

Электрохимия, 2020, T. 56, № 5, стр. 407-415
Электровосстановление производных N,N'-диоксидов феназина и хиноксалина в неводных средах и в присутствии донора протонов средней силы
Л. В. Михальченко a, *, Д. В. Насыбуллина a, М. Ю. Леонова a, М. А. Сыроешкин a, В. П. Гультяй a
a Институт органической химии им. Н.Д. Зелинского, РАН
119991 Москва, Ленинский просп., 47, Россия
* E-mail: mlv@ioc.ac.ru
Поступила в редакцию 22.01.2019
После доработки 19.03.2019
Принята к публикации 06.09.2019
Аннотация
Методами циклической вольтамперометрии, хроноамперометрии и электролиза при контролируемом потенциале изучено электровосстановление (ЭВ) бензо[a]феназин-7,12-диоксида (1) и 2-этоксикарбонил-3-метил-хиноксалин-1,4-диоксид (2) в ДМФА на электроде из стеклоуглерода. В апротонной среде исследованные соединения восстанавливаются с образованием сравнительно устойчивых анион-радикалов. В присутствии СН3СООН соединения 1 и 2 образуют комплексы, которые зафиксированы как на кривых циклической вольтамперометрии, так и в УФ-спектрах. Показано, что дезоксигенирование производных N,N′-диоксидов феназина и хиноксалина происходит в результате распада радикала, образованного либо при ЭВ комплексов этих соединений с СН3СООН, либо в результате протонирования анион-радикалов. В случае соединения 2 наблюдается конкуренция реакций распада и ЭВ этого радикала.
ВВЕДЕНИЕ
Производные N,N′-диоксидов феназина и хиноксалина часто рассматриваются [1–3] в качестве потенциальных лекарственных средств для лечения “твердых” опухолей, имеющих большие участки гипоксии, куда почти не проникает кислород. Клетки в условиях гипоксии становятся устойчивыми к радиации и химиотерапии. Обычные химиотерапевтические препараты действуют на быстрорастущие канцерогенные клетки, препятствуя делению, и поэтому малоэффективны для клеток в условиях гипоксии, поскольку последние делятся медленно. Новые подходы к воздействию на опухоль разработаны на основе использования так называемых “предлекарств” [4, 5], которые способны восстанавливаться энзимами in vivo в условиях гипоксии, превращаясь в лекарства, поражающие канцерогенные клетки. Именно к таким биовосстанавливаемым соединениям относятся производные гетероароматических N-оксидов. Одним из первых введенных в клиническую практику препаратов, проявляющих высокую селективную цитотоксичность по отношению к клеткам, находящимся в условиях гипоксии, относится препарат тирапазамин – 1,3-амино-1,2,4-бензотриазин-1,4-N-диоксид [6, 7]. С использованием многих биологических методов исследования показано [8, 9], что одноэлектронное восстановление тирапазамина энзимами приводит к высоко реакционноспособному радикалу •ОН, разрушающему ДНК клеток опухоли. Принято считать, что дезоксигенирование предлекарства наиболее эффективно именно в условиях недостатка кислорода, так как кислород предотвращает восстановительный процесс в тканях. Вопросы о механизме окислительно-восстановительных реакций предлекарств и взаимодействия образующихся радикалов с ДНК до сих пор активно обсуждаются в литературе [9].
Последние годы наиболее перспективными соединениями с точки зрения получения предлекарств с высокой противоопухолевой активностью рассматриваются производные феназина [10, 11]. Образование радикала •ОН в результате биовосстановления производных диоксидов феназина, так же как и тирапазамина, связывают с активностью этих диоксидов как биологических агентов. Однако рассмотрение поведения производных диоксидов феназина с различными заместителями привело к выводу, что этот радикал образуется не во всех случаях [4]. Для систематизации знаний о свойствах потенциально активных соединений предпринимались попытки установить связь между противоопухолевой активностью и физико-химическими свойствами соединений, в частности с потенциалом восстановления [12, 13]. Далеко не все изученные структуры замещенных N,N′-диоксидов феназина подчинялись линейности этой корреляции, вероятно механизм реакций восстановления (или действия на клетки) для структур с различными заместителями может существенно различаться. Поскольку образование радикальных частиц связано с окислительно-восстановительными свойствами гетероароматических N,N-диоксидов, изучение стадий переноса электрона и приэлектродных химических реакций электрохимическими методами может быть полезным при определении критериев для оценки перспективности синтезируемых органических соединений.
В данной работе для получения данных о механизме восстановления ароматических диоксидов использованы циклическая вольтамперометрия (ЦВА), хроноамперометрия (ХА) и электролиз при контролируемом потенциале (ЭКП). В качестве модельных соединений для изучения электровосстановления (ЭВ) производных N,N′-диоксидов феназина и хиноксалина служили бензо[а]феназин-7,12-диоксид и 2-этоксикарбонил-3-метил-хиноксалин-1,4-диоксид. Поскольку в живой клетке осуществляются реакции не только переноса электронов, но и переноса протонов, представлялось необходимым выяснение влияния реакций протонирования на механизм действия предлекарств. Целью данного исследования является установления механизма дезоксигенирования исследованных соединений в модельных условиях: в апротонной среде (ДМФА) и в присутствии доноров протонов (CH3COOH).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Бензо[a]феназин-7,12-диоксид был синтезирован по методике [14]. Физико-химические характеристики полученного соединения аналогичны известным из литературы данным [15]. 2‑Этоксикарбонил-3-метил-хиноксалин-1,4-диоксид был получен из бензофуроксана и ацетоуксусного эфира [16] и идентифицирован по результатам масс-спектрометрии, 1Н и 13С ЯМР-спектрам сравнением с известными данными [17].
Хроноамперограммы и циклические вольтамперограммы были получены при помощи потенциостатов IPC“Pro”MF (“Эконикс”, Россия) и Р‑4 (“Элинс”, Россия). В качестве фонового электролита использовали растворы перхлората или тетрафторбората тетрабутиламмония (“Acros Organics”) в ДМФА (категории “extra dry”). Поляризационные кривые записывали с использованием трехэлектродной схемы. В качестве рабочего применяли электрод из стеклоуглерода (d = = 1.7 мм), вспомогательным электродом служила спираль из платиновой проволоки, а электродом сравнения – насыщенный каломельный электрод (нас. к. э.), соединенный с раствором через мостик с пористой керамической диафрагмой, заполненный фоновым электролитом. Для удаления кислорода из исследуемого раствора, через него пропускали аргон высокой чистоты перед записью каждой кривой ХА и ЦВА. Во время эксперимента над поверхностью раствора подавали аргон для предотвращения его контакта с воздухом. Поверхность рабочего электрода полировали перед съемкой каждой кривой. ЭКП проводили с использованием потенциостата по трехэлектродной схеме. Рабочий электрод – графитовый, площадью 4 см2, вспомогательный – платиновый, площадью 2 см2, электрод сравнения – нас. к. э. Катодное и анодное пространства были разделены стеклянной мембраной. Электролиз проводили при постоянном потенциале первой (–0.8 В) стадии ЭВ соединения 1 или при потенциале появляющейся в присутствии CH3COOH стадии при –1.0 В до исчерпания деполяризатора. Контроль содержания исходного соединения осуществляли по ЦВА-кривым. После окончания электролиза раствор разбавляли в 3–5 раз водой, выпавший осадок отфильтровывали и сушили. Выделенные продукты анализировали методами 1Н ЯМР-спектроскопии и масс-спектрометрии.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Как следует из сказанного во введении, противоопухолевая активность “биовосстанавливаемых агентов” напрямую связана с реакционной способностью продуктов их одноэлектронного восстановления. Поэтому наибольшее внимание было уделено электрохимическим и химическим реакциям, протекающим при потенциалах первой стадии ЭВ модельных соединений 1 и 2. Вначале мы рассмотрели электрохимическое поведение этих соединений в неводном растворителе, т.е. в условиях, когда на первой стадии они образуют достаточно стабильные анион-радикалы (АР). Результаты электрохимических измерений в апротонной среде представлены в табл. 1.
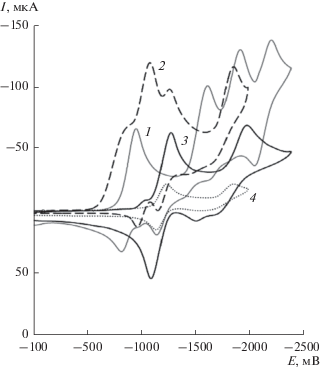
Первый катодный пик на кривых ЦВА соединения 1 обратим (рис. 1). Разность потенциалов между катодным и соответствующим ему анодным пиком ΔEp = Epc – Epa имеет значение (63 мВ), близкое к теоретическому (57 мВ) для обратимых реакций. Величина тока первого катодного пика соединения 1 почти равна значению тока одноэлектронного окисления ферроцена при тех же условиях (nцва = 0.81). По данным ХА величина предельного тока ЭВ соединения 1 на первой стадии отвечает переносу одного электрона на молекулу соединения 1 (nХА = 0.99). Отношение величин анодного и катодного токов на ЦВА-кривой первой стадии ЭВ соединения 1 ipa/ipc близко к единице при скорости наложения потенциалов v ≥100 мВ с–1.
Рис. 1.
ЦВА-кривые 5.0 мМ соединения 1 при 100 мВ с–1 в ДМФА (0.1 М Bu4NClO4) при различных потенциалах реверса.

Значение Ipc линейно изменяется с ростом концентрации соединения 1 в интервале от 3.9 до 18.8 мМ и корня квадратного из скорости наложения потенциалов $({{v}^{{1{\text{/}}2}}})$ до 1 В с–1. Графики этих зависимостей проходят через начало координат, что свидетельствует о диффузионном контроле образования на первой стадии ЭВ соединения 1 сравнительно стабильного в апротонной среде АР 1.
Следует отметить, что литературных данных о механизме ЭВ соединения 1 в апротонной среде не обнаружено. Опубликованы [13, 17] значения потенциалов ЭВ в ДМСО некоторых ароматических N,N′-диоксидов близкого строения, в частности замещенных бензо[a]феназин-7,12-диоксидов. Несколько подробнее изучено электрохимическое поведение феназин-N,N′-диоксида [18, 19]. При его ЭВ образуются сравнительно стабильные в апротонных органических растворителях АР, которые были зафиксированы методом ЦВА- и ЭПР-спектроскопией [18, 19]. Потенциал первой стадии ЭВ феназин-N,N′-диоксида в ДМФА, равный –0.85 В [19], не слишком отличается от определенного нами значения для соединения 1 (–0.91 В).
В условиях нескольких циклов в интервале от 0 до –1.2 В на последующих кривых ЦВА токи первого катодного пика и соответствующего ему анодного остаются приблизительно такими же, как и при первом сканировании, но около –1 В появляется сравнительно небольшой по величине новый катодный пик, который, как будет показано далее, совпадает по потенциалу с пиком ЭВ бензофеназин-N-монооксида – продукта восстановления соединения 1 в присутствии доноров протонов. На этом основании можно предположить, что в апротонных средах АР соединения 1 очень медленно протонируется следовыми количествами протонодонорных примесей в растворе с образованием электрохимически активных продуктов.
На второй стадии ЭВ соединения 1 (Е2 = –1.50 В) (рис. 1) происходит перенос второго электрона на молекулу 1 и образование соответствующего дианиона. Эта стадия ЭВ необратима, что связано с большей основностью дианиона по сравнению с АР. Дианион соединения 1 даже в практически безводном ДМФА быстро протонируется, что приводит к образованию электрохимически активных промежуточных продуктов, пики окисления которых наблюдаются на реверсивной части кривой ЦВА (рис. 1). Пики ЭВ при –1.78 и –2.00 В связаны с восстановлением скелета гетероароматической системы соединения 1.
Для выяснения роли доноров протонов при ЭВ 1 к раствору исследуемого соединения добавляли известное количество CH3COOH и снимали кривые ЦВА и ХА. При добавлении кислоты на кривых ЦВА пик окисления АР 1 при –0.85 В уменьшается (рис. 2).
Рис. 2.
ЦВА-кривые 2.2 мМ соединения 1 при $v$ = 100 мВ с–1 с добавлением CH3COOH. Концентрация CH3COOH, мМ: 0 (1), 0.94 (2), 1.9 (3), 2.8 (4), 4.4 (5).

Если концентрация кислоты меньше, чем концентрация соединения 1, то пик окисления его АР еще фиксируется на кривой ЦВА. Эквивалентное содержание донора протонов приводит к исчезновению этого пика. При содержании CH3COOH в количестве 1–2 эквивалента по отношению к концентрации исходного соединения 1, катодный ток первой стадии его ЭВ в данных условиях (–0.81 В) практически не меняется и составляет величину, приблизительно равную току первого катодного пика в апротонном ДМФА растворе. Иначе говоря, присутствие донора протонов не приводит к образованию электрохимически активных продуктов при потенциалах первой стадии ЭВ соединения 1. Однако при добавлении CH3COOH уже в количестве 1/4 эквивалента по отношению к исходной концентрации соединения 1 на кривых ЦВА регистрируются новые пики: Е = = –0.81, –1.05 и –1.25 В. Их токи растут с увеличением концентрации CH3COOH в растворе. При этом ток пика при потенциалах переноса второго электрона на молекулу деполяризатора Е = = ‒1.5 В, уменьшается (рис. 2, кривые 1–3), а в условиях равных концентраций донора протонов и соединения 1 эта стадия ЭВ на кривой ЦВА исчезает вовсе.
На рис. 3 (кривая 1) представлено изменение тока пика при –0.81 В на кривых ЦВА при добавлении CH3COOH.
Рис. 3.
Зависимость значения наблюдаемого числа электронов (n), участвующих в ЭВ соединений 1 (1) и 2 (2) при потенциалах первой стадии от концентрации CH3COOH по отношению к деполяризатору в растворе ДМФА при $v$ = 100 мВ с–1.

Величина тока нормирована (n = ipc/i0) на значение тока первой стадии ЭВ соединения 1 в апротонном ДМФА (i0), отвечающей току одноэлектронного переноса, а значение концентрации CH3COOH в растворе отнесено к концентрации исходного соединения. Как видно из рисунка, значение n вплоть до 4-кратного избытка кислоты близко к единице. Аналогичные данные получены методом ХА в результате обработки хроноамперограмм, полученных при потенциалах предшествующей стадии, растворов соединения 1 с добавлением 1–4 эквивалентов CH3COOH.
Полученные данные позволяют предполагать, что пик, появляющийся в присутствии донора протонов при –0.81 В, вероятно, отвечает ЭВ комплекса соединения 1 с уксусной кислотой. Известно [20], что образование донорно-акцепторных комплексов (молекулярных комплексов) и даже твердых солей с донорами протонов характерно для гетероароматических N-оксидов. Прочное связывание соединения 1 с CH3COOH за счет водородных связей, вероятно, приводит к снижению энергии низшей вакантной орбитали молекулярного комплекса и, следовательно, снижению потенциала его ЭВ. Образование комплекса феназин-N,N′-диоксида, являющегося близким по структуре к рассмотренному выше соединению 1, с кислотами (HClO4, H2SO4 и CF3COOH) предполагалось в [21] при изучении его электроокисления в CH3CN.
С целью получения экспериментальных данных о возможном взаимодействии соединения 1 с кислотами были сняты УФ-спектры ДМФА растворов соединения 1 с добавлением CCl3COOH и CH3COOH.
Как видно из спектра (рис. 4), максимум поглощения при λ = 472 нм уменьшается с ростом содержания добавленной CCl3COOH, а интенсивность поглощения 284–290 нм при этом растет.
Рис. 4.
УФ-спектры 1 в ДМФА в присутствии CCl3COOH в соотношение концентраций: 1 : 0 (1); 1 : 1 (2); 1 : 2 (3); 1 : 3 (4); 1 : 4 (5); 1 : 5 (6). Стрелками показано направление изменения величины поглощения с ростом концентрации кислоты в растворе.

В случае добавления CH3COOH эффекты на спектрах качественно те же, но гораздо менее выражены, вероятно, из-за меньшей силы кислоты CH3COOH по сравнению с CCl3COOH. Подобный характер изменения УФ-спектров в присутствии донора протонов наблюдали [22] в случае феназин-N,N′-диоксида в CH2Cl2. На основании данных УФ-спектроскопии растворов соединения 1 в ДМФА можно предполагать образование комплекса за счет донорно-акцепторного взаимодействия этого соединения как с CCl3COOH, так и с CH3COOH. Совокупность результатов, полученных методами УФ-спектроскопии и ЦВА, дает основание полагать, что при потенциалах первой стадии ЭВ соединения 1 в присутствии CH3COOH основной реакцией является восстановление комплекса исходного с донором протонов:
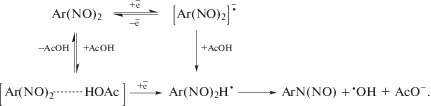
Конкурирующая реакция протонирования АР соединения 1 при незначительном избытке CH3COOH не приводит к электрохимически активным при данных потенциалах продуктам. Образующийся на первой стадии в этих условиях бензофеназин-N-монооксид обратимо восстанавливается при –1.05 В. С увеличением концентрации CH3COOH выше эквивалентного по отношению к исходному соединению 1 появляется на кривой и растет (рис. 2, кривые 4 и 5) пик при Е = –0.96 В, по-видимому отвечающий ЭВ комплекса бензофеназин-N-монооксида с CH3COOH.

Другими словами, дезоксигенирование соединения 1 протекает последовательно при разных потенциалах сначала по одной, а потом и по второй N–O-группе.
С целью установления природы пиков, появляющихся на ЦВА-кривых ЭВ соединения 1 в присутствии невысоких концентраций донора протонов, были проведены электролизы при контролируемом потенциале первой и второй стадии ЭВ 1 в присутствии СН3СООН. Электролиз раствора, содержащего 10 мМ соединения 1 и эквивалентное количество СН3СООН, провели при потенциале –0.8 В. После пропускания одного F электричества в расчете на исходную концентрацию соединения 1 ток пика при –0.91 В на кривых раствора после электролиза практически исчез (рис. 5).
Рис. 5.
Кривые ЦВА 1 (10.7 мМ) в присутствии CH3COOH (10 мМ) до электролиза (1); 2 – раствор после электролиза (1 F); 3 – кривая ЦВА 1 в ДМФА.

При этом появился обратимый пик ЭВ при ‒1.05 В. Величина тока этого пика близка по величине к значению тока исходного соединения 1. Согласно данным HRMS, молярная масса основного продукта ЭВ 1, выделенного после электролиза, оказалась равной 247.06 г/моль (+М (Н)), а спектр 1Н ЯМР соответствовал структуре бензо[a]феназин-N-монооксида. Эти факты позволяют утверждать, что в описанных условиях при ЭВ соединения 1 образуется именно монооксид бензо[a]феназина.
Как уже упоминалось выше, в присутствии СН3СООН свыше одного эквивалента по отношению к 1 на ЦВА-кривых наблюдается еще один последующий обратимый пик при –1.25 В (рис. 2). Предполагая, что при этом потенциале восстанавливается продукт полного дезоксигенирования 1, т.е. бензо[a]феназин, мы провели ЭКП при постоянном Е = –1.0 В. Как видно из рис. 6, в растворе после исчерпывающего электролиза в этих условиях присутствует продукт ЭВ соединения 1, имеющий два катодных пика: –1.28 и –1.98 В, потенциалы которых близки значениям потенциалов двух пиков ЭВ феназина в ДМФА (–1.2 и ‒2.01 В) [23]. Масс-спектр и спектр Н1 ЯМР выделенного после электролиза из католита основного продукта ЭВ соединения 1 соответствуют характеристикам бензо[a]феназина. На основании полученных данных можно утверждать, что именно он является основным продуктом ЭВ соединения 1 при потенциале –1.0 В в данных условиях. Концентрация бензо[a]феназина в католите после электролиза, судя по кривой ЦВА (рис. 6), практически равна концентрации исходного соединения перед электролизом. То есть, бензо[a]феназин при электролизе образуется с количественным выходом.
Рис. 6.
ЦВА-кривые 10 мМ соединения 1 в ДМФА (1); тот же раствор с добавлением 20 мМ уксусной кислоты до электролиза (2) и после электролиза при Е = ‒1.0 В (3). Кривая 4 – ЭВ бензо[a]феназина (2.9 мМ), выделенного после электролиза из католита.
Результаты исследования ЭВ соединения 1 в ДМФА в присутствии СН3СООН показывают, что преимущественным направлением его превращений на электроде является ЭВ комплекса диоксида 1 с донором протонов. Гомолитический разрыв связи N–OH в радикале, образующимся при ЭВ этого комплекса, приводит к количественному образованию моно-N-оксида и к отщеплению частицы •OH. Моно-N-оксид также образует комплекс с добавленным в раствор донором протонов и восстанавливается до бензо[a]фензина, образуя эквивалентное количество активного радикала •OH.
Чтобы определить сходство и различие в механизмах ЭВ производных бензофеназинов и хиноксалинов провели исследование механизма ЭВ соединения 2. На рис. 7 представлены кривые ЦВА соединения 2 в ДМФА для сравнения с кривыми ЭВ соединения 1 (рис. 1).
Рис. 7.
ЦВА-кривые 2 (4.6 мМ) на электроде из СУ при различных потенциалах реверса в ДМФА (0.1 М Bu4NBF4), $v$ = 100 мВ с–1.
Электрохимическое поведение соединения 2 на первой стадии в апротонной среде аналогично поведению соединения 1 (табл. 1). Ток первого пика ЭВ соединения 2 линейно меняется с изменением как концентрации деполяризатора, так и величины v1/2, что свидетельствуют об образовании относительно стабильного АР соединения 2. Последующая стадия ЭВ на ЦВА соединения 2 в апротонном безводном ДМФА представляет сумму пиков с близкими потенциалами: около –1.65 и –1.76 В. Причем при изменении направления развертки потенциала с –1.85 В на реверсивной кривой отсутствуют пики продуктов дезоксигенирования дианиона 2 (рис. 7). Иначе говоря, в апротонной среде основным направлением протонирования дианиона 2, вероятно, является не связь N–O, как при ЭВ 1, а гетероароматическое кольцо молекулы 2. В результате протонирования дианиона соединения 2 образуется моноанион, способный восстанавливаться при –1.76 В и окисляться при –0.30 В на реверсивной кривой (рис. 7).
Таблица 1.
Значения потенциалов пиков на ЦВА-кривой и количество электронов (n), участвующих в ЭВ соединений 1 и 2 на первой стадии в ДМФА на фоне 0.1 М Bu4NClO4. Электрод из стеклоуглерода, скорость развертки v = 100 мВ с–1
| № | Соединение | Ер, В | ΔEp = Epc – Epa, мВ | nцва | NХА |
|---|---|---|---|---|---|
| 1 |  |
–0.91 | 63 | 0.81 | 0.99 |
| 2 |  |
–1.12 | 67 | 0.86 | 0.97 |
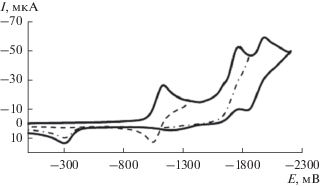
В присутствии СН3СООН эти пики на кривых ЦВА соединения 2 исчезают (рис. 8), но при этом так же, как и в случае соединения 1, появляются и растут пики ЭВ продуктов дезоксигенирования 2.
Рис. 8.
ЦВА-кривые 2 (4.6 мМ) в ДМФА в присутствии СН3СООН, cСН3СООН, мМ = 0 (1), 0.93 (2), 1.90 (3), 2.86 (4), 3.85 (5), 4.86 (6) при $v$ = 100 мВ с–1.
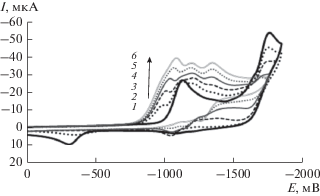
Как видно из рис. 8, добавление СН3СООН так же, как в случае ЭВ соединения 1, приводит к появлению предшествующей стадии на ЦВА-кривых соединения 2. Потенциал этой стадии равен –1.0 В. Ток этого пика увеличивается с ростом концентрации кислоты заметно в большей степени, чем в случае 1 (ср. рис. 3). Вероятно, радикал, образующийся при потенциалах этой предшествующей стадии из комплекса соединения 2 с СН3СООН, частично восстанавливается на электроде.

АР соединения 2 образуется на 200 мВ отрицательнее, чем АР соединения 1, поэтому, принимая во внимание результаты корреляции [24] основности АР ароматических соединений и потенциала их образования, можно утверждать, что АР 2 вступает в реакции протонирования с более высокой скоростью, чем АР соединения 1.
ЭВ протонированного АР соединения 2, вероятно, приводит к гидрированию гетероароматического кольца в молекуле 2, поскольку распределение электронной плотности неспаренного электрона происходит в большей степени по гетероароматической части хиноксалинового скелета [25]. Подтверждением этому может служить также и тот факт, что окисление кислородом продукта одноэлектронного восстановления хиноксалин-диоксидов протекает не всегда обратимо, а с образованием соответствующего гидроксипроизводного [26].
ЗАКЛЮЧЕНИЕ
Полученные экспериментальные данные свидетельствуют о том, что в апротонной среде образующиеся на электроде анион-радикалы N,N′-диоксидов 1 и 2 достаточно устойчивы. Дезоксигенирование этих соединений протекает лишь в присутствии доноров протонов, и в результате восстановление N–O-групп осуществляется в две последовательные одноэлектронные стадии. Обе стадии сопровождаются отщеплением радикала •ОН и образованием на первой стадии моно-N-оксида соответствующего производного и, соответственно, на второй – бензофеназина или хиноксалина. Согласно результатам, полученным электрохимическими методами, возможны два пути образования продуктов. Первый заключается в том, что восстановлению подвергается комплекс деполяризатора с молекулой донора протонов (схема реакций (1)). Такой механизм наиболее вероятен для соединения 1. Вторым направлением превращений является протекание реакции протонирования АР диоксида с последующим распадом радикала (схема 2 ). Если радикал Аr(N–O)2H• распадается с высокой скоростью, то обе реакции практически количественно приводят к генерированию активного радикала •OH. В случае ЭВ соединения 2 радикал Аr(N–O)2H• успевает частично восстановиться на электроде, это проявляется в увеличении тока появляющегося предпика выше тока одноэлектронного уровня, и, следовательно, количество активных радикалов •OH снижается. Таким образом, для характеристики активности N,N′-диоксидов в реакциях генерирования активных кислородных частиц необходимо учитывать совокупность основных окислительно-восстановительных и сопутствующих им других химических реакций.
Список литературы
Da Cunha, J., Lavaggi, M.L., Abasolo, M.I., Cerecetto, H., and Gonzalez M., 2D- and 3D-Quantitative Structure-Activity Relationship Studies for a Series of Phenazine N,N'-Dioxide as Antitumour Agents, Chem. Biol. Drug Des., 2011, vol. 78, p. 960.
Pachon, O. G., Azqueta, A., Lavaggi, M.L., Lopez de Cerain, A., Creppy, E., Collins, A., Cerecetto H., Gonzalez, M., Centelles, J.J., and Cascante, M., Antitumoral Effect of Phenazine N5,N10-Dioxide Derivatives on Caco-2 Cells, Chem. Res. Toxicol., 2008, vol. 21, p. 1578.
Gonda, M., Nieves, M., Nunes, E., Lopez de Cerain, A., Monge, A., Lavaggi, M. L., Gonzalez, M., and Cerecetto, H., Phenazine N,N0-dioxide scaffold as selective hypoxic cytotoxin pharmacophore. Structural modifications looking for further DNA topoisomerase II-inhibition activity, Med. Chem. Commun., 2013, no. 4, p. 595.
Lavaggi, M.L., Cabrera, M., Pintos, C., Arredondo, C., Pachon, G., Rodrıguez, J., Raymondo, S., Pacheco, J.P., Cascante, M., Olea-Azar, C., Lopez de Cerain, A., Monge, A., Cerecetto, H., and Gonzalez, M., Novel Phenazine 5,10-Dioxides Release •OH in Simulated Hypoxia and Induce Reduction of Tumour Volume In Vivo, International Scholarly Research Network ISRN Pharmacology, Volume 2011, Article ID 314209, https://doi.org/10.5402/2011/314209
Chowdhury, G., Sarkar, U., Pullen, S., Wilson, W.R, Rajapakse, A., Fuchs-Knotts, T., and Gates, K.S., DNA Strand Cleavage by the Phenazine Di-N-oxide Natural Product Myxin under Both Aerobic and Anaerobic Conditions, Chem. Res. Toxicol., 2012, vol. 25, p. 197.https://doi.org/10.1021/tx2004213
Fuchs, T., Chowdhury, G., Barnes, Ch.L., and Gates, K. S., 3-Amino-1,2,4-benzotriazine 4-Oxide: Characterization of a New Metabolite Arising from Bioreductive Processing of the Antitumor Agent 3-Amino-1,2,4-benzotriazine 1,4-Dioxide (Tirapazamine), J. Org. Chem., 2001, vol. 66, p. 107.
Anderson, R.F., Shinde, S.S., Hay, M.P., Gamage, S.A., and Denny, W.A., Activation of 3-Amino-1,2,4-benzotriazine 1,4-Dioxide Antitumor Agents to Oxidizing Species Following Their One-Electron Reduction, J. Am. Chem. Soc., 2003, vol. 125, p. 748.
Yin, J., Glaser, R., and Gates, K.S., On the Reaction Mechanism of Tirapazamine Reduction Chemistry: Unimolecular N−OH Homolysis, Stepwise Dehydration, or Triazene Ring-Opening, Chem. Res. Toxicol., 2012, vol. 25, p. 634. https://doi.org/10.1021/tx200546u
Shen, X., Laber, Ch.H., Sarkar, U., Galazzi, F., Johnson, K.M., Mahieu, N.G., Hillebrand, R., Fuchs-Knotts, T., Barnes, Ch.L., Baker, G.A., and Gates, K.S., Exploiting the Inherent Photophysical Properties of the Major Tirapazamine Metabolite in the Development of Profluorescent Substrates for Enzymes That Catalyze the Bioreductive Activation of Hypoxia-Selective Anticancer Prodrugs, J.Org.Chem.,2018, vol. 83, no. 6, p. 3126. https://doi.org/10.1021/acs.joc.7b03035
Cimmino, A., Evidente, A., Mathieu, V., Andolfi, A., Lefranc, F., Kornienko, A., and Kiss, R., Phenazines and cancer, Nat. Prod. Rep., 2012, vol. 29, p. 487. https://doi.org/10.1039/c2np00079b
Cerecetto, H., Gonzalez, M., Lavaggi, M.L., Azqueta, A., Lopez de Cerain, A., and Monge, A., Phenazine 5,10-Dioxide Derivatives as Hypoxic Selective Cytotoxins, J. Med. Chem., 2005, vol. 48, p. 21.
Hay, M.P., Gamage, S.A., Kovacs, M.S., Pruijn, F.B., Anderson, R.F., Patterson, A.V., Wilson, W. R., Brown, J. M., and Denny, W.A., Structure-Activity Relationships of 1,2,4-Benzotriazine 1,4-Dioxides as Hypoxia-Selective Analogues of Tirapazamine, J. Med. Chem., 2003, vol. 46, p. 169.
Lavaggi, M.L., Nieves, M., Cabrera, M., Olea-Azar, C., Lopez de Cerain, A., Monge, A., Cerecetto, H., and Gonzalez, M., Structural modifications on the phenazine N,N′-dioxide-scaffold looking for new selective hypoxic cytotoxins, Europ. J. Med. Chem., 2010, vol. 45, p. 5362.
Abu El-Haj, M. J., Dominy, B. W., Johnston, J. D., Haddadin, M.J., and Issidorides, C.H., A New Route to Phenazine 5,10-Dioxides and Related Compounds, J. Org. Chem., 1978, vol. 7, no. 4, p. 589.
Lavaggi, M.L., Cabrera, M., Aravena, M. de los A., Olea-Azar, C., Lypez de Cerain, A., Monge, A., Pachyn, G., Cascante, M., Bruno, A.M., Pietrasanta, L.I., Gonzalez, M., and Cerecetto, H., Study of benzo[a]phenazine 7,12-dioxide as selective hypoxic cytotoxin-scaffold. Identification of aerobic-antitumoral activity through DNA fragmentation, Bioorg.Med. Chem., 2010, vol. 18, p. 4433.
El-Gogary, S.R., Waly, M.A., Ibrahim, I.T., and El-Sepelgy, O.Z., Synthesis and UV absorption of new conjugated quinoxaline1,4-dioxide derivatives anticipated as tumor imaging and cytotoxic agents, Monatsh.Chem., 2010, vol. 141, p. 1253. https://doi.org/10.1007/s00706-010-0386-1
Romeiro, N.C., Aguirre, G., Hernandez, P., Gonzalez, M., Cerecetto, H., Aldana, I., Perez-Silanes, S., Monge, A., Barreiro, E.J., and Lima, L.M., Synthesis, trypanocidal activity and docking studies of novel quinoxaline-N-acylhydrazones, designed as cruzain inhibitors candidates, Bioorg. Med. Chem. 2009, vol. 17, p. 641.
Кулаковская, С.И., Кривенко, А.Г., Комарова, Н.С., Куликов, А.В., Шестаков, А.Ф. Электрохимическое и ЭПР исследование механизма окисления феназин-ди-N-оксида в присутствии циклогексанола на электродах из стеклоуглерода и одностенных углеродных нанотрубок. Электрохимия. 2014. Т. 50. С. 3. [Kulakovskaya, S.I., Krivenko, A.G., Komarova, N.S., Kulikov, A.V., and Shestakov, A.F., Electrochemical and ESR study of the mechanism of oxidation of phenazine-di-N-oxide in the presence of cyclohexanol on glassy carbon and single-walled carbon nanotube electrodes, Russ. J. Electrochem., 2014, vol. 50, p. 1.]https://doi.org/10.7868/S042485701401006X
Miyazaki, H., Matsuhisa, Y., and Kubota, T., Cyclic voltammetry of aromatic amine N-oxides in nonaqueous solvents and the stability of the free radicals produced, Bull. Chem. Soc. Jpn., 1981, vol. 54, p. 3850.
Рыжаков, А.В., Алексеева, О.О., Родина, Л.Л. Новые тенденции в химии молекулярных комплексов гетероароматических N-оксидов. Вестн. СПбГУ. Сер. 4. 2009. Вып. 1. С. 67. [Ryzhakov, A.V., Alekseeva, S.A., and Rodina, L.L., New trends in the chemistry of molecular complexes of heteroaromatic N-oxides, Vestnik SPbGU, Ser. 4. 2009, Issue. 1, p. 67 (in Russian).]
Колдашева, Е.М., Шестаков, А.Ф., Гелетий, Ю.В., Шилов, А.И. Образование и редокс-свойства комплекса феназин-ди-N-оксида с протоном. Изв. АН СССР. Сер. хим. 1992. № 4. С. 845. [Koldasheva, E.M., Shestakov, A.F., Geletii, Yu.V., and Shilov, A.E., Formation and redox properties of a complex of phenazine di-N-oxide with a proton, Bull. Russ. Acad. Sci. Division Chem. Sci., 1992, vol. 41, no. 4, p. 655.]
Колдашева, Е.М., Стрелец, В.В., Це, Ю.-Х., Гелетий, Ю.В., Шестаков, А.Ф. Катион-радикал феназин- ди-N-оксида и его реакции с углеводородами. Изв. АН СССР. Сер. хим. 1996. 8. 1992. [Koldasheva, E.M., Strelets, V.V., Tse, Y.K., Geletii, Yu.V., and Shestakov, A. F., Phenazine di-N-oxide radical cation and its reaction with hydrocarbons, Russ. Chem. Bull., 1996, vol. 45, no. 8, p. 1889.]
Tabner J. and Yandle J. R., A Correlation of Half-wave Reduction Potentials with Theoretical Calculations for Some Nitrogen-containing Heteromolecules in Dimethylformamide, J. Chem. SOC. (A), 1968, p. 381.
Mendkovich, A.S., Syroeshkin, M.A., Mikhalchenko, L.V.,Mikhailov, M.N., Rusakov, A.I., and Gul’tyai, V.P., Integrated Study of the Dinitrobenzene Electroreduction Mechanism by Electroanalytical and Computational Methods, Intern. J. Electrochem., vol. 2011, Article ID 346043. https://doi.org/10.4061/2011/346043
Chowdhury, G., Kotandeniya, D., Daniels, J.S., Barnes, Ch.L., and Gates, K.S., Enzyme-Activated, Hypoxia-Selective DNA Damage by 3-Amino-2-quinoxalinecarbonitrile 1,4-Di-N-oxide, Chem. Res. Toxicol., 2004, vol.17, no. 11, p. 1399. https://doi.org/10.1021/tx049836w
Ganley, B., Chowdhury, G., Bhansali, J., Daniels, J.S., and Gates K.S., Redox-Activated, Hypoxia-Selective DNA Cleavage by Quinoxaline 1,4-di-N-Oxide, Bioorg. Med. Chem., 2001, vol. 9, p. 2395.
Дополнительные материалы отсутствуют.