

Электрохимия, 2022, T. 58, № 11, стр. 766-778
Алгоритм теоретической оценки электрохимической устойчивости электролитов литий-ионных аккумуляторов на примере LiBF4 в смеси ЭК/ДМК
С. С. Борисевич a, b, Е. Ю. Евщик b, *, М. Г. Ильина a, Э. М. Хамитов a, Т. И. Мельникова c, Р. Ю. Рубцов b, О. В. Бушкова b, d, Ю. А. Добровольский b
a Уфимский институт химии УФИЦ РАН
Уфа, Россия
b Институт проблем химической физики РАН
Черноголовка, Россия
c Центр “Цифрового биодизайна и персонализированного здравоохранения” Первого МГМУ им. И.М. Сеченова
Москва, Россия
d Институт химии твердого тела УрО РАН
Екатеринбург, Россия
* E-mail: liza@icp.ac.ru
Поступила в редакцию 14.02.2022
После доработки 27.04.2022
Принята к публикации 16.05.2022
- EDN: ADMUKQ
- DOI: 10.31857/S0424857022110044
Аннотация
Устойчивость электролита к окислительному разложению на поверхности положительного электрода является одним из барьеров, затрудняющих разработку аккумуляторов с высокой плотностью энергии. Электрохимическая устойчивость электролита напрямую связана с составом и строением сольватных комплексов, образующихся при растворении соли. Используя совокупность методов молекулярной динамики и квантовой химии, можно создать алгоритм теоретической оценки устойчивости электролита к анодному окислению в зависимости от состава. В дальнейшем этот алгоритм можно использовать для подбора вариантов среди неисследованных смесей растворителей и солей лития с целью разработки новых электролитов, устойчивых до 5 и 6 В. В данной работе методы классической молекулярной динамики и квантовой химии были использованы для установления строения сольватных комплексов, образующихся в растворе LiBF4 в бинарной смеси этиленкарбонат (ЭК)/диметилкарбонат (ДМК). Квантово-химическая оценка термодинамической и окислительной стабильности сольватных комплексов позволила установить, какие именно комплексы вносят наиболее существенный вклад в электрохимическую устойчивость электролитной системы, и рассчитать аддитивное значение потенциала окисления электролита.
ВВЕДЕНИЕ
Литий-ионные аккумуляторы (ЛИА) широко используются в качестве источников тока в портативной электронике, электротранспорте, системах накопления энергии. Однако с ростом потребляемой мощности возрастают и требования к энергетическим параметрам аккумуляторов. С целью их повышения могут быть использованы разрабатываемые в настоящее время высоковольтовые материалы положительного электрода, позволяющие получить ЛИА класса 5 В и даже 6 В [1–5]. К сожалению, их практическое применение сдерживается недостаточно высокой электрохимической устойчивостью коммерчески доступных электролитов.
Используемые в производстве ЛИА электролиты представляют собой 1–1.5 М раствор LiPF6 в базовых смесях линейных и циклических карбонатов, в состав которых дополнительно введены функциональные добавки [6–10]. Это обусловлено сочетанием хорошей сольватирующей способности органических карбонатов по отношению к иону лития, их высокой электрохимической устойчивости и возможности формировать стабильный защитный твердоэлекролитный слой (solid electrolyte interface (SEI)) на поверхности углеродного электрода в первом (формировочном) цикле заряда аккумулятора из продуктов электровосстановления [8–11]. Поскольку потенциал окисления (Eox) таких растворов не превышает 4–4.5 В [12], то это дает возможность использовать в катодном полуэлементе такие соединения, как LiCoO2 (LCO), LiNi1 – x – yCoxAlyO2 (NCA), LiNixMnyCozO2 (NMC), LiFePO4 (LFP), LiMn2O4 (LMO) [13], но исключает их замену на новые материалы с более высоким потенциалом относительно Li/Li+.
К настоящему времени установлено, что помимо электрохимического формирования слоя SEI на поверхности отрицательного электрода, аналогичные процессы протекают и на поверхности положительного электрода [8, 9]. Помимо первичного химического взаимодействия, большой вклад вносят малоизученные процессы каталитического окисления электролитного раствора на сильно окисленных делитированных поверхностях гранул активного материала; в них принимают участие как соли лития, так и компоненты смешанного растворителя и функциональные добавки. Нерастворимые продукты химических и электрохимических взаимодействий локализуются на поверхности гранул, формируя слой CEI (cathode–electrolyte interface). В зависимости от состава электролита и природы активного материала, этот слой может выполнять или не выполнять защитные функции. Таким образом, вопрос устойчивости электролита к электрохимическому окислению на поверхности положительного электрода при его анодной поляризации тесно увязан с формированием высокоэффективного CEI, способного обеспечить длительное стабильное циклирование аккумулятора. К сожалению, эти проблемы пока не нашли широкого отражения в исследованиях электролитов.
Согласно литературным данным, к основным эмпирическим способам оптимизации состава электролитов для высоковольтовых ЛИА относятся перебор органических растворителей, использование специальных функциональных добавок, а также варьирование природы аниона литиевой соли и изменение ее концентрации в сторону увеличения. В качестве растворителей, стабильных при потенциалах ~5 В и выше, используют сульфоны [14, 15], нитрилы [16, 17], некоторые простые и сложные эфиры [8, 11, 12]. При этом эффекта расширения диапазона электрохимической устойчивости можно добиться путем добавления более устойчивого к анодному окислению растворителя к менее устойчивым, а также при использования альтернативных LiPF6 литиевых солей, обладающих повышенной электрохимической стабильностью (например, LiBF4, LiTFSI [18, 19] и др. [20]), или специальных добавок [21]. Однако возможности оптимизации электролита ограничены межфазными процессами в анодном полуэлементе: изменение базового состава электролитного раствора не должно приводить к ухудшению качества SEI на поверхности углеродного отрицательного электрода.
Для фундаментального понимания природы электрохимической стабильности электролитных систем и описания молекулярных механизмов электрохимических реакций разложения успешно используются теоретические подходы [22–34]. Методы квантовой химии позволяют описать строение образующихся в электролитном растворе локальных структур ближнего порядка (сольватированных ионных частиц), достоверно оценить энергии связи, термодинамические характеристики, окислительные и восстановительные потенциалы [22, 23, 27, 28, 32]. Чаще всего в таких исследованиях проводится моделирование при помощи ресурсоемких композитных методов (G4MP2) и методов теории функционала плотности, с последующим решением колебательной задачи [10, 32, 33, 35, 36]. При этом преимущественно используются методы учета неявной сольватации [37]; в качестве растворителя учитывают воду, ацетон или диэтиловый эфир [35, 38]. В работах Бородина [27, 32, 35] указывается, что диэлектрическая проницаемость ацетона (ε = 20.5) близка к диэлектрической проницаемости большинства используемых на практике растворов электролитов. Следует отметить, что корректность выбора метода расчета и/или способа учета растворителя может быть проверена только на основании результатов эксперимента. Масштабные атомно-молекулярные системы моделируют методами молекулярной динамики, основанными на молекулярных силовых полях, с прогнозированием структурных и транспортных свойств электролитных систем в объеме и во времени. Электрохимическое окисление исследуемой частицы (комплекса) может быть рассмотрено в рамках модели LOMO–HOMО [30] либо как процесс ионизации [10, 29, 31, 34]. Полученные расчетные значения потенциалов окисления представляют собой предельные величины; экспериментально определяемые значения потенциала окисления всегда ниже расчетных [39–43] и зависят от материала электрода [44, 45]. Вполне очевидно, что одновременное использование обоих подходов, квантово-химического и молекулярно-динамического, дает значительные преимущества. К сожалению, в большинстве работ используются либо методы квантовой химии [22, 27–31, 34, 36], либо методы классической молекулярной динамики [24, 25]. Работы, в которых используются оба метода, гармонично дополняющие друг друга, встречаются значительно реже [10, 32]. Кроме того, чаще всего исследуются системы на основе индивидуального растворителя, тогда как на практике они почти не применяются. Исследования электролитов со смешанным растворителем, даже бинарным, составляют скорее исключение [31].
В данной работе комплексом расчетных и экспериментальных методов исследована электрохимическая устойчивость 1 М раствора LiBF4 в широко используемой базовой смеси растворителей этиленкарбонат (ЭК)/диметилкарбонат (ДМК) (1 : 1, об.). Выбор такой системы обусловлен повышенной термической, гидролитической и электрохимической устойчивостью аниона ${\text{BF}}_{4}^{ - }$ по сравнению с ${\text{PF}}_{6}^{ - }$ [20]. Предложен алгоритм прогнозирования электрохимической устойчивости неводного электролита, основанный на сочетании квантово-химических и молекулярно-динамических расчетов и рассматривающий процесс электрохимического окисления электролитного раствора как процесс ионизации входящих в его состав ионных частиц. Адекватность результатов расчетов, выполненных с учетом ионной структуры раствора и ее изменения с ростом концентрации соли, оценена путем сравнения с экспериментально измеренными величинами потенциалов окисления электролитных растворов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для удаления следов воды из этиленкарбоната (battery grade, Sigma Aldrich) и диметилкарбоната (anhydrous, ≥99%, Sigma Aldrich) смесь растворителей выдерживали над молекулярными ситами 3А в течение 1 нед. LiBF4 (≥98%, Sigma Aldrich) сушили в вакуумном сушильном шкафу при 140°C в течение 4 дней. Растворы электролита с концентрацией 1 М готовили в перчаточном боксе с атмосферой аргона (остаточное содержание воды и кислорода не превышало 1 ppm). Полноту удаления воды из раствора электролита контролировали методом ИК-спектроскопии (Vertex 70, Bruker, Германия).
Окно электрохимической устойчивости электролита определяли методом пошаговой поляризации согласно методике, описанной в работах [46, 47]. Для измерений использовали двухэлектродные трехзондовые электрохимические ячейки с инертным платиновым рабочим электродом и обратимым литиевым противоэлектродом, который одновременно служил электродом сравнения. Суть методики заключается в поэтапной поляризации ячейки постоянной разностью потенциалов, заданной в интересующем диапазоне значений, с измерением стационарного тока (Iss). Рабочий электрод ступенчато поляризовали в диапазоне от 2.5 В (напряжение разомкнутой цепи) до 5.9 В относительно Li0/Li+ с шагом 215 мВ; для каждого заданного значения потенциала регистрировали зависимость тока от времени до достижения стационарного значения. Для определения величины Eox строили зависимости стационарного тока от потенциала. Для измерений использовали потенциостат P-20×8 (ООО “Элинс”, Россия).
Плотность раствора электролита определяли c помощью пикнометров с номинальным объемом 5 мл, предварительно откалиброванных с помощью бидистиллированной воды при температуре 25°С; измерения выполняли в трех параллелях.
ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
Молекулярно-динамические симуляции
Все молекулярно-динамические симуляции проводили, используя модуль Desmond [47] на платформе программного обеспечения Schrodinger Suite Release 2021-2, методами силового поля OPLS3/OPLS4 [48]. Для симуляции выбранных систем была построена модель, где пространство ограничено кубической решеткой с учетом периодических условий. Количество частиц в расчетных системах соответствовало составу исследуемого раствора (1 М LiBF4 в ЭК/ДМК (1 : 1, об.)). Протокол подготовки системы к симуляции включал предварительную минимизацию и уравновешивание компонентов в ансамбле NPT в течение 2 нс при 298 и при 400 К, последовательно. Период регистрируемой симуляции составил 50 нс в ансамбле NVT при 298 К. Для поддержания постоянства температуры систем использовался термостат Нозе–Хувера. Для описания состава и строения комплексов оценивали функцию радиального распределения g(R) и ее интегральную составляющую N(R).
Квантово-химические расчеты
Все квантово-химические расчеты проводили с использованием программного обеспечения GAUSSIAN 16 rev.C.01 [49]. Оптимизацию геометрических параметров атомно-молекулярных систем с последующим решением колебательной задачи проводили на уровне DFT с использованием функционала M052X [50] и базисного набора TZVP [51]. Учитывали эмпирическую поправку D3 [52]. Термодинамические параметры рассчитывали в приближении газовой фазы при 298 К и атмосферном давлении, а также с учетом неявной сольватации [53]. В качестве растворителя рассматривали ацетон с ε = 20.5, что близко к значению диэлектрической проницаемости большинства смесей электролитов [28].
Для оценки термодинамической стабильности исследуемых комплексов (сольватированных ионных частиц) рассматривали инкрементальные (ступенчатые) значения энергий образования $\left( {{{\Delta }_{{\text{f}}}}G_{{{\text{inc}}}}^{^\circ }} \right),$ как энергию Гиббса реакции последовательного присоединения молекулы растворителя к комплексам [46], исходя из второго следствия закона Гесса.
Адиабатический окислительный потенциал $E_{{{\text{abs}}}}^{^\circ }$ для каждого сорта комплексов оценивали согласно циклу Борна–Габера [28, 54].
Аддитивное значение окислительного потенциала, характеризующего систему в целом, оценивалось с использованием статистики Максвелла–Больцмана по формуле $\left( {E_{{{\text{additive}}}}^{^\circ }} \right){\text{:}}$
(1)
$E_{{{\text{additive}}}}^{^\circ } = \sum\limits_i {{{N}_{i}}{{{\left( {E_{{{\text{ads}}}}^{^\circ }} \right)}}_{i}},} $РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Потенциал окисления $E_{{{\text{ox}}}}^{^\circ }$ раствора электролита состава 1 М LiBF4 в смеси растворителей ЭК/ДМК измеряли в стандартных условиях (25°С, атмосферное давление). На рис. 1а представлена зависимость стационарного тока от приложенного потенциала E в диапазоне от 2.5 до 5.9 В. Потенциал окисления может быть определен электрохимически как потенциал, при котором Iss достигает заданного порогового значения. В данном исследовании пороговый ток принимали равным 5 мкА, что соответствует увеличению величины Iss по сравнению с плато в 50 раз (рис. 1а). В соответствии с этим критерием исследуемый раствор можно считать электрохимически устойчивым во всем интервале 2.5 до 5.9 В. Плотность исследуемого раствора электролита составила 1.27 г/см3.
Рис. 1.
Зависимость стационарного тока от приложенного потенциала для 1 М раствора LiBF4 в ЭК/ДМК (1 : 1, об.) (а) и система того же состава (б) для молекулярно-динамической симуляции (количество частиц соответствует заявленной концентрации соли и объемному соотношению растворителей).

В ходе проведения компьютерного эксперимента на всем протяжении молекулярно-динамической симуляции наблюдается постоянное значение плотности рассматриваемой системы. Расчетные данные разумно коррелируют с экспериментом (рис. 1б), что может свидетельствовать об адекватности модели.
Полученная на основании анализа результатов расчетов радиальная функция распределения частиц g(R) имеет осциллирующий характер (рис. 2), где первый пик (максимум) отвечает первой координационной сфере, второй пик (максимум) отвечает второй координационной сфере, третий пик (максимум) – третьей и т.д. Статический структурный фактор N(R) представляет собой интегральную характеристику (от функции радиального распределения g(R)), и тем самым он характеризует структурные особенности системы. Наиболее отчетливые локальные структурные особенности будут проявляться в значениях статического структурного фактора N(R).
Рис. 2.
Радиальная функция распределения частиц g(R) и статистический структурный фактор N(R) для системы, моделирующей 1 М раствора LiBF4 в ЭК/ДМК (1 : 1, об.).
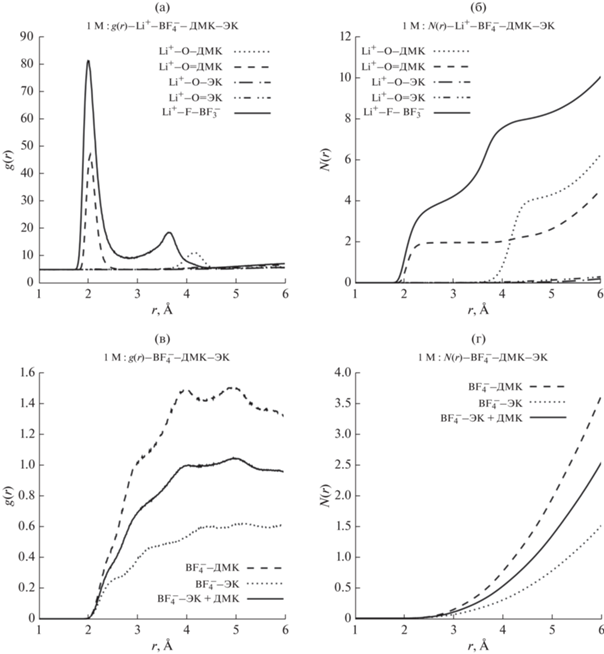
Из полученных результатов следует, что ближний порядок катиона лития реализуется в радиусе 2 Å. В течение всего времени симуляции катион лития чаще всего координируется рядом с атомами фтора аниона ${\text{BF}}_{4}^{ - }$ (об этом свидетельствует высота первого пика кривой Li+ – F–B–${\text{F}}_{3}^{ - }$ на рис. 2а, и соответствующий статистический структурный фактор N(R) на рис. 2б). Анализируя кривые на рис. 2, можно отметить, что в первую сольватную оболочку катиона лития с большой долей вероятности будет входить молекула ДМК, координированная к Li+ через атом кислорода карбонильной группы.
Анализ кривых на рис. 2в и 2г позволяет сделать вывод, что образование анионных сольватных комплексов маловероятно. Функция радиального распределения и статистический структурный фактор – это, прежде всего, статистические характеристики, которые накапливаются в течение всего времени симуляции. При этом в молекулярно-динамических расчетах учитывается образование любых комплексов, включая коротко живущие переходные формы, которые с точки зрения термодинамики могут быть просто нестабильными. Очевидно, что их вклад в электрохимическую устойчивость системы будет незначительным. Для того, чтобы ответить на вопрос, какие комплексы устойчивы, а какие – нет, и какие из них нужно принимать во внимание при оценке электрохимической стабильности электролитного раствора, в дополнение к результатам молекулярно-динамического моделирования следует использовать методы квантовой химии.
Растворы ионных солей в координирующих растворителях образуются в результате ион-молекулярных взаимодействий за счет сольватации ионных частиц молекулами растворителей. Набор образовавшихся комплексов и определяет электрохимическую устойчивость электролитного раствора. (В данной работе мы не рассматриваем поверхность положительного электрода и ее потенциальное влияние на эти процессы). Тогда устойчивость электролитного раствора к окислению может быть рассмотрена как некоторая аддитивная величина, которая складывается из устойчивости отдельных составляющих: изолированных молекул растворителя, их ассоциатов, анионных сольватных комплексов и несольватированных анионов, сольватированных и несольватированных ионных пар, а для концентрированных растворов – еще и ассоциатов более высокого порядка. Хотя в растворе литиевой соли обязательно присутствуют катионные сольватные комплексы (их образование – термодинамически выгодный процесс; такие частицы обнаружены во множестве экспериментальных и теоретических исследований, в том числе в нашей недавней работе [46]), в контексте данного исследования мы их не рассматриваем, так как положительно заряженные частицы очевидно не подвергаются электрохимическому окислению и, соответственно, не вносят вклад в окислительный потенциал электролитного раствора.
Как известно, молекулы многих диполярных растворителей склонны к самоассоциации и часто образуют димеры или более сложные комплексы [55], в которых возможно различное расположение молекул растворителя относительно друг друга. Возможные конфигурации молекул ЭК и ДМК, а также их димеров и гетеромолекулярных ассоциатов приведены на рис. 3.

Из рассчитанных для этих комплексов величин инкрементальной энергии образования Гиббса ${{\Delta }_{{\text{f}}}}G_{{{\text{inc}}}}^{^\circ },$ приведенных в табл. 1, следует, что образование подобных комплексов маловероятно, так как значения ${{\Delta }_{{\text{f}}}}G_{{{\text{inc}}}}^{^\circ }$ положительны. Оцененные методами квантовой химии значения их потенциалов окисления $E_{{{\text{abs}}}}^{^\circ }$ (табл. 1) попадают в интервал от 5.78 до 6.19 В. Это соответствует интервалу значений $E_{{{\text{abs}}}}^{^\circ }$ = 5.58–9.70 B (табл. 1), полученных для молекул растворителей и их ассоциатов с помощью различных квантово-химических методов и приведенных в научной литературе [28–31, 38, 57]. Подобный разброс рассчитанных значений $E_{{{\text{abs}}}}^{^\circ }$ связан прежде всего с методами расчета и способом учета растворителя. Неучет объема растворителя, или другими словами, расчеты в приближении газовой фазы сильно завышают значения $E_{{{\text{abs}}}}^{^\circ }$ по сравнению с Eox [28, 30]. Наиболее близкими к экспериментальным данным (обычно 5.5–6.7 В [39, 40]) являются значения $E_{{{\text{abs}}}}^{^\circ }$ [25], рассчитанные с помощью композитных методов, например G4MP2 [57]. Однако следует отметить, что подобные высокоточные расчеты требуют больших затрат вычислительных ресурсов и времени.
Таблица 1.
Термодинамические и окислительные параметры молекул растворителей и различных комплексов, существующих в системе 1 M LiBF4 в смеси ЭК/ДМК: ${{\Delta }_{{\text{f}}}}G_{{{\text{inc}}}}^{^\circ }$ – инкрементальная (пошаговая) энергия образования Гиббса; N – распределение по энергии Максвелла–Больцмана; $E_{{{\text{abs}}}}^{^\circ }$ – адиабатический окислительный потенциал, оцененный в данной работе (M052X/TZVP); $E_{{{\text{abs - calc}}}}^{{^\circ {\text{*}}}}$ – адиабатический окислительный потенциал, опубликованный ранее, оценка которого проводилась на основании квантово-химических расчетов, и $E_{{{\text{ox}}}}^{^\circ }$ – экспериментально измеренная величина потенциала окисления
| Растворитель или комплекс | ${{\Delta }_{{\text{f}}}}G_{{{\text{inc}}}}^{^\circ }$, эВ | N, % | $E_{{{\text{abs}}}}^{^\circ }$, В | $E_{{{\text{abs - calc}}}}^{{^\circ {\text{*}}}}$, В | $E_{{{\text{ox}}}}^{^\circ }$, В |
|---|---|---|---|---|---|
| Молекулы растворителей, их димеры и гетеромолекулярные ассоциаты | |||||
| ЭК | − | − | 7.13 | 5.58–8.50 [25–27, 38, 56] | 5.5–6.7 [40–43] |
| ДМК I | − | − | 7.42 | 5.62–7.60 [25, 26] | 5.3–6.7 [39–42] |
| ДМК II | − | − | 7.09 | 5.62–7.13 [25, 26] | − |
| (ЭК)2III | 0.33 | 5.9 | 6.19 | 5.94–6.00 [22] | − |
| (ЭК)2IV | 0.25 | 5.7 | 5.73 | 5.91–5.90 [25] | 6.0–6.2 [39] |
| (ДМК)2V | 0.13 | 6.2 | 6.15 | 5.84–6.13 [25] | − |
| (ЭК)1(ДМК)1VI | 0.16 | 6.1 | 6.04 | 5.95–7.70 [25] | − |
| Анионные сольватные комплексы | |||||
| Несольватированный анион ${\text{BF}}_{4}^{ - }$ | − | − | 8.54 | 8.00–8.57 [25] | − |
| ${\text{BF}}_{4}^{ - }$(ЭК)1VII | 0.24 | 6.3 | 6.16 | 6.07–6.39 [25, 27, 30] | − |
| ${\text{BF}}_{4}^{ - }$(ДМК)1VIII | 0.33 | 6.1 | 6.08 | 5.79–6.29 [25, 27] | − |
| ${\text{BF}}_{4}^{ - }$(ЭК)2IX | 0.24 | 6.3 | 5.65 | 6.30–6.46 [25] | − |
| ${\text{BF}}_{4}^{ - }$(ДМК)2X | 0.33 | 6.1 | 5.60 | − | − |
| ${\text{BF}}_{4}^{ - }$(ЭК)1(ДМК)1XI | 0.52 | 5.6 | 5.43 | − | − |
| Ионные пары | |||||
| Несольватированная ионная пара $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$ | − | − | 9.32 | − | − |
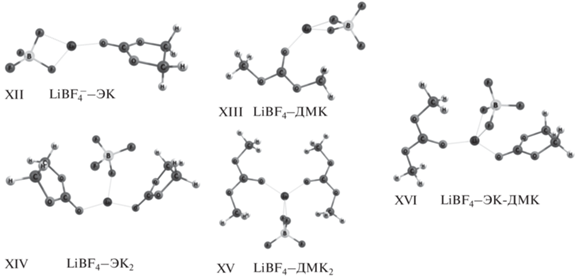
| $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1XII | –0.19 | 7.5 | 6.64 | 6.64–8.74 [25] | − |
| $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК) XIII | –0.26 | 7.7 | 6.69 | − | − |
| $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)2XIV (по реакции $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1 + + ЭК $ \rightleftarrows $$\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)2) |
–0.06 | 7.1 | 6.36 | 6.60–6.72 [25] | − |
| $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)2XV (по реакции $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)1 + + ДМК $ \rightleftarrows $$\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)2) |
–0.12 | 7.3 | 6.72 | − | |
| $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1(ДМК)1XVI (по реакции $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1 + + ДМК $ \rightleftarrows $$\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1(ДМК)1) |
–0.18 | 7.4 | 6.80 | − | |
| $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1(ДМК)1XVI (по реакции $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)1 + + ЭК $ \rightleftarrows $$\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1(ДМК)1) |
–0.11 | 7.2 | 6.80 | − | |
| Вся система 1 M LiBF4 в ЭК/ДМК (1 : 1) | |||||
| $E_{{{\text{additive}}}}^{^\circ }$ (расчет) = 6.24 В | $E_{{{\text{ox}}}}^{^\circ }$ (эксперимент) = 5.90 В | ||||
Как видно из табл. 1, образование сольватных комплексов аниона ${\text{BF}}_{4}^{ - }$ с молекулами ЭК и/или ДМК также маловероятно, так как, согласно расчетам, этот процесс носит эндотермический характер вне зависимости от того, какой из двух растворителей координируется рядом с анионом первым. При этом величина $E_{{{\text{abs}}}}^{^\circ }$ для всех анионных сольватных комплексов достаточно высока и укладывается в интервал от 5.60 до 6.16 В. Высокое значение $E_{{{\text{abs}}}}^{^\circ }$ характерно также и для несольватированного аниона ${\text{BF}}_{4}^{ - }{\text{.}}$ Все рассчитанные значения коррелируют с литературными данными для окислительного потенциала, рассмотрены анионные комплексы только с одной молекулой растворителя. Между тем анализ молекулярно-динамических симуляций указывает на возможность образования анионных сольватных комплексов с двумя и более молекулами растворителя (рис. 4). Безусловно, подобные комплексы – это нестабильные структуры с точки зрения термодинамики. Скорее всего, это короткоживущие переходные формы ионных частиц. Тем не менее, они также могут вносить свой вклад в устойчивость системы к окислительному разложению

В отличие от ассоциатов молекул растворителей и анионных сольватных комплексов, образование сольватированных и несольватированных ионных пар всегда протекает с выигрышем свободной энергии – инкрементальные значения энергии Гиббса образования комплекса во всех рассмотренных случаях являются отрицательными (табл. 1). Согласно результатам молекулярно-динамических симуляций, включение молекулы ЭК в первую сольватную оболочку катиона лития в составе ионной пары маловероятно – Li+ охотнее контактирует с молекулами ДМК. Это заключение подтверждается и данными квантово-химических расчетов. Например, образование комплекса $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)1 по вероятной реакции $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$ + ДМК $ \rightleftarrows $ $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)1 на 36% выгоднее образования $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1 по аналогичной реакции (табл. 1). Внедрение второй молекулы растворителя в первую сольватную оболочку в случае ДМК также более выгодный процесс, чем в случае ЭК. Образование сольватированной ионной пары со смешанной сольватной оболочкой $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1(ДМК)1 может протекать по двум разным реакциям: $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1 + ДМК $ \rightleftarrows $ $ \rightleftarrows $ $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1(ДМК)1 или $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)1 + + ЭК $ \rightleftarrows $ $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1(ДМК)1. При этом координация молекулы ДМК комплексом $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$ (ЭК)1 на 64% выгоднее, чем присоединение ЭК к комплексу $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)1.
Молекулы карбонатных растворителей довольно объемные, поэтому координационное число катиона лития в рассматриваемых комплексах близко к 4 (причем, две координационные позиции из четырех заняты атомами фтора) (рис. 5). Самое высокое значение адиабатического окислительного потенциала $E_{{{\text{abs}}}}^{^\circ }$ = 9.32 В было получено для несольватированной ионной пары $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$ (табл. 1). Однако учитывая тот факт, что такая ионная пара достаточно легко диссоциирует на ионы или формирует свою сольватную оболочку, ее вклад в аддитивное значение окислительного потенциала можно не рассматривать.
Исходя из термодинамических параметров каждого из возможных комплексов, с учетом их электрохимической стабильности мы оценили аддитивное значение окислительного потенциала всей исследуемой электрохимической системы 1 М LiBF4 в смеси растворителей ЭК/ДМК как равное 6.24 В (табл. 1). Другими словами, при достижении значения потенциала в 6.24 В рассматриваемая система может подвергаться реакциям электрохимического окисления, при которых возможны различного рода перегруппировки в составляющих ее комплексах с перестройкой молекул растворителей или аниона ${\text{BF}}_{4}^{ - }.$ Например, при окислении анионных сольватных комплексов ${\text{BF}}_{4}^{ - }$(ЭК)1 и ${\text{BF}}_{4}^{ - }$(ДМК)1 наблюдается существенное изменение состава и геометрических параметров частиц с одновременным образованием свободной молекулы HF (рис. 6а и 6б). Похожая ситуация имеет место и для сольватированных ионных пар в комплексах изолированных ионных пар $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ДМК)1 (рис. 6в) и $\left\{ {{\text{L}}{{{\text{i}}}^{{\text{ + }}}}{\text{BF}}_{4}^{ - }} \right\}$(ЭК)1(ДМК)1 (рис. 6г). Результаты, полученные в рамках данного теоретического исследования, хорошо согласуются с данными работ [28, 30], где также описывается возможность образования свободной молекулы HF.
Рис. 6.
Геометрические параметры анионных сольватных комплексов и сольватированных ионных пар, иллюстрирующие реакции электрохимического окисления с образованием свободной молекулы HF.

Рассчитанное значение адиабатического потенциала окисления, равное 6.24 В (табл. 1), ожидаемо превышает экспериментально измеренную величину $E_{{{\text{ox}}}}^{^\circ },$ равную 5.90 В (рис. 1а); расхождение составляет всего 6%. Таким образом, результаты теоретического исследования хорошо коррелируют с экспериментальными данными. Здесь необходимо принять во внимание, что $E_{{{\text{abs}}}}^{^\circ }$ – максимально возможное (предельное) значение потенциала, при котором начинаются необратимые изменения в электролите, связанные с его окислительным разложением, что позволяет говорить о правильности выбранного нами алгоритма.
ЗАКЛЮЧЕНИЕ
В рамках данной работы на примере раствора LiBF4 в смеси растворителей ЭК/ДМК предложен алгоритм теоретической оценки устойчивости электролита к окислительному разложению. Совокупность методов квантовой химии и молекулярной динамики позволяет описать строение образующихся в электролитном растворе комплексов. Оценка термодинамической и окислительной устойчивости комплексов позволяет оценить предельное значение потенциала, по достижению которого будет наблюдаться необратимое разрушение структур вследствие электрохимического окисления. Теоретические расчеты, приведенные в данной работе, коррелируют с результатами эксперимента. Используя описанный в данной работе подход, можно прогнозировать величину потенциала окисления в зависимости от концентрации соли и соотношения растворителей.
Список литературы
Fridman, K., Sharabi, R., Elazari, R., Gershinsky, G., Markevich, E., Salitra, G., Aurbach, D., Garsuch, A., and Lampert, J., A new advanced lithium-ion battery: Combination of high performance amorphous columnar silicon thin film anode, 5V LiNi0.5Mn1.5O4 spinel cathode and fluoroethylene carbonate-based electrolyte solution, Electrochem. commun., 2013, vol. 33, p. 31.
Hu, L., Amine, K., and Zhang, Z., Fluorinated electrolytes for 5-V Li-ion chemistry: Dramatic enhancement of LiNi0.5Mn1.5O4/graphite cell performance by a lithium reservoir, Electrochem. commun., 2014, vol. 44, p. 34.
Fan, X. and Wang, C., High-voltage liquid electrolytes for Li batteries: progress and perspectives, Chem. Soc. Rev., 2021, vol. 50, no.18, p. 10486.
Ling, J., Karuppiah, Ch., Krishnan, S.G., Reddy, M.V., Misnon, I.I., Ab Rahim, M.H., Yang, Ch.-Ch., and Jose, R., Phosphate Polyanion Materials as High-Voltage Lithium-Ion Battery Cathode: A Review, Energy & Fuels., 2021, vol. 35, no. 13, p. 10428.
Ji, X., Xia, Q., Xu, Y., Feng, H., Wang, P., and Tan, Q., A review on progress of lithium-rich manganese-based cathodes for lithium-ion batteries, J. Power Sources., 2021, vol. 487, p. 229362.
Han, J., Kim, K., Lee, Y., and Choi, N.-S., Scavenging Materials to Stabilize LiPF6 Containing Carbonate-Based Electrolytes for Li-Ion Batteries, Adv. Mater., 2019, vol. 31, no. 20, p. 1804822.
Zhao, W., Ji, Y., Zhang, Zh., Lin, M., Wu, Z., Zheng, Xi, Li, Qi, and Yang, Y., Recent advances in the research of functional electrolyte additives for lithium-ion batteries, Curr. Opin. Electrochem., 2017, vol. 6, no. 1, p. 84.
Xu, K., Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries, Chem. Rev., 2004, vol. 104, no. 10, p. 4303.
Xu, K., Electrolytes, and Interphases in Li-Ion Batteries and Beyond, Chem. Rev., 2014, vol. 114, no. 23, p. 11503.
Borodin, O., Challenges with prediction of battery electrolyte electrochemical stability window and guiding the electrode–electrolyte stabilization, Curr. Opin. Electrochem., 2019, vol. 13, p. 86.
Xu, K., Zhuang, G.V., Allen, J.L., Lee, U., Zhang, Sh.S., Ross, Ph.N., and Jow, T.R., Syntheses and Characterization of Lithium Alkyl Mono- and Dicarbonates as Components of Surface Films in Li-Ion Batteries, J. Phys. Chem. B, 2006, vol. 110, no. 15, p. 7708.
Nazri, M., Liquid Electrolytes: Some Theoretical and Practical Aspects, Lithium Batteries. Boston, MA: Springer US, 2009. p. 509–529.
Кулова, Т.Л., Скундин, А.М. Высоковольтовые материалы положительных электродов литий-ионных аккумуляторов (обзор). Электрохимия. 2016. Т. 52. С. 563.
Watanabe, Y., Kinoshita, Sh., Wada, S., Hoshino, K., Morimoto, H., and Tobishima, Sh., Electrochemical properties, and lithium-ion solvation behavior of sulfone–ester mixed electrolytes for high-voltage rechargeable lithium cells, J. Power Sources., 2008, vol. 179, no. 2, p. 770.
Abouimrane, A., Belharouak, I., and Amine K., Sulfone-based electrolytes for high-voltage Li-ion batteries, Electrochem. commun., 2009, vol. 11, no. 5, p. 1073.
Wang, X., Xue, W., Hu, K., Li, Y., Li, Y., and Huang, R., Adiponitrile as Lithium-Ion Battery Electrolyte Additive: A Positive and Peculiar Effect on High-Voltage Systems, ACS Appl. Energy Mater., 2018, vol. 1, no. 10, p. 5347.
Li, S., Zhao, D., Wang, P., Cui, X., and Tang, F., Electrochemical effect, and mechanism of adiponitrile additive for high-voltage electrolyte, Electrochim. Acta, 2016, vol. 222, p. 668.
Lu, Y., Xu, Sh., Shu, J., Aladat, W.I.A., and Archer, L.A., High voltage LIB cathodes enabled by salt-reinforced liquid electrolytes, Electrochem. commun., 2015, vol. 51, p. 23.
Xue, Z.-M., Zhao, B.-H., and Chen, C.-H., A new lithium salt with 3-fluoro-1,2-benzenediolato and lithium tetrafluoroborate for lithium battery electrolytes, J. Power Sources., 2011, vol. 196, no. 15, p. 6478.
Bushkova, O.V., Yaroslavtseva, T.V., and Dobrovolsky, Y.A., New lithium salts in electrolytes for lithium-ion batteries (Review), Russ. J. Electrochem., 2017, vol. 53, no. 7, p. 677.
Xu, M., Liu, Y., Li, B., Li, W., Li, X., and Hu, Sh., Tris(pentafluorophenyl) phosphine: An electrolyte additive for high voltage Li-ion batteries, Electrochem. commun., 2012, vol. 18, p. 123.
Xing, L. and Borodin, O., Oxidation induced decomposition of ethylene carbonate from DFT calculations – Importance of explicitly treating surrounding solvent, Phys. Chem. Chem. Phys., 2012, vol. 14, no. 37, p. 12838.
Xing, L., Vatamanu, J., Borodin, O., Smith, G.D., and Bedrov, D., Electrode/electrolyte interface in sulfolane-based electrolytes for Li ion batteries: A molecular dynamics simulation study, J. Phys. Chem. C, 2012, vol. 116, no. 45, p. 23871.
Narayanan, K.A., Oldiges, K., Winter, M., Heuer, A., Cekic-Laskovic, I., Holm, Ch., and Smiatek, J., Electrolyte solvents for high voltage lithium-ion batteries: Ion correlation and specific anion effects in adiponitrile, Phys. Chem. Chem. Phys. Royal Soc. Chem., 2018, vol. 20, no. 40, p. 25701.
Borodin, O., Behl, W., and Jow, T.R., Oxidative Stability and Initial Decomposition Reactions of Carbonate, Sulfone, and Alkyl Phosphate-Based Electrolytes, J. Phys. Chem. C., 2013, vol. 117, no. 17, p. 8661.
Zhang, X., Pugh, J.K., and Ross, P.N., Computation of Thermodynamic Oxidation Potentials of Organic Solvents Using Density Functional Theory, J. Electrochem. Soc., 2001, vol. 148, no. 5, p. E183.
Borodin, O. and Jow, T.R., Quantum Chemistry Studies of the Oxidative Stability of Carbonate, Sulfone and Sulfonate-Based Electrolytes Doped with ${\text{BF}}_{4}^{ - },$ ${\text{PF}}_{6}^{ - }$ Anions, ECS Trans, 2019, vol. 33, no. 28, p. 77.
Han, Y.-K., Yoo, J., and Yim, T., Computational screening of phosphite derivatives as high-performance additives in high-voltage Li-ion batteries, RSC Adv., 2017, vol. 7, no. 32, p. 20049.
Yu, Z., Wang, H., Kong, X., Huang, W., Tsao, Y., Mackanic, D.G., Wang, K., Wang, X., Huang, W., Choudhury, S., Zheng, Yu, Amanchukwu, Ch.V., Hung, S.T., Ma, Yu, Lomeli, E.G., Qin, J., Cui, Yi, and Bao, Zh., Molekular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries, Nat. Energy., 2020, vol. 5, no. 7, p. 526.
Borodin, O., Olguin, M., Spear, C.E., Leiter, K.W., and Knap, J., Towards high throughput screening of electrochemical stability of battery electrolytes, Nanotechnology, 2015, vol. 26, no. 35, p. 354003.
Zhang, J., Yang, J., Yang, L., Lu, H., Liu, H., and Zheng, B., Exploring the redox decomposition of ethylene carbonate-propylene carbonate in Li-ion batteries, Mater. Adv., 2021, vol. 2, no. 5, p. 1747.
Seo, D.M., Borodin, O., Balogh, D., O’Connell, M., Ly, Q., Han, S.-D., Passerini, St., and Henderson, W.A., Electrolyte Solvation, and Ionic Association III. Acetonitrile-Lithium Salt Mixtures–Transport Properties, J. Electrochem. Soc, 2013, vol. 160, no. 8, p. A1061.
Hou, T., Yang, G., Rajput, N.N., Self, J., Park, S.-W., Nanda, J., and Persson, K.A., The influence of FECon the solvation structure and reduction reaction of LiPF6/ECelectrolytes and its implication for solid electrolyte interphase formation, Nano Energy, 2019, vol. 64, p. 103881.
Cheeseman, J.R., Trucks, G.W., Keith, T.A., and Frisch, M.J., A comparison of models for calculating nuclear magnetic resonance shielding tensors, J. Chem. Phys. 1996, vol, 104, no. 14, p. 5497.
Borodin, O., Behl, W., and Jow, T.R., Oxidative stability and initial decomposition reactions of carbonate, sulfone, and alkyl phosphate-based electrolytes, J. Phys. Chem. C. 2013, vol. 117, no. 17, p. 8661.
Delp, S.A., Borodin, O., Olguin, M., Eisner, C.G., Allen, J.L., Jow, T., and Richard, et al., Importance of Reduction and Oxidation Stability of High Voltage Electrolytes and Additives, Electrochim. Acta, 2016, vol. 209, p. 498.
Tomasi, J., Mennucci, B., and Cammi, R., Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, vol. 105, no. 8, p. 2999.
Zhang, J., Yang, J., Yang, L., Lu, H., Liu, H., and Zheng, B., Exploring the redox decomposition of ethylene carbonate–propylene carbonate in Li-ion batteries, Mater. Adv., 2021, vol. 2, no. 5, p. 1747.
Abe, K., Hattori, T., Kawabe, K., Ushigoe, Y., and Yoshitake, H., Functional Electrolytes, J. Electrochem. Soc, 2007, vol. 154, no. 8, p. A810.
Abe, K., Miyoshi, K., Hattori, T., Ushigoe, Y., and Yoshitake, H., Functional electrolytes: Novel type additives for cathode materials, providing high cycleability performance, J. Power Sources, 2006, vol. 153, no. 2, p. 328.
Xu, K., Ding, S.P., and Jow, T.R., Toward Reliable Values of Electrochemical Stability Limits for Electrolytes, J. Electrochem. Soc., 1999, vol. 146, no. 11, p. 4172.
Azcarate, I., Yin, W., Méthivier, Ch., Ribot, Fr., Laberty-Robert, Ch., and Grimaud, A., Assessing the Oxidation Behavior of EC:DMC Based Electrolyte on Non-Catalytically Active Surface, J. Electrochem. Soc., 2020, vol. 167, no. 8, p. 080530.
Abe, K., Miyoshi, K., Hattori, T., Ushigoe, Y., and Yoshitake, H., Functional electrolytes: Synergetic effect of electrolyte additives for lithium-ion battery, J. Power Sources, 2008, vol. 184, no. 2, p. 449.
Mahmood, N., Tang, T., and Hou, Y., Nanostructured Anode Materials for Lithium-Ion Batteries: Progress, Challenge and Perspective, Adv. Energy Mater, 2016, vol. 6, no. 17, p. 1600374.
Smith, L. and Dunn, B., Opening the window for aqueous electrolytes, Science, 2015, vol. 350, no. 6263, p. 918.
Sanginov, E.A., Borisevich, S.S., Kayumov, R.R., Istomina, A.S., Evshchik, E.Yu., Reznitskikh, O.G., Yaroslavtseva, T.V., Melnikova, T.I., Dobrovolsky, Yu.A., and Bushkova, O.V., Lithiated Nafion plasticised by a mixture of ethylene carbonate and sulfolane, Electrochim. Acta, 2021, vol. 373, p. 137914.
Shaw, D.E., Schrödinger: Desmond Molecular Dynamics System: 2021–4. N.Y.: Schrödinger, 2021.
Lu, C., Wu, Ch., Ghoreishi, D., Chen, W., Wang, L., Damm, W., Ross, G.A., Dahlgren, M.K., Russell, E., Von Bargen, Ch.D., Abel, R., Friesner, R.A., and Harder, E.D., OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space, J. Chem. Theory Comput., 2021, vol. 17, no. 7, p. 4291.
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G.A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A.V., Bloino, J., Janesko, B.G., Gomperts, R., Mennucci, B., and Hratch D.J., GAUSSIAN: Revision C.01. Wallingford CT: Gaussian, Inc., 2016.
Zhao, Y., Schultz, N.E., and Truhlar, D.G., Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2006, vol. 2, no. 2, p. 364.
Schäfer, A., Huber, C., and Ahlrichs, R., Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, vol. 100, no. 8, p. 5829.
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, vol. 132, no. 15, p. 154104.
Marenich, A.V., Cramer, C.J., and Truhlar, D.G., Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B., 2009, vol. 113, no. 18, p. 6378.
Morris, D.F.C. and Short, E.L., The Born-Fajans-Haber Correlation, Nature, 1969, vol. 224, no. 5223, p. 950.
Перелыгин, И.С. Инфракрасные спектры и сольватация ионов. В кн.: Ионная сольватация. М.: Наука, 1987. С. 100–199.
Han, Y.-K., Lee, K., Yoo, J., and Huh, Y.-S., Virtual screening of borate derivatives as high-performance additives in lithium-ion batteries, Theor. Chem. Acc., 2014, vol. 133, no. 10, p. 1562.
Curtiss, L.A., Redfern, P.C., and Raghavachari, K., Gaussian-4 theory, J. Chem. Phys., 2007, vol. 126, no. 8, p. 084108.
Дополнительные материалы отсутствуют.