. Эта версия рассматривалась в работе [8]. В этой работе предполагалось, что продукты реакции обмена ${\text{Au--O}} \leftrightarrow {\text{O--Au}}$ (${\text{O--Au}}$) являются двумерными зародышами оксида золота, которые способны быстро (автокаталитически) расти и образовывать пассивные островки на поверхности электрода. Присутствие сульфит-ионов в растворе уменьшает вероятность образования пассивных островков, так как они вступают в химическую реакцию: $({\text{Au--O}}) + {\text{SO}}_{3}^{{2 - }} \to {\text{SO}}_{4}^{{2 - }} + {\text{Au}}$ и уменьшают количество частиц (${\text{O--Au}}$) на поверхности электрода. Динамическое равновесие этих разнонаправленных процессов, которое зависит от величины pH, концентрации сульфит-ионов и потенциала электрода, определяет степень блокировки поверхности. При этом скорость, с которой блокируется поверхность электрода, контролируется скоростью образования зародышей и скоростью роста площади электрода, которая занята пассивными островками оксида золота. С помощью этого механизма авторы [8] объяснили резкое уменьшение величины измеряемого тока, которое происходит вблизи 1.3 В при образовании компактной пленки оксида золота. С точки зрения указанного механизма, противоположный процесс резкого разрушения оксидной пленки может быть ответственным за всплески тока на инверсном участке измеряемой j,E‑кривой, которые наблюдаются в кислых растворах сульфита натрия (рис. 11). Вероятно также, что аномально резкие всплески тока окисления сульфит-ионов, которые наблюдаются в сильнощелочных растворах (рис. 7, 12), связаны с быстрым разрушением пассивных участков золотого электрода при достаточно высокой (критической) концентрации сульфита натрия. Однако следует отметить, что для выяснения более полной или альтернативной версии специфического механизма электрохимического окисления сульфитных частиц на золотом электроде необходимы дальнейшие исследования.
ЗАКЛЮЧЕНИЕ
В настоящей работе, благодаря применению метода обновления поверхности путем среза тонкого слоя материала электрода непосредственно в растворе, был получен большой объем воспроизводимых и новых экспериментальных результатов. Из их числа отметим несколько, интерпретация которых требует, с нашей точки зрения, более полного изучения. Прежде всего, это относится к тому факту, что величина тока для плато j,E-кривой в сульфатных и буферных растворах не зависит от кислотности раствора (рис. 1–3). Наиболее простое объяснение, которое, вслед за [24], приводится в данной работе, состоит в том, что скорость формирования α-оксидов лимитируется скоростью реакции обмена местами между атомами кислорода и золота в адсорбционном слое: ${\text{Au--O}} \leftrightarrow {\text{O--Au}}.$ Однако детальное понимание механизма этой реакции, по нашему мнению, все еще не достигнуто. Проведенные эксперименты дают основание предположить, что в сильнощелочных (pH > 12.5) растворах электрохимическое окисление сульфит-ионов протекает с образованием сульфат-ионов, тогда как в более кислых приводит к образованию дитионат-ионов. Этот новый результат может быть подтвержден независимыми методами. Экспериментально наблюдается, что изменение указанных выше механизмов реакции окисления сульфитных частиц происходит в узком диапазоне изменения величины pH раствора (между pH 11 и pH 12.5), а также при достижении определенной критической концентрации сульфита натрия в растворе и сопровождается аномально резким изменением величин фарадеевского тока. Эти результаты можно объяснить, если допустить, что в сильнощелочных растворах сульфита натрия оксиды золота частично блокируют поверхность электрода. Причем импульсный характер поведения плотности тока в этой области pH может быть связан с быстрым (автокаталитическим) увеличением или уменьшением площади пассивных участков поверхности электрода, которые при критической степени заполнения мгновенно блокируют или деблокируют поверхность электрода. Такая интерпретация указанного эффекта, по-видимому, не является единственно возможной, так как оставляет неиспользованными известные сведения о склонности сульфит-ионов хемосорбироваться на золоте. Интересный и новый экспериментальный факт, который наблюдался в настоящем исследовании, – это отсутствие зависимости скорости окисления сульфитных частиц от парциального состава электролита (${\text{SO}}_{3}^{{2 - }}$ при pH 10 или ${{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{3}}$ и ${\text{HSO}}_{{\text{3}}}^{ - }$ при pH 2). Более детальное изучение и обсуждение этого феномена будет способствовать более полному пониманию механизма окисления указанных кислородсодержащих соединений S(IV) на золотом электроде.
Список литературы
O’Brien, J.A., Hinkley, J.T., Donne, S.W., and Lindquist, S.-E., The electrochemical oxidation of aqueous sulfur dioxide: A critical review of work with respect to the hybrid sulfur cycle, Electrochim. Acta, 2010, vol. 55, p. 573.
Seo, E.T. and Sawyer, D.T., Electrochemical oxidation of dissolved sulphur dioxide at platinum and gold electrodes, Electrochim. Acta, 1965, vol. 10, p. 239.
Samec, Z. and Weber, J., Study of the oxidation of SO2 dissolved in 0.5 M H2SO4 on a gold electrode. I. Stationary electrode, Electrochim. Acta, 1975, vol. 20, p. 403.
Samec, Z. and Weber, J., Study of the oxidation of SO2 dissolved in 0.5 M H2SO4 on a gold electrode. II. A rotating disc electrode, Electrochim. Acta, 1975, vol. 20, p. 413.
Varga, K., Baradlai, P., and Vertes, A., In-situ radiotracer studies of sorption processes in solutions (bi)sulfite ions. I. Polycrystalline gold, Electrochim. Acta, 1997, vol. 42, p. 1143.
Quijada, C., Morallon, E., Vazquez, J.L., and Berlouis, L.E.A., Electrochemical behaviour of aqueous SO2 at polycrystalline gold electrodes in acidic media. A voltammetric and in-situ vibrational study. Part II. Oxidation of SO2 on bare and sulphur-modified electrodes, Electrochim. Acta, 2000, vol. 46, p. 651.
Allen, J.A., Rowe, G., Hinkley, J.T., and Donne S.W., Electrochemical aspects of the Hybrid Sulfur Cycle for large scale hydrogen production, Intern. J. Hydrogen Energy, 2014, vol. 39, p. 11376.
Tolmachev, Y.V. and Scherson, D.A., The electrochemical oxidation of sulfite on gold: UV-Vis reflectance spectroscopy at rotating disk electrode, Electrochim. Acta, 2004, vol. 49, p. 1315.
Quijada, C., Huerta, F.J., Morallón, E., Vázquez, J.L., and Berlouis, L.E.A., Electrochemical behaviour of aqueous SO2 at polycrystalline gold electrodes in acidic media: a voltammetric and in-situ vibrational study. Part I. Reduction of SO2: deposition of monomeric and polymeric sulfur, Electrochim. Acta, 2000, vol. 45, p. 1847.
Nicol, M.J., The Anodic Behaviour of Gold. Part I. Oxidation in acidic solution, Gold Bulletin, 1980, vol. 13, p. 46.
Nicol, M.J., The Anodic Behaviour of Gold. Part II. Oxidation in alkaline solution, Gold Bulletin, 1980, vol. 13, p. 105.
Burke, L.D. and Nugent, P.F., The Electrochemistry of Gold: I. The Redox Behaviour of the Metal in aqueous Media, Gold Bulletin, 1997, vol. 30(2), p. 43.
Petrović, Ž., Metikoš-Huković, M., Babić, R., Katić, J., and Milun, M., A multi-technique study of gold oxidation and semiconducting properties of the compact α‑oxide layers, J. Electroanal. Chem., 2009, vol. 629, p. 43.
Burke, L.D., McCarthy, M.M., and Roche, M.V.C., Influence of solution pH on monolayer and multilayer oxide formation processes on gold and palladium, J. Electroanal. Chem., 1984, vol. 167, p. 291.
Zelinsky, A.G. and Pirogov, B.Ya., Electrochemical oxidation of sulfite and sulfur dioxide at a renewable graphite electrode, Electrochim. Acta, 2017, vol. 231, p. 371.
Zelinsky, A.G., Features of Sulfite Oxidation on Gold Anode, Electrochim. Acta, 2016, vol. 188, p. 727.
Zelinsky, A.G. and Novgorodtseva, O.N., Effect of thiourea on the rate of anodic processes at gold and graphite electrodes in thiosulfate solutions, Electrochim. Acta, 2013, vol. 109, p. 482.
Angerstein-Kozlowska, H., Conway, B.E., Barnett, B., and Mozota, J., The role of ion adsorption in surface oxide formation and reduction at noble metals: General features of the surface process, J. Electroanal. Chem., 1979, vol. 100, p. 417.
Watanabe, T. and Gerischer, H., Photoelectrochemical studies on gold electrodes with surface oxide layers, J. Electroanal. Chem., 1981, vol. 117, p. 185.
Jusys, Z. and Bruckensteine, S., Electrochemical quartz crystal microgravimetry of gold in perchloric and sulfuric acid solution, Electrochem. and Solid-State Letters, 1998, vol. 1 (2), p. 74.
Tian, M., Pell, W.G., and Conway, B.E., Nanogravimetry study of the initial stages of anodic surface oxide film growth at Au in aqueous HClO4 and H2SO4 by means of EQCN, Electrochim. Acta, 2003, vol. 48, p. 2675.
Baten, S.M.A., Taylor, A.G., and Wilde, C.P., Second harmonic generation studies of the oxidation of metal electrodes: compact and hydrous oxide growth at gold electrodes in acid solutions, Electrochim. Acta, 2008, vol. 53, p. 6829.
Giron, R.G.P. and Ferguson, G.S., Development of cathodic silence in an oxide film on gold electrode, Electrochim. Acta, 2015, vol. 180, p. 560.
Reddy, A.K.N., Genshaw, M.A., and Bockris, J.O’M., Ellipsometric study of oxygen-containing films on platinum anodes, J. Chem. Phys., 1968, vol. 48, p. 671.
Vetter, K.J. and Shultze, J.W., The kinetics of the electrochemical formation and reduction of monomolecular oxide layers on platinum in 0.5 M H2SO4. Part I. Potentiostatic pulse measurement, J. Electroanal. Chem., 1972, vol. 34, p. 131.
Vetter, K.J. and Shultze, J.W., The kinetics of the electrochemical formation and reduction of monomolecular oxide layers on platinum in 0.5 M H2SO4. Part II. Galvanostatic pulse measurement and the model of oxide growth, J. Electroanal. Chem., 1972, vol. 34, p. 141.
Xia, S.J. and Birss, V.I., A multi-technique study of compact and hydrous Au oxide growth in 0.1 M sulfuric acid solution, J. Electroanalyt. Chem., 2001, vol. 500, p. 562.
Нечаев, И.В., Введенский, А.В. Квантово-химическое моделирование адсорбции гидроксид-иона на металлах IВ группы из водных растворов. Физикохимия поверхности и защита металлов. 2009. № 4 (45). С. 358. [Nechaev, I.V. and Vvedenskii, A.V., Quantum chemical modeling of hydroxide ion adsorption on group IB metals from aqueous solution, Protection of metals and physical chemistry of surface, 2009, vol. 45, p. 391.]
Ogura, K., Haruyama, S., and Nagasaki, R., The electrochemical oxidation and reduction of Gold, J. Electrochem. Soc., 1971, vol. 118, p. 531.
Burke, L.D., Lyons, M.E., and Whelan, D.P., Influence of solution pH on the reduction of thick anodic oxide films on gold, J. Electroanal. Chem., 1982, vol. 139, p. 131.
Hunger, T., Lapicque, F., and Storck, A., Electrochemical oxidation of sulphite ions at graphite electrodes, J. Appl. Electrochem., 1991, vol. 21, p. 588.
Glasstone, S. and Hickling, A., Studies in electrolytic oxidation. Part III. The formation of dithionate by the electrolytic oxidation, J. Chem. Soc., 1933, vol. 67, p. 829.
Katagiri, A. and Matsubara, T., Electro-oxidation of sulfite to dithionate in presence of copper ion, J. Electrochem. Soc., 1988, vol. 135, p. 1709.
Bancroft, W.D., Anode reactions, J. Chem. Soc., 1937, vol. 71, p. 195.
Дамаскин, Б.Б., Петрий, О.А. Введение в электрохимическую кинетику. М.: Высшая школа, 1975. С. 219. [Damaskin, B.B. and Petrii, O.A., Introduction to Electrochemical Kinetics (in Russisn), Moscow: Higher School, 1975. p. 219].
O’Brien, J.A., Hinkley, J.T., and Donne, S.W., Observed electrochemical oscillations during the oxidation of aqueous sulfur dioxide on sulfur modified platinum electrode, Electrochim. Acta, 2011, vol. 56, p. 4224.
Cho, B.W., Yun, K.S., and Chung, I.J., A study on anodic oxidation of iodide-mediated sulfur dioxide solution, J. Electrochem. Soc., 1987, vol. 134, p. 1664.
Zelinsky, A.G., Anode current on gold in mixed thiosulfate-sulfite electrolytes, Electrochim. Acta, 2015, vol. 154, p. 315.
Lu, J., Dreisinger, D.B., and Cooper, W.C., Anodic oxidation of sulphite ions on graphite anodes in alkaline solution, J. Appl. Electrochem., 1999, vol. 29, p. 1161.
Brevett, C.A.S. and Jonson, D.C., Anodic oxidations of sulfite, thiosulfate, and dithionite at doped PbO2-film electrodes, J. Electrochem. Soc., 1992, vol. 139, p. 1314.
Дополнительные материалы отсутствуют.