

Электрохимия, 2019, T. 55, № 4, стр. 463-472
Вольтамперометрическое определение цефуроксима аксетила в фармацевтических препаратах с применением модифицированного оксидом графена стеклоуглеродного электрода и ртутного капельного электрода
Севилей Эрдоган Каблан a, Нуран Езалтин a, *
a Отделение аналитической химии, Фармацевтический факультет, Университет Хаджеттепе
06100 Хаджеттепе, Турция
* E-mail: nozaltin@hacettepe.edu.tr
Поступила в редакцию 19.08.2018
После доработки 20.10.2018
Принята к публикации 09.11.2018
Аннотация
Два новых, экономичных, быстрых, простых, чувствительных и селективных метода квадратно-волновой вольтамперометрии (КВВ) разработаны для определения цефуроксима аксетила (ЦЕФА) в фармацевтических препаратах. Электрохимическое восстановление и окисление вещества на ртутном капельном электроде и модифицированном оксидом графена стеклоуглеродном электроде исследовали методами квадратно-волновой вольтамперометрии, в которых использованы скорости развертки до 1000 мВ/с или более, что обеспечивает значительно более быстрое определение. Для ртутного капельного электрода четко определенный пик наблюдали при –1.06 В относительно Ag/AgCl/4.6 M KCl в 0.1 M фосфатно-боратном буферном растворе при pH 7.0, предел обнаружения (ПО), предел количественного определения (ПКО) и диапазон линейности составляли 0.09, 0.26 и 0.26–15 мкг/мл, соответственно. Для модифицированного стеклоуглеродного электрода четко определенный пик наблюдали при 1.30 В относительно Ag/AgCl/4.6 M KCl в буферном растворе Бриттона–Робинсона при pH 2.0; ПО, ПКО и диапазон линейности составляли 2.70, 8.20 и 8.20–45 мкг/мл, соответственно. Согласно валидационным исследованиям, разработанные методы КВВ оказались точными, прецизионными, специфичными, чувствительными, воспроизводимыми, устойчивыми и надежными. Разработанные и валидированные методы КВВ применяли для определения ЦЕФА в фармацевтических препаратах. Результаты сравнивали с данными, полученными с помощью опубликованного метода ультрафиолетовой спектроскопии, и не обнаружили статистических различий.
ВВЕДЕНИЕ
Цефуроксима аксетил (ЦЕФА), (1RS)-1-[(ацетил)окси]этил(6R,7R)-3-[(амино карбонилокси)метил]-7-[(Z)-2-(фуран-2-ил)-2-(метоксиимино)ацетил]амино]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоксилат [1], представляет собой цефалоспориновый антибиотик второго поколения и абсорбируемый в полости рта предшественник лекарственного средства цефуроксима (CEF). Химическая структура ЦЕФА представлена на рис. 1. Этот сложноэфирный продукт ЦЕФ повышает липофильность и биодоступность в полости рта исходного соединения [2]. Его применяют, чтобы лечить острое воспаление среднего уха, инфекции костей и суставов, менингит, фарингит и тонзиллит, инфекции дыхательных путей, заражение крови, инфекции кожи и кожных структур [3].
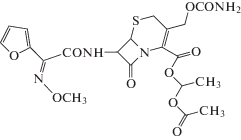
Описано несколько методов для определения ЦЕФА, присутствующего индивидуально или в сочетании с различными лекарственными средствами, такие как спектрофотометрия [4–11], высокоэффективная жидкостная хроматография (ВЭЖХ) [12–16], высокоэффективная тонкослойная хроматография (ВЭТСХ) [17], тонкослойная хроматография [18], капиллярный электрофорез [19, 20]. Однако большинство из этих методов являются относительно дорогостоящими и трудоемкими, расходуют большое количество токсичных органических растворителей и редко применяются в аналитических целях. Эти недостатки можно устранить посредством вольтамперометрических методов, области применения которых расширились в последние годы. Вольтамперометрические методы имеют ряд преимуществ. В случае ртутного капельного электрода (РКЭ) непрерывно обновляется поверхность металла, и поэтому данный метод обеспечивает воспроизводимость результатов. Получение образца осуществляется легче и дешевле, процедуры разделения и экстракции не требуются, что снижает стоимость. Вольтамперометрические методы являются наиболее подходящими для исследования окислительно-восстановительных свойств лекарственных средств. Благодаря высокой чувствительности, низкой стоимости и относительно короткой продолжительности анализа, вольтамперометрические процедуры представляют собой важные методы для количественного анализа следов многих органических и неорганических веществ [21–25]. Получение образца осуществляется легко, процедуры разделения и экстракции не требуются. Квадратно-волновая вольтамперометрия (КВВ) в последнее время привлекает все большее внимание в качестве вольтамперометрической процедуры для рутинных количественных анализов. КВВ представляет собой высокоамплитудную дифференциальную процедуру, в которой волновая форма, состоящая из симметричной квадратной волны, прилагается к рабочему электроду. Измерение тока осуществляют дважды в течение каждого квадратно-волнового цикла, первый раз в конце прямого импульса и второй раз в конце обратного импульса. Получаемый в результате ток пика является пропорциональным концентрации анализируемого вещества. Превосходная чувствительность обусловлена тем, что чистый ток превышает прямые и обратные компоненты. В сочетании с эффективным подавлением тока заряда могут быть достигнуты очень низкие пределы обнаружения. Сравнение квадратно-волновой и дифференциально-импульсной вольтамперометрии для обратимых и необратимых случаев показало, что превышение квадратно-волнового тока является 4- и 3.3-кратным, по сравнению с аналогичным дифференциально-импульсным откликом [26]. Основное преимущество метода КВВ представляет собой его высокая скорость. КВВ можно использовать для осуществления эксперимента значительно быстрее, чем в случае обычных и дифференциально-импульсных методов, которые работают при скоростях развертки, составляющих, как правило, от 1 до 10 мВ/с. КВВ использует скорости развертки вплоть до 1000 мВ/с или выше, что обеспечивает значительное ускорение анализов. Электродная кинетика и константы образования комплексов исследованы методом КВВ [27–30]. Что касается вольтамперометрического определения ЦЕФА, в литературе существует лишь единственная работа [31]. Алексис и др. исследовали вольтамперометрическое поведение трех цефалоспоринов, включая цефтазидим, цефуроксим-аксетил и цефтриаксон, методом дифференциально-импульсной вольтамперометрии (ДИВ) с применением РКЭ. На основании данного исследования был разработан метод ДИВ, чтобы определять цефтазидим при pH 2.0, цефуроксим-аксетил при pH 3.5 и для цефтриаксон при pH 8.0.
Не найдены литературные данные в отношении определения ЦЕФА методом КВВ в нейтральных растворах на РКЭ, где реализуется иной механизм восстановления, а также производится повышенный сигнал тока. Кроме того, до настоящего времени не было описано определение ЦЕФА на основе электроокисления с применением немодифицированного стеклоуглеродного электрода (СУЭ) и модифицированного оксидом графена стеклоуглеродного электрода (ОГ/СУЭ). Оксид графена (GO) представляет собой слоистый материал, получаемый посредством окисления графита [32]. Он содержит многочисленные кислородные функциональные группы, такие как эпоксидные, спиртовые и карбоксильные [33]. Кроме того, ОГ имеет большую площадь поверхности и превосходную электропроводность, которые делают его идеальным кандидатом для применения в электрохимических датчиках [34]. Еще большее значение имеет способность ОГ легко расслаиваться в воде на отдельные листы, из которых затем удобно получать непрерывным пленки, представляющие собой эффективные модифицированные электродные материалы [35]. Кроме того, эти материалы можно далее использовать для изготовления электронного оборудования, такого как химические датчики и биодатчики. Недавно были разработаны электрохимические датчики на основе ОГ для чувствительного анализа фармацевтических препаратов [36–38].
Таким образом, цель настоящего исследования заключалась в том, чтобы разработать два метода КВВ для определения ЦЕФА в фармацевтических препаратах на основе электровосстановления на РКЭ и электроокисления на ОГ/СУЭ. Валидационные параметры, такие как устойчивость, линейность, чувствительность, точность, прецизионность, степень извлечения, надежность и добротность, оценивали согласно руководству Международной конференции по гармонизации (МКГ) [39]. Оба метода были успешно применены для фармацевтических препаратов, содержащих ЦЕФА. Результаты сравнивали с данными, полученными методом УФ-спектрофотометрии, приведенным в литературе [4], и не обнаружили никакого значительного статистического различия.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Аппаратура
Все эксперименты осуществляли с применением электрохимического анализатора BAS 100 B/W от компании Bioanalytical System (США). Электрод сравнения представлял собой электрод Ag/AgCl с насыщенным раствором KCl (4.6 M), платиновую проволоку использовали в качестве вспомогательного электрода, РКЭ (BAS MF-9058, площадь поверхности 3.34 × 10−2 см2) и ОГ/СУЭ использовали в качестве рабочего электрода для восстановления и окисления, соответственно.
Химические вещества и реагенты
Рабочий стандарт ЦЕФА любезно предоставлен компанией Fargem A.Ş. HNO3, H3PO4, H3BO3, K2HPO4, Na2B4O7 · 2H2O, MeOH, ацетонитрил (ACN), HCl и NaOH были приобретены у компании Merck. Оксид графена, используемый для модификации, получен методом Хаммера [40]. Деионизированную воду, полученную из системы очистки воды Milli-Q, использовали для приготовления растворов. Стандартный концентрированный раствор ЦЕФА (1 г/л) получали в MeOH. Его хранили при температуре +4°C, при которой он сохраняет устойчивость в течение по меньшей мере двух месяцев. Рабочие стандартные растворы получали ежедневно, разбавляя концентрированный раствор деионизированной водой.
Получение фоновых электролитных растворов
Фосфатно-боратный (ФБ) буферный раствор при pH 7.0 использовали в качестве фонового электролита для восстановления ЦЕФА на РКЭ. Для получения ФБ буферного раствора навески 4.35 г K2HPO4 и 9.53 г Na2B4O7, 2H2O отдельно растворяли в 250 мл воды, затем смешивали растворы в объемном соотношении 1 : 1 и доводили pH до 7.0, используя раствор 0.1 M HCl. Буферный раствор Бриттона–Робинсона (БР) при pH 2.0 использовали в качестве фонового электролита для окисления ЦЕФА на ОГ/СУЭ. Его получали, растворяя 2.47 г H3BO3 в смеси 2.7 мл H3PO4 и 2.3 мл ледяной CH3COOH, разбавляя до 1000 мл деионизированной водой, и pH регулировали с помощью раствора 0.1 M NaOH.
Получение раствора таблеток
Десять таблеток взвешивали и тонко измельчали в ступке. Количество порошкообразной массы, эквивалентное одной таблетке, точно взвешивали и помещали в мерную колбу объемом 100 мл. Добавляли 50 мл MeOH и обрабатывали ультразвуком в течение 15 мин, затем доводили объем до метки, используя MeOH. Аликвоту данного раствора центрифугировали при 5000 об./мин в течение 10 мин. Прозрачную надосадочную жидкость помещали в другую колбу. Соответствующие аликвоты раствора образца надлежащим образом разбавляли водой для достижения желательных концентраций. Содержание лекарственного средства в расчете на одну таблетку определяли с помощью калибровочной кривой.
Получение синтетических таблеточных препаратов
Синтетические таблетки получали, смешивая обычные неактивные ингредиенты (бензоат Na, пропиленгликоль, метилпарабен, аэросил, TiO2, натриевую соль кроскарамеллозы, микрокристаллическую целлюлозу) и заданное количество ЦЕФА. Затем смесь помещали в мерную колбу объемом 50 мл, добавляли 25 мл MeOH и обрабатывали ультразвуком в течение 15 мин, затем доводили объем до метки, используя MeOH, и соответствующие растворы получали согласно описанию для раствора таблеток.
Получение модифицированного оксидом графена стеклоуглеродного электрода
Навеску синтезированного оксида графена растворяли в деионизированной воде, обрабатывали ультразвуком и центрифугировали. Необходимый объем прозрачного раствора, соответствующий 16 мкг оксида графена, наносили с помощью микропипетки на полированную поверхность СУЭ. Затем на поверхность воздействовали инфракрасным излучением в течение 1.5 ч. ОГ/СУЭ использовали во всех исследованиях окисления.
Методика
Для электровосстановления на РКЭ 3.0 мл фонового электролита (ФБ буферный раствор, pH 7.0) деоксигенировали предварительно очищенным азотом в течение 15 мин. После снятия вольтамперограммы этого раствора соответствующий объем рабочего стандартного раствора ЦЕФА добавляли с помощью микропипетки в фоновый электролит. Повторно снимали вольтамперограмму. Все измерения осуществляли при комнатной температуре. Такую же процедуру осуществляли в буферном растворе БР при pH 2.0 с применением ОГ/СУЭ для электроокисления ЦЕФА.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Влияние фонового электролита и pH
Вольтамперограммы ЦЕФА на РКЭ исследовали в диапазоне pH от 2.0 до 10.0, причем механизмы восстановления были подробно описаны в предыдущей работе авторов. Было установлено, что электрохимическое восстановление ЦЕФА при pH 7.0 обусловлено восстановлением карбонильной группы аксетила, а не метоксииминогруппы в структуре ЦЕФА [41].
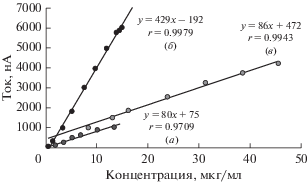
Как представлено на рис. 2, потенциал пика не зависит от pH, а ток пика при pH 7.0 превышает в 6 раз ток пика, полученный при pH 2.0. Чтобы показать различную чувствительность при pH 2.0 и pH 7.0, для этих значений pH строили калибровочные кривые. Как представлено на рис. 3, наклон кривой при pH 7.0 (б) выше, чем при pH 2.0 (a). Очевидно, что буферный раствор, имеющий pH 7.0, является наилучшим для определения ЦЕФА методом КВВ. Таким образом, было решено осуществлять исследование при pH 7.0 вследствие более высокой чувствительности; кроме того, ранее не был описан вольтамперометрический метод для определения ЦЕФА в нейтральной среде с применением РКЭ.
Рис. 2.
Вольтамперограммы КВВ 8.0 мкг/мл ЦЕФА при различных значениях pH в 0.1 M ФБ буферном растворе с применением РКЭ: (a) фоновый электролит при pH 2.0, (б) ЦЕФА при pH 2.0, (в) фоновый электролит при pH 7.0, (г) ЦЕФА при pH 7.0.
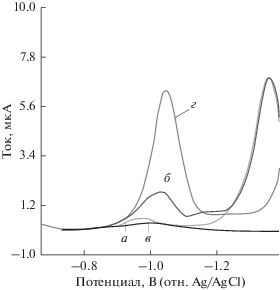
Рис. 3.
Калибровочные кривые ЦЕФА (n = 6): (a) 0.1 M буферный раствор ФБ при pH 2.0 с применением РКЭ, (б) 0.1 M буферный раствор ФБ при pH 7.0 с применением РКЭ, (в) буферный раствор БР при pH 2.0 с применением ОГ/СУЭ.
Были исследованы буферные растворы различных типов (БР и фосфатно-боратный или ФБ) при pH 7.0, и лучшие пики были получены в буферном растворе ФБ. Буферный раствор БР в диапазоне pH от 2.0 до 10.0 исследовали в качестве фонового электролита для электроокисления ЦЕФА на ОГ/СУЭ, и единственный и наиболее высокий ток пика наблюдали около 1.30 В при pH 2.0 [41].
Влияние приборных параметров
Были оценены приборные параметры, влияющие на ток пика в методе КВВ. Таким образом, частоту, шаг потенциала и амплитуду импульсов исследовали для 3.0 мкг/мл ЦЕФА в буферном растворе ФБ при pH 7.0. Наиболее высокий и четко определенный пик восстановления получали при –1.06 В относительно Ag/AgCl/4.6 M KCl, когда использовали начальный потенциал –0.7 В, частоту 40 Гц, шаг потенциала 5 мВ и амплитуду импульса 35 мВ на РКЭ. Четко определенный пик окисления получали при 1.30 В относительно Ag/AgCl/4.6 M KCl, когда использовали начальный потенциал +0.8 В, частоту 45 Гц, шаг потенциала 5 мВ и амплитуду импульса 60 мВ на ОГ/СУЭ при pH 2.0 в буферном растворе БР.
В присутствии оксида графена, вследствие увеличенной площади поверхности и повышенной электропроводности, ток пика (Iп) увеличивался в 7 раз для модифицированного СУЭ, по сравнению с немодифицированным СУЭ. Наиболее высокий ток пика получали при модификации СУЭ, на который наносили 16 мкг оксида графена [41]. Электрохимические измерения осуществляли с помощью модифицированного электрода, на который наносили 16 мкг оксида графена, при pH 2.0 в буферном растворе БР. Рисунок 4 представляет типичные полученные методом СЭМ изображения немодифицированного СУЭ (а) и модифицированного оксидом графена СУЭ (б). На рис. 4б можно наблюдать структуру пленочного слоя графена, который образуется на поверхности СУЭ.

Тип тока и обратимость
Тип тока и обратимость электродных реакций были исследованы с применением методов CV, CA и CC в предыдущей работе авторов. Полученные результаты показали, что ЦЕФА претерпевает необратимое (для полного восстановления) и контролируемое диффузией восстановление, на которое влияет адсорбция в нейтральной среде (pH 7.0), в то время как необратимое и контролируемое диффузией окисление происходит в кислой среде (pH 2.0) [41].
Валидация предложенных методов
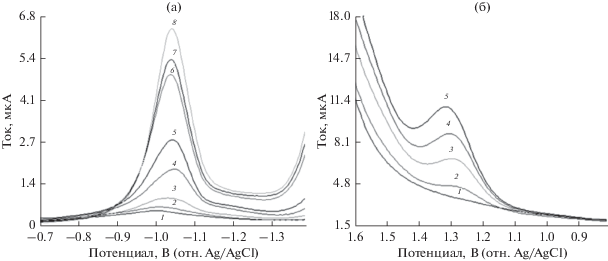
На рис. 5 представлены квадратно-волновые вольтамперограммы при повышении концентрации ЦЕФА, снятые с применением РКЭ и ОГ/СУЭ в оптимальных условиях.
Рис. 5.
Квадратно-волновые вольтамперограммы с увеличением концентрации ЦЕФА: (а) 0.1 M буферный раствор ФБ при pH 7.0 на РКЭ: (1) фоновый электролит, (2) 0.33, (3) 1.32, (4) 3.23, (5) 6.25, (6) 10.45, (7) 12.28, (8) 14.29 мкг/мл ЦЕФА; (б) буферный раствор БР при pH 2.0 на ОГ/СУЭ: (1) фоновый электролит, (2) 3.31, (3) 12.99, (4) 23.81, (5) 38.46 мкг/мл ЦЕФА.
Рабочие характеристики были определены посредством валидационных исследований разработанных аналитических методов. Валидационные параметры, такие как устойчивость, диапазон линейности, ПО, ПКО, точность, прецизионность, степень извлечения, селективность, воспроизводимость, надежность и добротность, оценивали согласно руководству МКГ [39].
Устойчивость. Стандартные концентрированные растворы ЦЕФА хранили в двух различных условиях, в том числе при 4°C в течение 60 сут для определения долгосрочной устойчивости и при комнатной температуре в течение 24 ч для определения краткосрочной устойчивости. После окончания данного периода растворы анализировали методом КВВ и не наблюдали никаких изменений потенциала пика и тока пика ЦЕФА. Это показывает высокую устойчивость ЦЕФА в течение данного периода.
Калибровочная кривая и диапазон линейности. Чтобы продемонстрировать соотношение между током пика и концентрацией ЦЕФА строили калибровочные кривые с применением стандартных растворов ЦЕФА при различных концентрациях. Ток пика линейно увеличивался при повышении концентрации ЦЕФА (рис. 3б, 3в). Данные калибровочных кривых для предложенных методов представлены в табл. 1. ПО определяли, оценивая минимальное количество анализируемого вещества, которое можно обнаружить, но не обязательно количественно определить как точное значение. ПКО определяли, устанавливая минимальное количество анализируемого вещества, которое может быть количественно определено с подходящей прецизионностью и точностью. Как видно в табл. 1, значения ПО и ПКО показывают, что предложенные методы можно считать достаточно чувствительными для определения ЦЕФА в фармацевтических препаратах.
Таблица 1.
Данные калибровочных кривых методов КВВ для определения ЦЕФА (n = 6).
| РКЭ | ОГ/СУЭ | |
|---|---|---|
| Уравнение регрессии | Iп (нА) = (429 ± 14) c (мкг/мл) –192 ± 11 | Iп (нА) = (86 ± 5) c (мкг/мл) + 472 ± 31 |
| Коэффициент корреляции (r) | 0.9979 | 0.9986 |
| Диапазон линейности, мкг/мл | 0.26–15 | 8.20–45 |
| ПО, мкг/мл | 0.09 | 2.70 |
| ПКО, мкг/мл | 0.26 | 8.20 |
Точность и прецизионность. Точность и прецизионность методов исследовали посредством внутрисуточного и межсуточного анализа. Три различные концентрации ЦЕФА (2.00, 6.00, 11.00 мкг/мл для РКЭ и 8.20, 16.13 и 31.25 мкг/мл для ОГ/СУЭ) были выбраны в линейном диапазоне. Для внутрисуточных исследований шесть различных серий стандартных растворов ЦЕФА получали и анализировали в тот же день при трех различных уровнях концентрации. Для РКЭ полученные значения ОСО и погрешности (%) определяли в диапазонах от 0.30 до 0.81% и от 0.66 до 1.34%, соответственно. Для ОГ/СУЭ полученные значения ОСО и погрешности (%) определяли в диапазонах от 0.66 до 1.09% и от 0.74 до 0.78%, соответственно. Для межсуточных исследований стандартные растворы ЦЕФА получали и анализировали в течение шести дней подряд при трех различных уровнях концентрации. Для РКЭ полученные значения ОСО и погрешности (%) определяли в диапазонах от 0.44 до 1.44% и от 1.13 до 1.30%, соответственно. Для ОГ/СУЭ полученные значения ОСО и погрешности (%) определяли в диапазонах от 0.73 до 0.98% и от 0.63 до 0.97%, соответственно. Результаты в табл. 2 демонстрируют удовлетворительную точность и прецизионность предложенных методов.
Таблица 2.
Точность и прецизионность методов КВВ для определения ЦЕФА (n = 6)
| Электрод | Добавка, мкг/мл | Внутрисуточные значения | Межсуточные значения | ||||
|---|---|---|---|---|---|---|---|
| найдено, мкг/мл | точность, погрешность, % | прецизион-ность RSD, % | найдено, мкг/мл | точность, погрешность, % | прецизион-ность RSD, % | ||
| РКЭ | 2.00 | 2.01 ± 0.01 | 0.66 | 0.81 | 2.00 ± 0.01 | 1.13 | 1.44 |
| 6.00 | 5.94 ± 0.02 | 0.82 | 0.67 | 5.97 ± 0.04 | 1.19 | 1.44 | |
| 11.00 | 11.15 ± 0.01 | 1.34 | 0.30 | 11.14 ± 0.02 | 1.30 | 0.44 | |
| ОГ/СУЭ | 8.20 | 8.15 ± 0.02 | 0.75 | 0.66 | 8.16 ± 0.03 | 0.78 | 0.90 |
| 16.13 | 16.13 ± 0.07 | 0.78 | 1.09 | 16.02 ± 0.06 | 0.97 | 0.98 | |
| 31.25 | 31.20 ± 0.09 | 0.74 | 0.74 | 31.13 ± 0.09 | 0.63 | 0.73 | |
Степень извлечения. Полученные из синтетических образцов растворы (содержащие 601.4 мг ЦЕФА) анализировали, используя разработанные методы. Оценивали результаты определения степени извлечения. Согласно литературе [42], результаты определения степени извлечения должны находиться в диапазоне 100 ± 2%. Значения степени извлечения находились в диапазоне от 99.83 до 100.35% с RSD < 2.00. Степень извлечения разработанных методов допонительно подтвердила точность методов.
Селективность. Селективность предложенных методов исследовали, сравнивавая вольтамперограммы растворов синтетического препарата, таблетки и стандарта ЦЕФА в одинаковой концентрации. Вольтамперограммы были идентичными, и никакие посторонние пики не наблюдались (рис. 6а, 6б). Это означает, что помехи от вспомогательных веществ были незначительными для предложенных методов. Сделан вывод, что разработанные методы отличаются высокой селективностью.
Рис. 6.
Вольтамперограммы КВВ растворов стандарта, таблетки и синтетического препарата: (а) применение РКЭ с содержанием 3.13 мкг/мл ЦЕФА, (б) применение ОГ/СУЭ с содержанием 16.13 мкг/мл ЦЕФА.
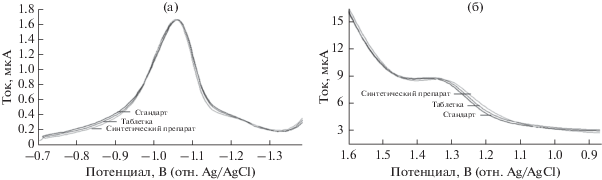
Чтобы оценить влияние присутствия вспомогательных веществ на предложенные методы, был также применен метод стандартных добавок. Для этой цели соответствующий объем раствора таблеток добавляли в фоновый электролит. После снятия вольтамперограммы добавляли три стандартных раствора с возрастающей концентрацией ЦЕФА, и вольтамперограммы снимали после каждой добавки. Измеренные токи пика наносили на график зависимости от концентрации ЦЕФА. Концентрацию образца вычисляли в результате экстраполяции прямой в отрицательную область на оси абсцисс. Уравнение средней регрессии метода стандартных добавок представляло собой y = = 430x + 1137, r = 0.9999 (n = 6) для восстановления на РКЭ и y = 87x + 600, r = 0.9993 (n = 6) для окисления на ОГ/СУЭ. Здесь y представляет собой ток пика (нА), x представляет собой концентрацию (мкг/мл) стандартной добавки ЦЕФА, и r представляет собой коэффициент корреляции. Близкий наклон калибровочной кривой и метод стандартных добавок показали отсутствие значительного влияния вспомогательных веществ, содержащихся в таблетированных лекарственных средствах. Таким образом, разработанные методы оказались способными к определению ЦЕФА в присутствии вспомогательных веществ и могут считаться селективными. Метод калибровочной кривой, который осуществляется легче и быстрее, чем метод стандартных добавок, использовали в количественном анализе ЦЕФА.
Надежность. Надежность аналитического метода представляет собой устойчивость метода в отношении небольших, но неслучайных изменений технологических параметров метода. Параметры, включая pH фоновых электролитов (±0.1) и начальный потенциал (±0.1 В), были исследованы для 8.0 мкг/мл ЦЕФА для восстановления на РКЭ и 16.13 мкг/мл ЦЕФА для окисления на ОГ/СУЭ (табл. 3). В каждом эксперименте изменялся только один параметр. Значения ОСО (%) составляли от 0.19 до 1.18%. Полученные результаты после каждого изменения сравнивали с результатами в оптимальных условиях. Эти незначительные изменения, которые могут происходить в процессе осуществления эксперимента, не влияют на ток пика. Результаты сравенивали, используя U-критерий Манна–Уитни, и не наблюдали существенного различия между результатами (p > 0.05). Таким образом, разработанные методы можно считать надежными для определения ЦЕФА.
Таблица 3.
Параметры надежности предложенных методов КВВ (n = 6)
| pH | Начальный потенциал | Найдено, мкг/мл | ОСО, % | U-критерий Манна–Уитни* | |
|---|---|---|---|---|---|
| 8.0 мкг/мл ЦЕФА, восстановление на РКЭ | 7 | –0.7 В (оптимальные условия) | 8.04 ± 0.01 | 0.19 | – |
| 7 | –0.69 В | 7.99 ± 0.02 | 0.68 | Uвычисл = 37 | |
| 7 | –0.71 В | 8.00 ± 0.02 | 0.83 | Uвычисл = 27 | |
| 6.9 | –0.7 В | 8.08 ± 0.01 | 0.40 | Uвычисл = 27 | |
| 7.1 | –0.7 В | 7.98 ± 0.02 | 0.69 | Uвычисл = 32 | |
| 16.13 мкг/мл ЦЕФА, окисление на ОГ/СУЭ | 2 | 0.8 В (оптимальные условия) | 16.08 ± 0.07 | 1.18 | – |
| 2 | 0.79 | 16.30 ± 0.04 | 0.70 | Uвычисл = 35 | |
| 2 | 0.81 | 16.20 ± 0.06 | 1.05 | Uвычисл = 31 | |
| 1.9 | 0.80 | 16.12 ± 0.03 | 0.46 | Uвычисл = 28 | |
| 2.1 | 0.80 | 16.12 ± 0.05 | 0.86 | Uвычисл = 29 |
Добротность. Добротность предложенного метода оценивали, используя разработанные процедуры для анализа ЦЕФА, два различных исследователя в различные дни, в одинаковых оптимизированных условиях, с применением одинаковых приборов (n = 7). Полученные результаты двух различных исследователей сравнивали, используя U-критерий Манна–Уитни, и не обнаружили значительного различия между этими результатами (p > 0.05). Сделан вывод, что разработанные методы являются добротными для определения ЦЕФА.
Анализ фармацевтических препаратов
Чтобы проверить применимость предложенных методов, товарные таблетированные препараты, содержащие ЦЕФА в двух различных дозах (601.4 мг на таблетку и 300.7 мг на таблетку), анализировали в оптимальных условиях. Содержание ЦЕФА в таблетках вычисляли согласно уравнениям регрессии для методов КВВ. Метод УФ-спектрофотометрии [4] использовали в качестве сравнительного для оценки валидности разработанных методов. Таблица 4 представляет результаты, полученные тремя методами для определения ЦЕФА в фармацевтических препаратах. Результаты сравнивали, используя критерий Краскела–Уоллиса, и не обнаруживали значительного различия между методами КВВ и методом УФ.
Таблица 4.
Результаты, полученные методами КВВ и методом УФ для таблеток CEFAKS® (n = 10)**
| 601.4 мг ЦЕФА на таблетку | 300.7 мг ЦЕФА на таблетку | |||||
|---|---|---|---|---|---|---|
| методы КВВ | метод УФ (4) | методы КВВ | метод УФ (4) | |||
| РКЭ | ОГ/СУЭ | РКЭ | ОГ/СУЭ | |||
| X ± СП | 601.0 ± 0.34 | 601.0 ± 0.38 | 599.8 ± 0.59 | 300.7 ± 0.18 | 299.7 ± 0.52 | 300.0 ± 0.54 |
| СО | 1.078 | 1.191 | 1.878 | 0.564 | 1.637 | 1.713 |
| ОСО, % | 0.180 | 0.198 | 0.313 | 0.187 | 0.546 | 0.571 |
ВЫВОДЫ
Два простых, быстрых, селективных, точных и прецизионных метода КВВ разработаны и полностью валидированы для определения ЦЕФА в фармацевтических препаратах. Это первое исследование методом квадратно-волновой вольтамперометрии для определения ЦЕФА, которое основано на восстановлении карбонильной группы в структуре аксетила при РКЭ в нейтральной среде и на основе электроокисления ЦЕФА на ОГ/СУЭ. В предыдущем исследовании [41] авторы заявили, что механизмы восстановления в нейтральной и в кислой средах различаются. Разработанные методы проще, быстрее и требуют менее дорогостоящего оборудования, чем хроматографические методы. Суммарное время анализа данными методами является весьма коротким (менее одной минуты). За счет применения процедуры КВВ к данным методам также уступают по скорости хроматографические и другие вольтамперометрические методы (полярография, ДИВ). Предложенные методы являются достаточно точными, прецизионными, и они были успешно применены для анализа ЦЕФА в соответствующих таблетированных препаратах. Анализ осуществляли без помех со стороны вспомогательных веществ, присутствующих в таблетках. Это быстрая одностадийная процедура, для которой требуется лишь простая обработка образца без применения токсичных органических растворителей. Преимущества предложенных методов по сравнению с методом ДИВ, описанным в литературе, представляют собой более высокая скорость и иной механизм восстановления, в котором производится более интенсивный сигнал тока. Можно сделать вывод, что разработанные методы КВВ окажутся более успешными и надежными для быстрого рутинного анализа в лабораториях контроля качества в целях анализа ЦЕФА в нерасфасованной форме и фармацевтических препаратах.
Список литературы
http://www.rxlist.com/cgi/generic/cefuro.htm, 2015 (accessed 13.08.15).
Finn, A., Straughn, A., Meyer, M., and Chubb, J., Effect of dose and food on the bioavailability of cefuroxime axetil, Biopharm. Drug. Dispos., 1987, vol. 8, p. 519.
Dollery, C.S., Therapeutic drugs, Churchill Livingstone, 1999.
Amir, S.B., Hossain, M., and Mazid, M., Development and Validation of UV Spectrophotometric Method for the Determination of Cefuroxime Axetil in Bulk and Pharmaceutical Formulation, J. Sci. Res., 2013, vol. 6, p. 133.
Pritam, J., Manish, P., and Sanjay, S., Development and validation of UV-Spectrophotometric method for determination of Cefuroxime Axetil in bulk and in Formulation, Int. J. Drug. Dev. & Res., 2011, vol. 3, p. 318.
Chaudhari, S., Karnik, A., Adhikary, A., Tandale, R., and Vavia, P., Simultaneous UV spectrophotometric method for the estimation of cefuroxime axetil and probenecid from solid dosage forms, Indian. J. Pharm. Sci., 2006, vol. 68, p. 59.
Game, M., Sakarkar, D., Gabhane, K., and Tapar, K., Validated spectrophotometric methods for the determination of cefuroxime axetil in bulk drug and tablets, Int. J. Chem. Tech. Res., 2010, vol. 2, p. 1259.
Ingale, P.L., Dalvi, S.D., Jadav, D.D., Gudi, S.V., Patil, L.D., and Kadam, Y.A., Simultaneous estimation of cefuroxime axetil and potassium clavulanate-analytical method development and validation, Der. pharma chem., 2013, vol. 5, p. 35.
Pavankumar, K.P.T., Jagadeeswaran, M., Caroline, Grace A., Sivakumar, T., Spectrophotometric determination for the analysis of cefuroxime axetil in pharmaceutical dosage forms, Anal. Chem., 2013, vol. 13, p. 347.
Sengar, M.R., Gandhi, S.V., Rajmane, V., Patil, U.P., and Gandhi B.B., Simultaneous Determination of Cefuroxime Axetil and Potassium Clavulanate in Tablet Dosage Form by Spectrophotometry, Research J. Pharmacy and Technology, 2010, vol. 3, p. 260.
Shelke, S., Dongre, S., Rathi, A., Dhamecha, D., Maria, S., and Dehghan, M.H.G., Development and validation of UV spectrophotometric method of Cefuroxime Axetil in bulk and pharmaceutical formulation, Asian J. Research in Chemistry, 2009, vol. 2, p. 222.
Can, N.Ö., Altiokka, G., and Aboul-Enein, H.Y., Determination of cefuroxime axetil in tablets and biological fluids using liquid chromatography and flow injection analysis, Anal Chim Acta., 2006, vol. 576, no. 2, p. 246.
Kumar, P.S., Jayanthi, B., Abdul, K., Prasad, U., Kumar, Y.N., and Sarma, P., A Validated High performance liquid chromatography (HPLC) Method for the Estimation of Cefuroxime axetil, Research J. Pharmaceutical, Research J. Pharmaceutical, Biological and Chemical Sciences, 2012, vol. 3, p. 223.
Sengar, M.R., Gandhi, S.V., Patil, U.P., and Rajmane, V.S., Reverse phase high performance liquid chromatographic method for simultaneous determination of Cefuroxime Axetil and potassium clavulanate in tablet dosage form, Int. J. Chem. Tech. Res., 2009, vol. 1, p. 1105.
Ranjane, P.N., Gandhi, S.V., Kadukar, S.S., and Ranher, S.S., Simultaneous determination of cefuroxime axetil and ornidazole in tablet dosage form using reversed-phase high performance liquid chromatography, Chin. J. Chromatogr., 2008, vol. 26, p. 763.
Ingale, P.L., Dalvi, S.D., Jadav, D.D., Gudi, S.V., Patil, L.D., and Kadam, Y.A., Simultaneous Determination of Cefuroxime Axetil and Potassium Clavulanate in Pharmaceutical Dosage Form By RP-HPLC, Int. J. Pharmacy and Pharm. Sci., 2013, vol. 5, p. 179.
Ranjane, P.N., Gandhi, S.V., Kadukar, S.S., and Bothara, K.G., HPTLC determination of cefuroxime axetil and ornidazole in combined tablet dosage form, J. Chromatogr. Sci., 2010, vol. 48, p. 26.
Krzek, J. and Dąbrowska-Tylka, M., Simultaneous determination of cefuroxime axetil and cefuroxime in pharmaceutical preparations by thin-layer chromatography and densitometry, Chromatographia, 2003, vol. 58, p. 231.
Altria, K. and Rogan, M., Reductions in sample pretreatment requirements by using high-performance capillary electrokinetic separation methods, J. Pharm. Biomed. Anal., 1990, vol. 8, p. 1005.
Raj, K.A., Determination of cefixime trihydrate and cefuroxime axetil in bulk drug and pharmaceutical dosage forms by electrophoretic method, Int. J. ChemTech. Res., 2010, vol. 2, p. 337.
Beltagi, A., Determination of the antibiotic drug pefloxacin in bulk form, tablets and human serum using square wave cathodic adsorptive stripping voltammetry, J. Pharm. Biomed. Anal., 2003, vol. 31, p. 1079.
Razak, O.A., Electrochemical study of hydrochlorothiazide and its determination in urine and tablets, Pharm. Biomed. Anal., 2004, vol. 34, p. 433.
El-Hefnawey, G., El-Hallag, I., Ghoneim, E., and Ghoneim, M., Voltammetric behavior and quantification of the sedative-hypnotic drug chlordiazepoxide in bulk form, pharmaceutical formulation and human serum at a mercury electrode, J. Pharm. Biomed. Anal., 2004, vol. 34, p. 75.
Al-Ghamdi, A.F. and Bani-Yaseen, A.D., Electrochemical reduction of ciprofloxacin at the mercury electrode and its voltammetric determination in tablet and urine, Russ. J. Electrochem., 2014, vol. 50, p. 355.
El Mhammedi, M.A., Achak, M., and Bakasse, M., Square wave voltammetry for analytical determination of cadmium in natural water using Ca10(PO4)6(OH)2-modified platinum electrode, Am. J. Analyt. Chem., 2010, vol. 1, p. 150.
Wang, J., Analytical electrochemistry, John Wiley & Sons, 2006.
O’Dea, J.J., Osteryoung, J., and Osteryoung, R.A., Theory of square wave voltammetry for kinetic systems, Anal. Chem., 1981, vol. 53, p. 695.
Krause, M.S., Jr. and Ramaley, L., Analytical application of square wave voltammetry, Anal. Chem., 1969, vol. 41, p. 1365.
Stojek, Z. and Osteryoung, J., Direct determination of chelons at trace levels by one-drop square-wave polarography, Anal. Chem., 1981, vol. 53, p. 847.
Lin, S.R. and Feng, Q.S., Determination of chemical reaction rate constants preceding or following electron transfer by mechanical square wave polarography, Anal. Chem., 1982, vol. 54, p. 1362.
Aleksić, M.M., Lijeskić, N., Pantić, J., and Kapetanović, V.P., Electrochemical behavior and differential pulse voltammetric determination of ceftazidime, cefuroxime-axetil and ceftriaxone, FU Phys. Chem. Technol., 2013, vol. 11, p. 55.
Stankovich, S., Dikin, D.A., Dommett, G.H., Kohlhaas, K.M., Zimney, E.J., Stach, E.A., Piner, R.D., Nguyen, S.T., and Ruoff, R.S., Graphene-based composite materials, Nature, 2006, vol. 442, p. 282.
Lerf, A., He, H., Forster, M., and Klinowski, J., Structure of graphite oxide revisited, J. Phys. Chem. B., 1998, vol. 102, p. 4477.
Huang, H.-P. and Jun-Jie, Z., Preparation of novel carbon-based nanomaterial of graphene and its applications electrochemistry, Chin. J. Anal. Chem., 2011, vol. 39, p. 963.
Wang, X., Zhi, L., and Müllen, K., Transparent, conductive graphene electrodes for dye-sensitized solar cells, Nano Lett., 2008, vol. 8, p. 323.
Li, J., Kuang, D., Feng, Y., Zhang, F., Xu, Z., and Liu, M., A graphene oxide-based electrochemical sensor for sensitive determination of 4-nitrophenol, J. Hazard. Mater., 2012, vol. 201, p. 250.
Wang, Y., Li, Y., Tang, L., Lu, J., and Li, J., Application of graphene-modified electrode for selective detection of dopamine, Electrochem. commun., 2009, vol. 11, p. 889.
Wu C., Sun, D., Li, Q., and Wu, K., Electrochemical sensor for toxic ractopamine and clenbuterol based on the enhancement effect of graphene oxide, Sens. Actuator B-Chem., 2012, vol. 168, p. 178.
Guideline, I.H.T., Validation of analytical procedures: text and methodology, Q2 (R1), 1 (2005).
Hummers, W.S., Jr. and Offeman, R.E., Preparation of graphitic oxide, J. Am. Chem. Soc., 1958, vol. 80, 1339.
Kablan, S.E. and Özaltın, N., Investigation of electrochemical behaviour of cefuroxime axetil using hanging mercury drop electrode and graphene oxide modified glassy carbon electrode, J. Electroanal. Chem., 2017, vol. 785, p. 144.
Green, J.M., Peer reviewed: a practical guide to analytical method validation, Anal. Chem., 1996, vol. 68, p. 305A.
Дополнительные материалы отсутствуют.