

Электрохимия, 2020, T. 56, № 4, стр. 308-316
Электросинтез моно- и дисульфидов на основе циклоалканов C5–C8, сероводорода и изомерных дибутилдисульфидов
Е. В. Шинкарь a, *, А. В. Швецова a, А. О. Охлобыстин a, Н. Т. Берберова a, **
a ФГБОУ ВО “Астраханский государственный технический университет”
414056 Астрахань, ул. Татищева, 16, Россия
* E-mail: elenshin@rambler.ru
** E-mail: nberberova@gmail.com
Поступила в редакцию 02.04.2019
После доработки 26.04.2019
Принята к публикации 13.07.2019
Аннотация
Разработан метод получения органических моно-, ди- и полисульфидов на основе электрохимических реакций незамещенных и алкилзамещенных циклоалканов C5–C8 с ди(н-бутил)дисульфидом (ди(трет-бутил)дисульфидом) и сероводородом. Трехкомпонентный электросинтез проводили в хлористом метилене при атмосферном давлении и комнатной температуре в условиях анодной активации H2S до катион-радикала, фрагментирующегося на протон и тиильный радикал. Предложенный подход с применением окислительного инициирования превращений позволил получить моно-, дисульфиды асимметричного строения и симметричные дисульфиды. Выход биологически активных органических производных серы зависит от продолжительности электросинтеза, строения изомерных дибутилдисульфидов, размера и степени насыщенности алицикла.
ВВЕДЕНИЕ
В последнее время потребность в создании препаративно доступных методов синтеза органических моно- и дисульфидов, представляющих особенный интерес ввиду высокой биологической активности, заметно возросла [1]. Большую роль данные соединения играют в фармацевтической промышленности как составляющие лекарственных препаратов разнообразного спектра действия и специфических лечебных косметологических средств [2]. Так, диалилдисульфид защищает организм от апоптоза сердечной мышцы, который индуцируется высоким содержанием глюкозы в организме [3]. Органические соединения серы используют в синтезе N-ацетилцистеина, глутатиона и таурина, являющихся эффективными средствами при болевых синдромах, спортивных травмах и нарушениях функции почек и мочевого пузыря [4]. Моно- и дисульфиды обладают химиопревентивными и/или химиотерапевтическими свойствами, что повышает на них спрос из-за перспективности применения в синтезе новых противораковых агентов, способствующих не только останавливать, но и предотвращать развитие онкологического процесса [5–8]. Важной особенностью указанных соединений является обеспечение защитного действия организма против окислительного повреждения клеток за счет их способности генерировать газ-трансмиттер – H2S, который относят к внутриклеточным сигнальным молекулам, выполняющим специфические регуляторные функции [9–12].
Известные методы получения органических моно- и дисульфидов как правило каталитические и реализуются при повышенной температуре. Диарилсульфиды получают взаимодействием арилгалогенидов с тиолами в присутствии катализатора Сu/Fe при 100°С [13]. Синтез дибензилсульфида на основе бензилхлорида с участием H2S и моноэтаноламина проводят в условиях жидкофазного катализа [14]. В последнее время особый интерес представляет поиск новых путей синтеза сульфидов, применяемых в качестве прекурсоров при получении сульфоксидов, тиоэфиров и сульфонов, на основе которых изготавливают лекарственные препараты антиневротического, антиастматического и антиаллергенного действия [15, 16]. Авторами [17] предложен способ синтеза алкиларилсульфидов взаимодействием алканов с арилсульфонилхлоридами в присутствии соли рутения (100°С). Новым подходом к получению сульфидов асимметричного строения является использование пероксидов в качестве иницииаторов радикальных превращений. Разработан новый способ получения алкиларилсульфидов путем прямого тиолирования алканов арилсульфонилгидразидами с использованием ди-трет-бутилпероксида в качестве окислителя [18]. Трансформация C–H-связи алканов в C–S-связь эффективно катализируется ацетилацетонатом палладия при нагревании реакционной смеси до 120°C [19]. Каталитическое арил(алкил)тиолирование замещенных аренов в присутствии Pd(II) ведет к получению различных асимметричных диарил(алкил)сульфидов благодаря генерированию электрофильного сернистого реагента из сукцинимида в трифторуксусной кислоте [20]. Достаточно удобным способом получения бис-(2-фенилэтил)сульфида в условиях межфазного катализа является взаимодействие H2S с 2-бромэтилбензолом в водном растворе N‑метилдиэтаноламина в присутствии ионной жидкости – хлорида тригексилтетрадецилфосфония [21].
Указанные примеры демонстрируют актуальность разработки нового метода синтеза моно- и дисульфидов с участием доступного и дешевого сернистого реагента (H2S) в мягких электрохимических условиях. Предлагаемый в работе подход с использованием окислительной активации H2S до нестабильного катион-радикала и тиильного радикала (25°С, 1 атм) в реакциях с органическими соединениями характеризуется слабым негативным воздействием на окружающую среду и отвечает основным принципам современной “зеленой” химии [22].
Ранее нами был разработан метод электросинтеза тиолов, симметричных моно-, ди- и полисульфидов на основе циклоалканов C5–C8 (циклоалкенов C5, C6) и сероводорода, активированного различными способами (редокс-активацией и с применением медиаторов) [23–29]. Информация об электрохимических способах получения асимметричных моно- и дисульфидов на основе циклоалканов C5–C8 с участием H2S и изомерных дибутилдисульфидов в современных литературных источниках отсутствует.
Цель настоящих исследований – разработка способа синтеза органических моно- и дисульфидов на основе реакций циклоалканов C5–C8 с ди(н-бутил)-/ди(трет-бутил)дисульфидом и анодно-активируемым H2S, а также установление зависимости выхода полученных органических производных серы от реакционной способности циклоалканов C5–C8 различного строения.
МЕТОДИКА ЭКСПЕРИМЕНТА
В работе использовали коммерчески доступные реактивы: циклопентан, циклогексан, циклогептан, циклооктан, метилциклогексан, 1,2‑диметилциклогексан, этилциклогексан, ди(н-бутил)дисульфид, ди(трет-бутил)дисульфид (98%, “Aldrich”), гексан (95%, “Alfa Aesar”) без дополнительной очистки. Сероводород получали по методике [30]. Очистку CH2Cl2 (“х. ч.”) осуществляли по известной методике [31]. Фоновый электролит – 0.15 М Bu4NClO4 (99%, “Acros”), дважды перекристаллизованный из водного EtOH и высушенный в вакууме (48 ч) при 50°С.
Метод циклической вольтамперометрии (ЦВА) использовали для анализа смеси продуктов электролиза и определения редокс-потенциалов соединений [32]. Анализ проводили с помощью потенциостата “IPC-Pro” (Россия) в бездиафрагменной трехэлектродной ячейке, в среде аргона, по методикам, описанным в [29, 30]. Рабочий электрод – стационарный дисковый Pt-электрод (D = 3 мм); вспомогательный электрод – платиновая пластина (S = 36 мм2); электрод сравнения (Ag/AgCl/KCl) с водонепроницаемой диафрагмой. Скорость развертки потенциала 0.2 В с–1.
Микроэлектролиз смеси (циклоалкан + H2S + + (t-C4H9)2S2 ((п-C4H9)2S2)) (1.5–2.0 ч) проводили в потенциостатическом режиме на платиновых электродах (S = 36 мм2) в бездиафрагменной трехэлектродной ячейке (2 мл), в CH2Cl2, при 25°C в анаэробной среде (аргон). Мольное соотношение сероводород : циклоалкан : дибутилдисульфид – 5.0 : 2.5 : 1.0. Содержание сероводорода в электрохимической ячейке составляло (ν(H2S) = 30 ммоль). Сероводород вводили в реакционную среду через 0.5 ч в виде насыщенного раствора в CH2Cl2 (20 мкл). Концентрацию H2S определяли гравиметрическим методом по реакции с Pb(CH3COO)2. Значение потенциала электролиза составляло 1.90 В (соответствует потенциалу окисления H2S).
Препаративный электролиз (12 мл) смеси (циклоалкан + H2S + (t-C4H9)2S2 ((п-C4H9)2S2)) в CH2Cl2 проводили на платиновых электродах (S = = 60 мм2) в течение 3 ч. Скорость подачи сероводорода составляла 2–3 мл/мин, что обеспечивало заданную концентрацию H2S в электрохимической ячейке. В ходе электролиза плотность тока поддерживали в диапазоне 10 мА см–2. Реакционную смесь после электролиза дегазировали током аргона в течение 30 мин, далее концентрировали в вакууме. Фоновый электролит осаждали гексаном. Смесь органических производных серы и непрореагировавший дибутилдисульфид выделяли трехступенчатой экстракцией гексаном, далее экстракт концентрировали в вакууме.
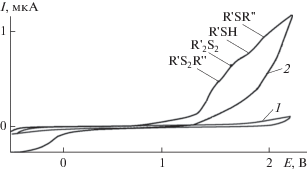
Для идентификации полученных органических соединений серы использовали методы циклической вольтамперометрии, ИК-спектроскопии, хроматомасс-спектрометрии и рентгено-флуоресцентного анализа. На циклических вольтамперограммах окисления продуктов электролиза фиксировали четыре анодных пика: R'S2R" (1.52–1.56 В), ${\text{R}}_{{\text{2}}}^{'}$S2 (1.66–1.69 В), R'SH (1.78–1.82 В), R'SR" (1.95–1.99 В). В трехкомпонентных реакциях расчет выхода продуктов электролиза проводили на прореагировавший сероводород. Неорганические полисульфаны (H2Sn) с различной молекулярной массой регистрировали методом ЦВА в диапазоне потенциалов 0.2–1.3 В; серу контролировали в катодной области (–1.2 В).
ИК-спектры продуктов электролиза регистрировали на ИК-фурье-спектрометре ФСМ-1201 (Россия) в таблетках KBr в диапазоне от 400 до 4000 см–1. В ИК-спектрах фиксировали валентные колебания связей: S–S (507–520 см–1), C–S (690–710 см–1) и S–H (2550–2600 см–1). Анализ смеси продуктов реакции проводили методом газовой хроматомасс-спектрометрии на приборе GCMS-QP2010 Ultra (Shimadzu, Япония) с детектором (EI, 70 эВ). Капиллярная колонка SPB-1 SULFUR (30 м × 0.32 мм), tmax = 320°С. Газ-носитель – гелий. Температурный режим колонки программировали в диапазоне от 30 до 280°С. В масс-спектрах фиксировали молекулярные ионы m/z (I, %):
С5Н9SH (m/z = 102 (M+, 40), 75 (15), 69 (100), 53 (20), 41 (60));
С6Н11SH (m/z = 116 (M+, 25), 83 (23), 67 (40), 55 (100), 45 (20));
С7Н13SH (m/z = 130 (M+, 20), 97 (35), 81 (50), 67 (45), 55 (100), 39 (30));
С8Н15SH (m/z = 144 (M+, 15), 142 (25), 110 (25), 82 (40), 69 (100), 55 (35), 41 (35));
(С5Н9)2S2 (m/z = 202 (M+, 11), 134 (21), 69 (100));
(С6Н11)2S2 (m/z = 230 (M+, 14), 147 (16), 83 (100), 55 (27));
(С7Н13)2S2 (m/z = 260 (M+, 13), 163 (17), 130 (10), 97 (100));
n-C4H9SC5H9 (m/z = 159 (M+, 48), 134 (26), 115 (23), 91 (20), 82 (100), 67 (78), 53 (20), 41 (68));
n-C4H9SC6H11 (m/z = 172 (M+, 52), 129 (18), 115 (32), 91 (20), 82 (100), 67 (78), 55 (75), 41 (66)).
n-C4H9S2C5H9 (m/z = 191 (M+, 44), 122 (49), 115 (42), 91 (20), 87 (5), 67 (78), 57 (100), 41 (33));
t-C4H9S2C6H11 (m/z = 204 (M+, 56), 126 (18), 115 (27), 91 (20), 87 (8), 67 (78), 57 (100), 41 (66)).
Содержание общей серы определяли рентгено-флуоресцентным методом на спектрометре АСЭ-1 (Россия).
Квантовохимические расчеты проводили с использованием программы Hyper Chem 8.0 методом функционала плотности (B3LYP/6-31++G(d,p)). Влияние растворителя (CH2Cl2) учитывали с помощью модели поляризуемого континуума (PCM). Энергетические эффекты реакций (ΔH) рассчитывали как разность полных энергий конечных и исходных структур.
Компьютерный прогноз потенциальной биологической активности полученных органических соединений серы производили с помощью программы PASS.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Электрохимические реакции сероводорода с незамещенными и алкилзамещенными циклоалканами C5–C8I–VII (схема 1 ) проводили в присутствии ди(н-бутил)дисульфида (ди(трет-бутил)дисульфида) в CH2Cl2.
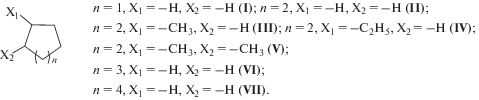
Схема 1 .
Алициклические соединения I–VII инертны в исследуемом диапазоне анодных потенциалов (до 2.4 В). В связи с этим в условиях электролиза трехкомпонентной смеси при потенциале 1.9 В анодной активации подвергаются реагенты – сероводород (1.70 В) и изомерные дисульфиды: (t‑С4Н9)2S2 (1.42 В), (n-С4Н9)2S2 (1.54 В). Окисление Н2S протекает в одноэлектронную стадию с образованием нестабильного катион-радикала, который фрагментируется с отщеплением протона и образованием тиильного радикала (схема 2 ).
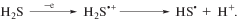
Схема 2 .
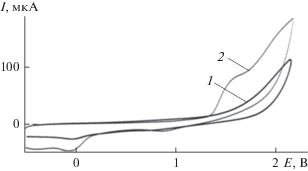
Для дибутилдисульфидов также характерно одноэлектронное окисление (схема 3 , рис. 1).
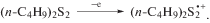
Схема 3 .
Рис. 1.
ЦВА-кривая окисления: 1 – n-Bu4NClO4 (c = = 0.015 М), 2 – (п-C4H9)2S2 (c = 0.005 М) (CH2Cl2, Pt‑анод, Ag/AgCl, v = 0.2 В с–1).
Этот факт позволил предположить возможность катион-радикалов изомерных дибутилсульфидов выступать в роли медиаторов окисления H2S (схема 4 ).
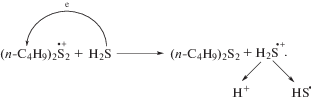
Схема 4 .
Ранее нами было обнаружено, что при прямом и непрямом (с применением медиаторов) электрохимическом окислении H2S образуются неорганические сульфаны H2Sn (n = 2–8), которые с увеличением продолжительности электролиза подвергаются циклизации в элементную серу [33, 34].
После электролиза смеси (H2S + (t-C4H9)2S2 ((п-C4H9)2S2)) при потенциале окисления дисульфидов, в отсутствие циклоалканов I–VII (1.5 ч), не фиксировали сульфанов и серы. На циклических вольтамперограммах (ЦВА-кривых) окисления продуктов электролиза наблюдали пики восстановления сероцентрированных катионов при потенциале –(0.48–0.52) В в зависимости от строения бутильного радикала.
Следовательно, ввиду неустойчивости катион-радикалов изомерных дибутилсульфидов и их способности к димеризации, генерирование тиильных радикалов из H2S с участием (t-C4H9)2S2 ((п-C4H9)2S2) затруднено.
Проведение реакций циклоалканов I–VII с (t‑C4H9)2S2 ((п-C4H9)2S2) в условиях электролиза при потенциале окисления дибутилсульфидов показало отсутствие бутилциклоалкилсульфидов. На следующем этапе исследования было изучено инициирование исследуемых трехкомпонентных реакций тиильным радикалом, образующимся при прямом одноэлектронном окислении сероводорода.
Электролиз (1.5–3 ч) смеси (H2S + циклоалкан + + (t-C4H9)2S2 ((п-C4H9)2S2)) проводили при 25°С. Реакция протекала при недостатке дибутилсульфида по сравнению с H2S, что связано с участием (t-C4H9)2S2 ((п-C4H9)2S2) в инициировании промежуточной стадии превращений.
Реакции анодно-активируемого H2S и изомерных дибутилсульфидов с циклоалканами I–VII протекают по радикальному механизму с образованием смеси продуктов реакции (циклоалкантиола (R′SH), моно-(R'SR") и дисульфида (R'S2R") асимметричного строения и симметричного дисульфида (${\text{R}}_{{\text{2}}}^{'}$S2). Полученные соединения идентифицировали методами циклической вольтамперометрии (рис. 2), ИК-спектроскопии, и хроматомасс-спектрометрии.
Рис. 2.
ЦВА-кривые окисления: 1 – n-Bu4NClO4 (c = = 0.015 М), 2 – продуктов реакции H2S и ди(н-бутил)дисульфида с циклогептаном (τ = 3 ч, CH2Cl2, Pt‑анод, Ag/AgCl, v = 0.2 В с–1).
Электрохимические превращения, протекающие через ряд последовательно-параллельных стадий, аналогичны для всех изученных соединений I–VII и описываются следующим образом (схема 5 на примере (t-C4H9)2S2).
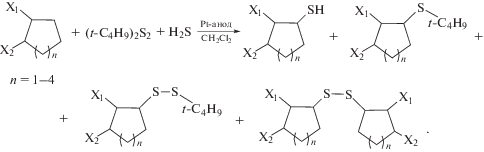
Схема 5 .
Генерируемый тиильный радикал атакует алицикл, что ведет к получению циклоалкантиолов как первичных продуктов реакции (схема 6 ).
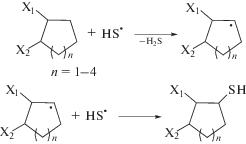
Схема 6 .
Полученные продукты тиолирования циклоалканов C5–C8 при потенциале электролиза способны к электрохимическому одноэлектронному окислению до дисульфидов симметричного строения (схема 7 ).
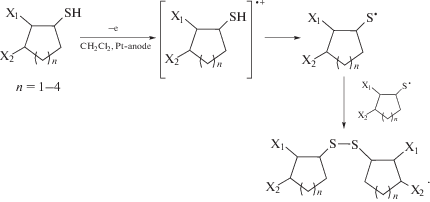
Схема 7 .
Образующийся циклоалкильный радикал далее взаимодействует с (t-C4H9)2S2, при этом генерируется трет-бутилтиильный радикал. Реакция диспропорционирования далее приводит к получению асимметричных моно- и дисульфидов (схема 8 ).
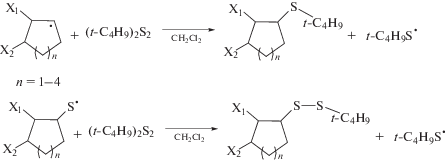
Схема 8 .
Полученные результаты исследований коррелируют с имеющимися данными по электрохимическим превращениям тиолов и дисульфидов [35].
Для подтверждения механизма изученных реакций (на примере циклогексана с H2S и (п‑C4H9)2S2/(t-C4H9)2S2) были проведены квантовохимические расчеты. В связи с тем, что тиильный радикал может, наряду с циклогексаном, атаковать дибутилдисульфид, была произведена оценка участия HS• в генерировании циклогексильного и бутилтиильного радикалов. Оказалось, что значения теплового эффекта в первом случае превышает расчетную величину для второго направления превращений на 58.1 ((п-C4H9)2S2) и 45.6 кДж/моль ((t-C4H9)2S2). Следовательно, инициирование трехкомпонентной реакции происходит за счет атаки циклогексана тиильным радикалом. Стадия генерирования бутилтиильного радикала при взаимодействии (п-C4H9)2S2 ((t-C4H9)2S2) с циклогексильным радикалом более термодинамически выгодна, чем реакция с циклогексилтиильным радикалом – на 44.0 (29.3) кДж/моль (схема 8 ). Это значение свидетельствует о том, что образование асимметричного дисульфида энергетически более затруднено по сравнению с формированием бутилциклогексилсульфида.
Результаты проведения электролиза смеси ((t‑С4H9)2S2 + H2S + циклоалкан С5–С8), позволившего получить R'S2R" (1.52–1.56 В), ${\text{R}}_{{\text{2}}}^{'}$S2 (1.66–1.69 В), R'SH (1.78–1.82 В), R'SR" (1.95–1.99 В), представлены в табл. 1.
Таблица 1.
Выход продуктов реакции H2S, ди(трет-бутил)дисульфида с циклоалканами С5–С8 (t = 25°С, CH2Cl2, τ = 180 мин, Еэл = 1.9 В)
| Соединения | Выход полученных соединений η, % | ||||
|---|---|---|---|---|---|
| R'S2R'' | ${\text{R}}_{2}^{'}$S2 | R'SH | R'SR'' | Σ | |
| I | 12.9 | 4.7 | 9.4 | 14.0 | 41.0 |
| II | 11.2 | 8.8 | 16.2 | 17.9 | 54.1 |
| III | 11.7 | 5.3 | 20.6 | 10.7 | 48.3 |
| IV | 14.9 | 5.6 | 14.5 | 25.8 | 60.8 |
| V | 6.1 | 5.8 | 8.9 | 7.7 | 28.5 |
| VI | 11.1 | 6.6 | 7.5 | 10.1 | 30.8 |
| VII | 2.5 | 6.6 | 9.9 | 5.3 | 24.3 |
Из табл. 1 следует, что размер алицикла влияет на активность субстрата и общий выход продуктов реакции. Наибольший суммарный выход серосодержащих соединений достигался в реакции H2S и (t-С4Н9)2S2 с этилциклогексаном, что объясняется протеканием параллельных реакций радикального замещения по алициклу и в заместителе. В ряду незамещенных циклоалканов по сравнению с их алкилпроизводными выход циклоклантиолов оказался ниже, при соизмеримом суммарном выходе серосодержащих продуктов реакции. В реакции тиолирования также активно участвовали незамещенные циклогексан и его монопроизводные. Достаточно высокий общий выход серосодержащих соединений в случае циклогексана был соизмеримым с данной величиной для метиллциклогексана. В случае диметилпроизводного циклогексана наблюдалось заметное снижение выхода продуктов реакции, что определяется стерическим фактором, определяющим реакционную способность субстрата при его атаке тиильным радикалом. Во всех рассмотренных взаимодействиях выход асимметричных сульфидов преобладал над концентрацией дисульфидов аналогичного строения. При этом выход дисульфидов симметричного строения для всех изученных соединений оказался ниже, чем содержание R'SR" и R'S2R". Для циклооктана наблюдался низкий выход продуктов димеризации циклоалкилтиильных радикалов и их рекомбинации с трет-бутильными радикалами. Следовательно, размер и степень насыщения алицикла влияют на выход полученных серосодержащих соединений.
Результаты электрохимического эксперимента согласуются с данными квантовохимических расчетов. Так, суммарный выход продуктов реакции (табл. 1) оказался выше для С5Н10 (41.0%) по сравнению с С7Н14 (30.8%) при одинаковом значении конверсии (t-C4H9)2S2 (≈79.0%), что подтверждается разницей значений теплового эффекта реакций (ΔН) тиильного радикала с соответствующими циклоалканами (5.7 кДж/моль) в пользу гомолога С5.
Степень превращения (п-C4H9)2S2 также варьируется примерно в аналогичных пределах – от 60 до 85%. Селективность реакции ди(трет-бутил)дисульфида с незамещенными циклоалканами С5–С8 в присутствии H2S по двум направлениям – образование асимметричных сульфида и дисульфида, также зависит от строения субстрата (рис. 3).
Рис. 3.
Зависимость селективности реакций образования асимметричных моно- и дисульфидов при взаимодействии H2S и ди(трет-бутил)дисульфида с циклоалканами C5–C8 от размера алицикла.
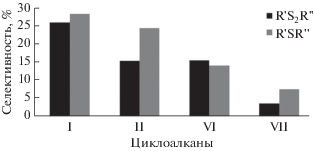
Как следует из диаграммы, с увеличением размера алицикла снижается вероятность образования моно- и дисульфидов асимметричного строения. Незамещенные циклоалканы по величине реакционной способности в рассматриваемых трехкомпонентных превращениях можно расположить в следующий ряд: С6Н12 > С5Н10 > С7Н14 > > С8Н16.
На примере реакции 1,2-диметилциклогексана с Н2S в присутствии ди(n-бутил)дисульфида изучена зависимость выхода и соотношения серосодержащих продуктов реакции от продолжительности электролиза (табл. 2).
Таблица 2.
Зависимость выхода продуктов реакции 1,2-диметилциклогксана с H2S, (n-С4Н9)2S2 от времени (t = 25°С, CH2Cl2, Еэл = 1.9 В)
| t, мин | Выход полученных соединений η, % | ||||
|---|---|---|---|---|---|
| R′S2R′′ | ${\text{R}}_{2}^{'}{{{\text{S}}}_{2}}$ | R′SH | R′SR′′ | Σ | |
| 60 | 2.1 | 2.5 | 5.2 | 3.1 | 12.9 |
| 120 | 5.5 | 4.0 | 8.3 | 4.8 | 22.7 |
| 180 | 7.8 | 4.6 | 10.0 | 5.4 | 27.8 |
Из табл. 2 видно, что в течение 180 мин суммарный выход продуктов реакции увеличивался. Наибольший выход характерен для стадии образования циклоалкантиола, являющегося промежуточным продуктом реакции. Заметное накопление R'SH в ходе электролиза свидетельствует о высокой скорости стадии тиолирования субстрата. Соотношения R'SH : ${\text{R}}_{{\text{2}}}^{'}$S2 (≈1.0 : 0.5) и R'SH : R'SR" (≈1.0 : 0.6) оставались во времени постоянными, что объясняется участием циклоалкильного радикала в обоих направлениях превращений циклоалкантиолов. В присутствии (n-С4Н9)2S2 доминировала реакция диспропорционирования с участием циклоалкилтиильных радикалов в отличие от димеризации последних. Для рассмотренной трехкомпонентной реакции увеличение продолжительности электросинтеза повышало выход асимметричных моно- и дисульфидов.
В ходе электролиза смеси (H2S + 1,2-диметилциклогексан) в присутствии (п-C4H9)2S2 соотношение продуктов реакции составляло – R'S2R" : : ${\text{R}}_{{\text{2}}}^{'}$S2 : R'SH : R'SR" ≈ 0.8 : 0.5 : 1 : 0.6. При использовании (t-С4Н9)2S2 баланс изменялся (0.7 : 0.7 : 1 : 0.9) по сравнению с применением (n-С4Н9)2S2 в пользу образования асимметричного сульфида, что, вероятно, связано с более высокой стабильностью трет-бутильного радикала. В случае (n‑С4Н9)2S2 соотношение R'S2R" и ${\text{R}}_{{\text{2}}}^{'}$S2 было смещено в пользу асимметричного дисульфида, а в реакции с участием (t-С4Н9)2S2 выход двух соединений оказался соизмеримым. Экспериментальные данные для соединения V и двух изомерных дисульфидов (табл. 1, 2) указывают на равноценный суммарный выход продуктов реакции, при этом наблюдалось различное соотношение компонентов смеси. В присутствии (t-С4Н9)2S2 селективность по каждому направлению с увеличением продолжительности реакции (от 1.5 до 2 ч) повышалась следующим образом: R'S2R'' – в 1.3 раза, ${\text{R}}_{{\text{2}}}^{'}$S2 – в 2.3 раза, R'SH – в 1.2 раза и R'SR" – в 1.6 раз. Таким образом, наибольшей селективностью отличаются два пути реакции – образование симметричного дисульфида и асимметричного сульфида. Этот факт объясняется тем, что оба соединения формируются за счет стадий димеризации или рекомбинации радикалов, достаточно выгодных с позиций энергетических затрат. Следует отметить, что степень превращения изомерных ди(трет-бутил)дисульфидов отличалась для реакций H2S с соединениями I–VII, что согласуется со значениями выхода полученных R'S2R" и R'SR".
При возрастании продолжительности электролиза смеси (циклоалкан + H2S + (t-C4H9)2S2 ((п-C4H9)2S2)) образуются неорганические полисульфаны Н2Sn (0.3–1.2 В) с различным содержанием атомов серы (n = 2–5) и элементная сера, как побочные продукты исследуемых превращений.
Среди полученных серосодержащих продуктов трехкомпонентных реакций, с точки зрения применения в качестве компонентов лекарственных препаратов, наибольший интерес вызывают асимметричные моно- и дисульфиды. В связи с этим, для данных соединений с помощью компьютерной программы PASS была произведена оценка их потенциальной биологической активности. Оказалось, что R'SR" и R'S2R" способны проявлять противоопухолевую активность, их возможно использовать в качестве мукомембранных протекторов, ингибиторов широкого ряда ферментов: супероксид-дисмутазы, тиоредоксина, ацилкарнитин-гидролазы, гастрина, cахар-фосфатазы и ацетил-эстеразы.
ЗАКЛЮЧЕНИЕ
Таким образом, электрохимическая окислительная активация H2S позволила провести трехкомпонентные реакции с ди(н-бутил)дисульфидом (ди(трет-бутил)дисульфидом) и циклоалканами С5–С8 в мягких условиях (атмосферное давление и комнатная температура). Используемый в работе прием генерирования реакционноспособного интермедиата – тиильного радикала окислением сероводорода на платиновом аноде, оказался идеальным инструментом для эффективного проведения одностадийного процесса тиолирования циклоалканов С5–С8. В результате изученных электрохимических реакций получены циклоалкантиолы, дибутилциклоалкилсульфиды и дициклоалкилдисульфиды. Суммарный выход органических производных серы для циклоалканов различного строения варьируется от ≈24 до 60%. В качестве наиболее перспективных соединений из ряда циклоалканов С5–С8 для целенаправленного синтеза асимметричных моно- и дисульфидов рекомендованы циклогексан и этилциклогексан. Эффективность разработанного метода электросинтеза определяется строением дибутилдисульфида, размером и степенью насыщения алицикла, а также продолжительностью реакции. Основными преимуществами предложенного подхода является его доступность и экологическая безопасность, что обеспечивается исключением использования химических инициаторов реакции (путем замены их на анодную активацию H2S) и снижением энергозатрат по сравнению с имеющимися синтетическими способами, реализуемыми при повышенной температуре.
Список литературы
Feng, M., Tang, B., and Liang, S., Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry, Curr. Top. Med. Chem., 2016, vol. 16, no 11, p. 1200. https://doi.org/10.2174/1568026615666150915111741
Parcell, S., Sulfur in human nutrition and applications in medicine, Altern Med Rev., 2002, vol. 7, no. 1, p. 22.
Hayashida, R., Kondo, K., Morita, S., Unno, K., Shintani, S., Shimizu, Y., Calvert, J.W., Shibata, R., and Murohara, T., Diallyl trisulfide augments ischemia-induced angiogenesis via an endothelial nitric oxide synthase-dependent mechanism, Circulation J., 2017, vol. 81, no. 6, p. 870. https://doi.org/10.1253/circj.CJ-16-1097
Marwan, S. and Al-Nimer, M., Hydrogen sulfide donors or related derivatives are the future medicines of renal diseases, Egypt. Pharmaceut. J., 2017, vol. 16, no. 1, p. 1. https://doi.org/10.4103/1687-4315.205827
St-Gelais A., Legault J., Mshvildadze V., and Pichette A., Dirchromones: Cytotoxic Organic Sulfur Compounds Isolated from Dirca palustris, J. Nat. Prod., 2015, vol. 78, no. 8, p. 1904. https://doi.org/10.1021/acs.jnatprod.5b00227
Jiang, X., Sulfur atom transfer (SAT) reaction, J. Phosphorus, Sulfur, and Silicon and the Related Elements, 2017, vol. 192, no. 2, p. 169. https://doi.org/10.1080/10426507.2016.1250762
Gu, X. and Zhu, Y.Z., Therapeutic applications of organosulfur compounds as novel hydrogen sulfide donors and/or mediators, Expert. Rev. Clin. Pharmacol., 2011, vol. 4, no. 1, p. 123. https://doi.org/10.1586/ecp.10.129
Cerella, C., Dicato, M., Jacob, C., and Diederich M., Chemical properties and mechanisms determining the anti-cancer action of garlic-derived organic sulfur compounds, Anti-Cancer Agents in Medic. Chem., 2011, V. 11, no. 3, p. 267. https://doi.org/10.2174/187152011795347522
Pluth, M.D., Bailey, T.S., Hammers, M.D., Hartle, M.D., Henthorn, H.A., and Steiger, A.K., Natural Products Containing Hydrogen Sulfide Releasing Moieties, Synlett, 2015, vol. 26, no. 19, p. 2633. https://doi.org/10.1055/s-0035-1560638
Hosgood, S.A. and Nicholson, M.L., Hydrogen sulphide ameliorates ischaemia-reperfusion injury in an experimental model of non-heart-beating donor kidney transplantation, British J. Surgery, 2010, vol. 97, no. 2, p. 202. https://doi.org/10.1002/bjs.6856
Misak, A., Grman, M., Bacova, Z.b, Rezuchova, I., Hudecova, S., Ondriasova, E., Krizanova, O., Brezova, V., Chovanec, and M., Ondrias, K., Polysulfides and products of H2S/S-nitrosoglutathione in comparison to H2S, glutathione and antioxidant Trolox are potent scavengers of superoxide anion radical and produce hydroxyl radical by decomposition of H2O2, Nitric Oxide – Biology and Chemistry, 2018, vol. 76, no. 1, p. 136. https://doi.org/10.1016/j.niox.2017.09.006
Kang, J., Xu, S., Radford, M.N., Zhang, W., Kelly, S.S., Day, J.J., and Xian, M., O→S Relay Deprotection: A General Approach to Controllable Donors of Reactive Sulfur Species, Angewandte Chemie – International Editionб, 2018, vol. 57, no. 20, p. 5893. https://doi.org/10.1002/anie.201802845
Kovácsa, S. and Novák, S., Oxidoreductive coupling of thiols with aryl halides catalyzed by copper on iron, Organic & Biomolec. Chem., 2011, vol. 9, p. 711. https://doi.org/10.1039/c0ob00397b
Singh, G., Nakade, P.G., Mishra, P., Jha, P., Sen, S., and Mondal, U., Kinetic investigation on liquid–liquid–solid phase transfer catalyzed synthesis of dibenzyl disulfide with H2S-laden monoethanolamine, J. Molec. Catal. A: Chemical, 2016, vol. 411, p. 78. https://doi.org/10.1016/j.molcata.2015.10.013
Yu, B., Diao, Z.-F., Liu, A.-H., Han, X., Li, B., He, L.-N., and Liu, X.-M., Selective Oxidation of Sulfides to Sulfoxides with Tert-Butylnitrite as an Alternative Oxidant, Curr. Organic Synthesis, 2014, vol. 11, no. 1, p. 156. https://doi.org/10.2174/1570179411999140304142430
Van, L.K., Hamilton, C.J., and Messens, J., Low-molecular-weight thiols in thiol-disulfide exchange, Antioxid Redox Signa, 2013, vol. 18, no. 13, p. 1642. https://doi.org/10.1089/ars.2012.4964
Guo, S., He, W., Xiang, J., and Yuan, Ya., Ruthenium-catalyzed direct thiolation of alkanes and ethers using arylsulfonyl chlorides as a sulfur source, Tetrahedron Lett., 2015, vol. 56, no. 17, p. 2159. https://doi.org/10.1016/j.tetlet.2015.01.068
Zhao, J., Fang, H., Song, R., Zhou, J., Han, J., and Pan, Y., Metal-free oxidative C(sp3)–H bond functionalization of alkanes and alkylation-initiated radical 1,2-aryl migration in α,α-diaryl allylic alcohols, Chem. Commun., 2015, vol. 51, no. 3, p. 599. https://doi.org/10.1039/C4CC07654K
Guo, S., He, W., Xiang, J., and Yuan, Ya., Palladium-catalyzed thiolation of alkanes and ethers with arylsulfonyl hydrazides, Chem. Commun., 2014, vol. 50, p. 8578. https://doi.org/10.1039/C4CC02876G
Saravanan, P. and Anbarasan, P., Palladium Catalyzed Aryl(alkyl)thiolation of Unactivated Arenes, Org. Lett., 2014, vol. 16, no. 3, p. 848. https://doi.org/10.1021/ol4036209
Mishra, P., Kumari, S., and Sen, S., Kinetic modeling on ionic liquid mediated bi-liquid phase transfer catalyzed synthesis of bis-(2-phenylethyl) sulfide with H2S-rich methyldiethanolamine, J. Molec. Liquids, 2018, vol. 271, p. 580. https://doi.org/10.1016/j.molliq.2018.09.038
Моисеев, И.И. “Зеленая химия”: траектория развития // Успехи химии. 2013. Т. 82. №7. С. 616. [Moiseev, I.I. “Green Chemistry”: the trajectory of development, Russ. Chem. Rev., 2013, vol. 82, no. 7, р. 616 (in Russian).] https://doi.org/10.1070/RC2013v082n07ABEH004393
Берберова, Н.Т., Шинкарь, Е.В., Смолянинов, И.В., Абдулаева, В.Ф. Анодная активация сероводорода в реакции с циклопентаном. Журн. общей химии. 2015. Т. 85. № 4. С. 697. [Berberova, N.T., Shinkar, E.V., Smolyaninov, I.V., and Abdulaeva, V.F., Anodic activation of hydrogen sulfide in reaction with cyclopentane, Russ. J. General Chem., 2015, vol. 85, no. 4, p. 998.] https://doi.org/10.1134/S1070363215040416
Шинкарь, Е.В., Швецова, А.В., Седики, Д.Б., Берберова, Н.Т. Редокс-активация сероводорода в реакции с циклогептаном. Электрохимия. 2015. Т. 51. № 11. С. 1182. [Shinkar, E.V., Shvetsova, A.V., Sediki, D.B., and Berberova, N.T., Redox activation of hydrogen sulfide in the reaction with cycloheptane, Russ. J. Electrochem., 2015, vol. 51, no. 11, p. 1046 (in Russian).] https://doi.org/10.1134/S1023193515110178
Берберова, Н.Т., Шинкарь, Е.В., Смолянинов, И.В., Швецова, А.В., Cедики, Д.Б. Электросинтез биологически активных дициклоалкилди- и трисульфидов с участием редокс-системы H2S–S8. Изв. РАН. Cер. хим. 2018. V. 67. № 1. С. 108. [Berberova, N.T., Shinkar, E.V., Smolyaninov, I.V., Shvetsova, A.V., and Sediki, D.B., Electrosynthesis of biologically active dicycloalkyl di- and trisulfides involving an H2S–S8 redox system, Russ. Chem. Bull., 2018, vol. 67, no. 1, p. 108 (in Russian).] https://doi.org/10.1007/s11172-018-2044-4
Берберова, Н.Т., Шинкарь, Е.В., Смолянинов, И.В., Пащенко, К.П. Редокс-медиаторы окисления сероводорода в реакциях с циклоалканами. Докл. Акад. наук. 2015. Т. 465 № 6. С. 1. [Berberova, N.T., Shinkar, E.V., Smolyaninov, I.V., and Pashchenko, K.P., Redox-mediators of hydrogen sulfide oxidation in reactions with cycloalkanes, Dokl. Chem., 2015, vol. 465, no. 2, p. 295 (in Russian).] https://doi.org/10.1134/S0012500815120058
Берберова, Н.Т., Шинкарь, Е.В., Смолянинов, И.В., Кузьмин, В.В., Швецова, А.В., Cедики, Д.Б. 3,6-Ди-трет-бутил-о-семихинолятные комплексы Сr(III), In(III) как редокс-медиаторы окисления сероводорода в реакциях с циклоалканами, Координационная химия. 2017. Т. 43. № 9. С. 540. [Berberova, N.T., Shinkar’, E.V., Smolyaninov, I.V., Kuz’min, V.V, Shvetsova, A.V., and Sediki, D.B., 3,6-Di-tert-butyl-o-seminquinolate complexes Cr(III), In(III) as redox-mediators of hydrogen sulfide oxidation in reactions with cycloalkanes, Russ. J. Coordinat. Chem., 2017, vol. 43, no. 9, p. 578 (in Russian).] https://doi.org/10.1134/S107032841707003X
Shinkar’, E.V., Kudryavtsev, D. A., Pashchenko, K.P., Berberova, N.T., and Okhlobystina, A.V., Thiolation of cycloalkenes C5, C6 by redox-activation of hydrogen sulfide, Mendeleev Commun., 2017, vol. 27, p. 1. https://doi.org/10.1016/j.mencom.2017.03.025
Берберова, Н.Т. Сероводород и алкантиолы в синтезе биологически активных органических соединений серы / Берберова, Н.Т., Шинкарь, Е.В., Смолянинов, И.В., Охлобыстина, А.В. Ростов н/Д: Изд-во ЮНЦ РАН. 2016. 260 с. [Hydrogen sulfide and alkanethiols in the synthesis of biologically active organic sulfur compounds / Berberova, N.T., Shinkar, Ye.V., Smolyaninov, I.V., and Okhlobystina, A.V., Rostov-on-Don: Southern Scientific Center of RAS, 2016, 260 p. (in Russian).]
Летичевская, Н.Н., Шинкарь, Е.В., Береберова, Н.Т., Охлобыстин, О.Ю. Катион-радикал сероводорода в роли сверхкислоты. Журн. общей химии. 1996. Т. 66(11). С. 1785. [Letichevskaya, N.N., Shinkar’, E.V., Berberova, N.T., and Okhlobystin, O.Yu., Radical Cation of Hydrogen Sulfide as a Superacid, Russ. J. General Chem., 1996, vol. 66, no. 11, p. 1739 (in Russian).]
Gordon, A.J., and Ford R.A., The chemistґs companion. New York: A Wiley interscience publication, 1972, 541 p.
Органическая электрохимия / Под ред. Байзера М.М. и Лунда Х. М: Химия, 1988. 1023 с. [Organic electrochemistry / Eds Baizer, M.M. and Lund, H., New York, Basel: Marcel Dekker Inc., 1983. 1166 p.]
Берберова, Н.T., Шинкарь, Е.В. Катион-радикал сероводорода и его участием в органических реакциях. Изв. РАН. Cер. хим. 2000. Т. 49. № 7. С. 1182. [Berberova, N.T., Shinkar’, E.V., The radical cation of hydrogen sulfide and related organic reactions, Russ. Chem. Bull., 2000, vol. 49, no. 7, p. 1178 (in Russian).] https://doi.org/10.1007/BF02495758
Вовлечение сероводорода, тиолов и полисульфанов в синтез органических соединений серы / Берберова, Н.Т., Шинкарь, Е.В., Смолянинов, И.В., Охлобыстин, А.О., Ростов н/Д: Изд-во ЮНЦ РАН, 2009. 256 с. [The involvement of hydrogen sulfide, thiols and polysulfans in the synthesis of organic sulfur compounds / Berberova, N.T., Shinkar, E.V., Smolyaninov, I.V., Okhlobystin, A.O., Rostov-on-Don: Southern Scientific Center of RAS, 2009, 256 p. (in Russian).]
Organic electrochemistry. / Eds Lund, H. and Hammerich, O. N.Y.: Marcel Dekker Inc., 4th ed. 2001. P. 621.
Дополнительные материалы отсутствуют.