

Электрохимия, 2022, T. 58, № 12, стр. 848-859
Электрохимическое соосаждение Sm и Co в дицианамидной ионной жидкости
Е. Б. Молодкина a, М. Р. Эренбург a, А. В. Руднев a, *
a Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, Россия
* E-mail: rudnev@phyche.ac.ru
Поступила в редакцию 18.01.2022
После доработки 11.07.2022
Принята к публикации 18.07.2022
- EDN: CKTQUU
- DOI: 10.31857/S0424857022120052
Аннотация
Электрохимическое осаждение лантаноидов и сплавов на их основе может быть реализовано при комнатной температуре из органических ионных сред, таких как ионные жидкости (ИЖ). В настоящей работе мы изучили электроосаждение Sm и Co и их соосаждение в ИЖ [BMP][DCA] (1-бутил-1-метилпирролидиний дицианамид) с контролируемым содержанием воды электрохимическими методами в сочетании со сканирующей зондовой микроскопией и рентгеновским анализом образующихся осадков. Показано, что как Co, так и Sm могут быть электрохимически осаждены из соответствующих однокомпонентных растворов в выбранной ИЖ, и потенциал их осаждения смещается в положительную сторону по мере увеличения концентрации воды (ускорение осаждения). В растворе ИЖ, содержащем ионы обоих металлов (Sm(III) и Co(II)), наблюдается соосаждение Sm–Co. При этом Sm соосаждается при потенциалах, существенно менее отрицательных (на 0.42 В), чем осаждение Sm из раствора ИЖ, содержащего только ионы Sm(III). Однако увеличение концентрации воды в растворе приводит к торможению соосаждения Sm–Co. Мы связываем торможение соосаждения Sm‒Co в присутствии воды с формированием оксидной пленки на поверхности осадка, препятствующей дальнейшему восстановлению ионов Co и, как следствие, соосаждению Sm–Co.
ВВЕДЕНИЕ
Разработка электрохимических методов получения функциональных материалов на основе лантаноидов является актуальной задачей для различных областей промышленности (микроэлектронная промышленность, медицина, хранение и конверсия энергии) [1]. Электрохимическое осаждение сплавов лантаноид–металл группы VIIIB (Fe, Co, Ni) привлекает большое внимание, поскольку эти материалы могут обладать превосходными магнитными свойствами. В частности, магниты на основе сплавов SmCo обладают высокой температурой Кюри (720–920°C) и термостойкостью, что обуславливает их перспективность для применения при высоких температурах [2]. Один из способов получения самария, как и других лантаноидов, – электроосаждение из расплавов солей, которое представляет собой довольно энергозатратный метод [3]. Поэтому важной задачей является поиск новых электрохимических сред, подходящих для осаждения лантаноидов и сплавов на их основе при низких температурах.
Электроосаждение лантаноидов и их соосаждение с другими металлами можно реализовать в ионных органических средах, таких как ионные жидкости (ИЖ) [4–9]. ИЖ обладают уникальным набором физико-химических свойств и позволяют электрохимически осаждать широкий спектр химических элементов, сплавов и оксидов [10, 11], а также композитных материалов [12]. Недавно в нескольких работах было продемонстрировано электрохимическое соосаждение лантаноидов с Fe, Co или Ni в различных безводных ионных средах [6], таких как ИЖ, глубокие эвтектические растворители (ГЭР) и низкотемпературные расплавы солей: Nd–Fe [4, 13], Sm–Co [14–20], Sm–Fe, Gd–Co, La–Ni [21]. При этом было показано, что электровосстановление Fe, Co или Ni (имеющих более положительный стандартный электрохимический потенциал, чем лантаноиды) приводит к ускорению соосаждения лантаноида, заметно сдвигая потенциал его осаждения в положительную сторону: этот сдвиг может превышать 0.3 В, как, например, в случае соосаждения Nd–Fe [4, 13]. В то же время, механизм соосаждения в безводных ионных средах до конца не ясен. Кроме того, поскольку многие ИЖ гигроскопичны, особый интерес представляет изучение влияния воды на процессы (со)осаждения металлов в ИЖ. Отдельные работы показали ускорение электроосаждения металлов, в том числе лантаноидов, в ИЖ при увеличении содержания воды в растворе [4–6, 8, 22, 23]. Тем не менее, систематические исследования этого явления пока отсутствуют.
Ранее нашей группой было изучено влияние добавок воды на соосаждение Nd и Fe в ИЖ [BMP][DCA] (1-бутил-1-метилпирролидиний дицианамид) [4]. Было показано, что, хотя добавление воды (вплоть до концентрации ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 3.092 М) ускоряет осаждение Fe и, в особенности, Nd из ИЖ, содержащей ионы только одного металла, соосаждение Nd–Fe, наоборот, тормозится. В данной работе мы исследовали процессы (со)осаждения Sm, Co и Sm–Co в [BMP][DCA], а также влияние добавок воды на данные процессы. Полученные результаты проанализированы в сравнении с данными по соосаждению Nd–Fe.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Растворы готовили на основе ИЖ [BMP][DCA] (Solvionic, ≥99.5%), высушенной в вакууме при 60–80°С, и безводных солей металлов Sm(OTf)3 (OTf – трифторметансульфонат, Alfa Aesar, >98%) и CoCl2 (Alfa Aesar, >99.7%). Взвешивание реагентов, приготовление и хранение растворов, заполнение ячейки проводили в перчаточном боксе в атмосфере Ar высокой чистоты (99.998%).
Для электрохимических измерений применяли герметичную стеклянную ячейку с одним отделением (объем ИЖ в ячейке 1.8 мл). Перед работой ячейку в разобранном виде обрабатывали горячим раствором 25% HNO3 с последующим кипячением и промыванием в воде Milli-Q (>18 МОм см, <5 ppb по углероду). Далее все части ячейки сушили при 105°C. Ячейку собирали в горячем состоянии и заполняли ИЖ в перчаточном боксе в атмосфере Ar. В качестве вспомогательного электрода использовали платиновую фольгу. В качестве электрода сравнения применяли герметичный Ag/AgCl-электрод сравнения (модель ET072, eDAQ, Австралия; концентрация KCl 3.4 М). До измерений через раствор продували Ar в течение 40 мин, а во время измерений Ar продували над раствором. Для работы использовали изготовленные по методу Клавилье [24] монокристаллы Pt(111) с площадью рабочих граней 0.030–0.040 см2.
Монокристаллические электроды отжигали в пламени горелки, а затем охлаждали в потоке Ar. Далее электрод вносили в ячейку и формировали мениск при потенциале E = 0.0 В, при котором отсутствовали фарадеевские процессы. Для осаждения РЗЭ с последующим анализом элементного состава осадка использовали поликристаллический электрод Pt, вырезанный из фольги (площадь 0.30–0.40 см2). Электрод Pt полировали механически с помощью частиц оксида алюминия (сначала частицы диаметром 1 мкм, затем 0.05 мкм для финишной предобработки). Далее электрод очищали обработкой в ультразвуковой ванне в воде Milli-Q, выдерживали в концентрированной H2SO4 в течение 5 мин, промывали водой Milli-Q и отжигали в пламени горелки непосредственно перед применением. Для электрохимических измерений использовали потенциостат и программное обеспечение, разработанные в Институте физической химии и электрохимии им. А.Н. Фрумкина РАН. Все измерения проводили при 24 ± 1°C.
Добавки воды вводили в ИЖ с помощью дозатора непосредственно в ячейку, далее раствор перемешивали, пропуская через него аргон. Содержание воды в ИЖ определяли с помощью кулонометрического титрования по Карлу Фишеру (917 Ti-Touch, Metrohm, Швейцария). Измерения показали, что в ИЖ после сушки концентрация воды обычно не превышает 200 ppm, что соответствует 0.012 М (молярная концентрация воды, ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$). Далее мы считаем, что в ИЖ без добавок воды ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.012 М. В ходе эксперимента невозможно исключить незначительные изменения концентрации воды вследствие продувки аргоном и кратковременных разгерметизаций ячейки для помещения или удаления рабочего электрода. Однако воспроизводимость ЦВА свежеотожженного электрода Pt(111) (в частности, характерного пика осаждения Sm, потенциал которого сильно зависит от концентрации воды) в течение нескольких часов указывает на то, что вероятные изменения в концентрации воды в растворе незначительны и не влияют на полученные результаты.
Все потенциалы в статье приведены относительно насыщенного хлоридсеребряного электрода (Ag/AgCl). Поскольку в электроде сравнения присутствовал водный раствор KCl, на границе раздела вода/ИЖ мог возникать потенциал жидкостного соединения, причем соответствующий скачок потенциала мог также зависеть от концентрации воды в ИЖ. По этой причине Ag/AgCl-электрод калибровали относительно редокс-пары ферроцен/ферроцений (Fc/Fc+). Формальный потенциал E1/2(Fc/Fc+) в [BMP][DCA] с добавками воды оценивали по циклическим вольтамперограммам (ЦВА) как полусумму потенциалов анодного и катодного пиков. Показано, что добавление самого большого количества воды (3.092 M) вызывало сдвиг E1/2(Fc/Fc+) в отрицательную сторону всего лишь на 13 мВ. Таким образом, можно принять, что дополнительный скачок потенциала, связанный с возникновением потенциала жидкостного соединения при добавлении воды в ИЖ, в наших экспериментах пренебрежимо мал. Потенциал электрода Ag/AgCl относительно E1/2(Fc/Fc+) составлял –0.400 В отн. Fc/Fc+. Подробности описаны в наших предыдущих публикациях [4, 5].
Осадки Sm, Co и Sm–Co получали в потенциостатическом режиме. Электроды с осадками промывали ацетоном (ос. ч.) и абсолютным спиртом (Merck) и далее сушили в потоке Ar. Морфологию поверхности осадков исследовали методом атомной силовой микроскопии (АСМ) на сканирующем зондовом микроскопе Solver Pro (НТ-МДТ, Россия) в полуконтактном режиме. Обработку изображений АСМ проводили с помощью программы WSxM [16]. АСМ-анализ выполняли как минимум для трех различных участков на поверхности электрода. В статье приведены АСМ-изображения, типичные для соответствующего образца. Отметим, что поверхность исходного электрода Pt(111) плоская и состоит из широких атомарно-гладких террас и различных структурных дефектов (ступеней, спиральных дислокаций и т.д.) [4, 5]. Таким образом, с помощью АСМ можно с легкостью идентифицировать образование осадка на плоских монокристаллических поверхностях.
Исследования методом рентгеновской фотоэлектронной спектроскопии (РФЭС) проводили ex situ. РФЭ-спектры были получены с использованием источника монохроматического рентгеновского излучения XM1000, установленного на спектрометре OMICRON ESCA+ с алюминиевым анодом (энергия излучения 1486.6 эВ, мощность 252 Вт). Нейтрализатор заряда CN-10 работал при токе эмиссии 4 мкА и энергии пучка 1 эВ для устранения локального заряда на анализируемой поверхности. Энергия пропускания анализатора была установлена на уровне 50 эВ для обзорного спектра и 20 эВ для отдельных элементов. Калибровку спектрометра проводили по линии Au4f7/2 при 84.1 эВ. Базовое давление в камере анализатора составляло не более 10–9 мбар. Все спектры накапливались не менее 3 раз, флуктуация положения пиков не превышала ±0.1 эВ. Травление поверхности проводили с помощью ионной пушки FDG150, работающей при энергии ионов 4 кэВ и ионном токе 14.5 мкА в течение 8 мин. Чистота газа Ar составляла 99.999%.
Элементный состав осадков оценивали с помощью энергодисперсионной рентгеновской спектроскопии (ЭДРС) на растровом электронном микроскопе (РЭМ) JSMU3, оборудованном анализатором WINEDS (Германия).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Электрохимическое окно потенциалов, отвечающее стабильности ИЖ, определяли на основе ЦВА [5]. Кривая 1 на рис. 1а, б демонстрирует рост катодного и анодного токов на Pt(111) при ‒1.83 и 1.54 В. Эти процессы, обозначенные как К0 и А0, соответствуют восстановлению и окислению компонентов ИЖ [25]. Стоит также отметить, что электрохимическое окно может определяться также и стабильностью подложки. Так, ранее с помощью сканирующей туннельной микроскопии (СТМ) мы показали, что электрод Au(111) подвергается электрохимическому растворению при потенциалах > 0.5 В и химическому травлению в области потенциалов от –0.4 до –0.8 В в [BMP][DCA] [26]. В то же время было обнаружено, что поверхность электрода Pt(111) в [BMP][DCA] не подвергается структурным изменениям в широком диапазоне потенциалов [27]. Катодный процесс К0 в литературе связывают с восстановлением катиона [BMP]+ до бутилена и водорода [28].
Рис. 1.
Первые циклы ЦВА Pt(111) в: (а) [BMP][DCA] (кривая 1) и [BMP][DCA] + 0.01 М Sm(OTf)3 (кривая 2), (б) [BMP][DCA] (кривая 1) и [BMP][DCA] + 0.01 М CoCl2 (кривая 3), (в) [BMP][DCA] + 0.01 М Sm(OTf)3 + 0.01 M CoCl2 (кривая 4); кривые 2 и 3 добавлены для сравнения. На вставке для лучшей видимости показаны увеличенные фрагменты ЦВА (кривые 2–4). Скорость развертки потенциала – 0.01 В/с. Содержание остаточной воды во всех растворах составляло 0.012 М (без добавки воды).
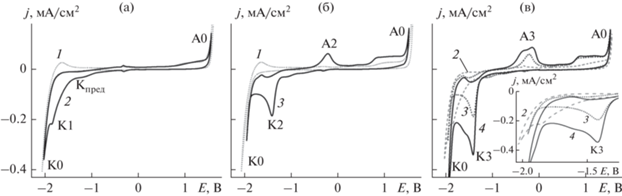
На ЦВА Pt(111) в растворе, содержащем ионы Sm(III) (рис. 1а), в отсутствие добавок воды (исходная концентрация воды в данном случае ~0.012 М), помимо процесса катодного разложения ИЖ (К0), наблюдается дополнительная катодная волна К1 при Е = –1.84 В, которую мы приписываем восстановлению Sm(III) до Sm(0). Этот катодный процесс протекает необратимо, поскольку не наблюдается анодного противопика. Следует также отметить, что волна К1 в большой степени перекрывается с К0, т.е. электросаждение Sm протекает параллельно с катодным разложением ИЖ. Более того, на втором последовательно измеренном цикле ЦВА волна К1 отсутствует, что вероятно, свидетельствует об образовании на поверхности осадка пленки продуктов разложения ИЖ и/или оксидов Sm, блокирующей дальнейшее осаждение. При потенциале около –1.2 В, т.е. положительнее К1, можно наблюдать предволну (обозначена как Кпред). Ранее эта волна была приписана восстановлению Sm(III) до Sm(II) [18, 29, 30]. Было показано, что при комнатной температуре предволна одноэлектронного восстановления лантаноидов выражена слабо, но становится более заметной при повышении температуры до 70–100°C [6, 31].
В случае осаждения Co наблюдаются четко выраженный катодный пик К2 при Е = –1.42 В и анодный противопик растворения осадка А2 (кривая 3 на рис. 1б). Потенциалы начала осаждения и растворения Co значительно разнятся (>0.6 В), что говорит о том, что осаждение Co происходит лишь при достижении значительного перенапряжения и/или растворение осадка сильно заторможено. Во втором цикле ЦВА осаждение Co начинается при более отрицательных потенциалах, что говорит о деактивации поверхности электрода по мере циклирования потенциала.
На ЦВА в растворе ИЖ, содержащей ионы обоих компонентов Sm(III) и Co(II) (кривая 4 на рис. 1в), наблюдается четко выраженный катодный пик К3, потенциал которого совпадает по потенциалам с пиком К2 (–1.42 В), наблюдаемым в растворе, содержащем только ионы Co(II). Однако пик К3 существенно выше К2. Учитывая то, что в случае процесса c диффузионными ограничениями высота пика увеличивается с концентрацией реагента, можно сделать вывод о соосаждении Sm с Co при потенциалах пика К3, т.е. примерно на 0.42 В положительнее, чем осаждение Sm из соответствующего однокомпонентного раствора. Подобное ускорение соосаждения лантаноида наблюдалось нами и для системы Nd–Fe [4]. На анодной развертке потенциала наблюдается анодный процесс А3, который можно приписать растворению Sm–Co.
Выводы, сделанные на основе ЦВА, подтверждаются с помощью исследований методами атомно-силовой микроскопии, ЭДРС и РФЭС осадков, полученных в потенциостатическом режиме в [BMP][DCA], содержащей соответствующие соли. На рис. 2 представлены АСМ-изображения осадков на Pt(111) и соответствующие потенциостатические транзиенты тока. Потенциалы осаждения были выбраны вблизи максимумов катодных пиков К1–К3 (рис. 1в), т.е. для осаждения Sm – вблизи К1, для осаждения Co – вблизи К2 и для соосаждения Sm–Co – вблизи К3. Осадки Sm имеют неравномерную зернистую структуру с перепадом высот от 100 до 300 нм (рис. 2б). Вероятно, неравномерность осадка связана с параллельным протеканием процесса катодного разложения ИЖ, при котором продукты разложения адсорбируются на поверхности и препятствуют дальнейшему росту осадка. Тем не менее, данные АСМ подтверждают формирование осадка при потенциале, близком к К1. При осаждении Co осадки получаются сплошными и равномерными, с мелкозернистой структурой (рис. 2в). Перепад высот обычно не превышает нескольких нанометров. На поле 10 × 10 мкм2 даже прослеживаются структурные дефекты (пакеты ступеней), характерные для поверхностей монокристаллических электродов Pt [32].
Рис. 2.
(а) Потенциостатические транзиенты тока Pt(111): 1 – в [BMP][DCA] + 0.01 М Sm(OTf)3 при –1.800 В, 2 – в [BMP][DCA] + 0.01 М CoCl2 при –1.380 В, 3 – в [BMP][DCA] + 0.01 M Sm(OTf)3 + 0.01 М CoCl2 при –1.385 В в течение 1200 с. АСМ-изображения осадков на (б–г) соответствуют транзиентам тока: 1 (б), 2 (в), 3 (г) . Для каждого осадка представлены два изображения: с большей (10 × 10 мкм2) и меньшей площадью сканирования (2 × 2 мкм2). Профили поперечного сечения, показанные справа, были получены вдоль белых пунктирных линий на соответствующих изображениях АСМ.

При соосаждении Sm–Co при потенциале вблизи максимума К3 (–1.445 В в течение 1200 с) осадки также сплошные и зернистые (рис. 2г) с перепадом высот от нескольких единиц до сотни нм. Таким образом, осадок Sm–Co обладает шероховатостью, промежуточной между осадками Sm и Co. Интересно, что транзиенты катодного тока осаждения Sm–Co проходят через максимум при ~10 с (кривая 3 на рис. 2а). Максимум тока обычно характерен для низких перенапряжений, когда зарождаются и растут отдельные кристаллиты в отсутствие диффузионных ограничений: катодный ток сначала растет за счет увеличения площади роста кристаллитов, а затем снижается вследствие перекрывания диффузионных зон. Однако в нашем случае потенциалы осаждения были достаточно отрицательными (вблизи максимума К3, т.е. в области существенных диффузионных ограничений) и морфология осадка на АСМ-изображениях не соответствует росту отдельных кристаллитов. Таким образом, наличие пика на транзиенте тока, вероятно, связано с механизмом соосаждения Sm–Co. В работе Xu и соавторов [13], исследовавших соосаждение Nd–Fe в [EMIm][DCA], предполагалось, что изначально формируется тонкий слой железа и уже затем происходит соосаждение Nd–Fe. Это предположение согласуется с формой транзиента тока соосаждения Sm–Co, на котором увеличение катодного тока при t > 5 с может быть связано с началом соосаждения Sm, в то время как пик при t = = 10 с – с диффузионными ограничениями (обеднением приэлектродного слоя как по ионам Co(II), так и по ионам Sm(III)).
Осадки для РФЭС и рентгеноэлементного анализа получали на поликристаллическом Pt-электроде в потенциостатическом режиме в течение 3600 с. На рис. 3 представлены ЭДР- и РФЭ-спектры Sm3d5/2 осадков Sm, полученных в растворе, содержащем только ионы Sm(III), при потенциале вблизи пика К1. ЭДР-спектр подтверждает наличие Sm в осадке (рис. 3а). Согласно литературным данным, РФЭС-сигналы при 1084.1 и 1081.7 эВ отвечают Sm3d5/2 в оксиде Sm2O3 и металлическому Sm [33–35] (обозначены вертикальными пунктирными линиями на рис. 3б). Разложение сигнала Sm3d5/2 на линии, отвечающей Sm(0) и Sm(III), указывает на то, что Sm в осадке находится как в металлической, так и в окисленной формах. Наши данные согласуются с данными РФЭС для осадка Sm, полученного в той же ИЖ в ранее опубликованной работе [29].
Рис. 3.
(а) ЭДР- и (б) РФЭ-спектры осадков Sm, полученных на Pt-фольге в течение 3600 с при –1.810 В в растворе [BMP][DCA] + 0.01 М Sm(OTf)3. На (б) представлены кривые: 1 – экспериментальный спектр после вычитания фоновой кривой; 2, 3 – спектральные компоненты; 4 – суммарная кривая. Вертикальными пунктирными линиями на (а) показаны положения пиков для различных состояний окисления Sm согласно литературным данным [33–35].

РФЭ-спектры Sm3d5/2 и Co2p3/2 для осадка Sm‒Co, полученного при потенциале вблизи максимума К3, показаны на рис. 4. Спектр Sm3d5/2 на рис. 4а схож со спектром, полученным для осадка Sm (рис. 3б). Разложение сигнала также указывает на то, что Sm находится в осадке в виде Sm(III) и Sm(0) (рис. 5а). Травление ионами Ar+ осадка Sm–Co не приводит к заметным изменениям в распределении пиков (рис. 5б), что свидетельствует о том, что и внутренние слои осадка содержат значительное количество Sm в окисленной форме.
Рис. 4.
РФЭ-спектры (а) Sm3d5/2 и (б) Co2p3/2 осадка Sm–Co на Pt-фольге, полученного в течение 3600 с при –1.445 В в растворе [BMP][DCA] + 0.01 М Sm(OTf)3 + 0.01 М CoCl2. РФЭ-спектры получены до (спектры 1) и после травления Ar+ (спектры 2). Вертикальными пунктирными линиями на (а) показаны положения пиков для различных состояний окисления Sm согласно литературным данным [33–35].

Рис. 5.
РФЭ-спектры Sm3d5/2 из рис. 3а: (а) до и (б) после травления. Кривые: 1, 1' – экспериментальные спектры после вычитания фоновой кривой; 2, 2 ', 3, 3 ' – спектральные компоненты; 4, 4 ' – суммарная кривая. Вертикальными пунктирными линиями показаны положения пиков для Sm согласно литературным данным [33–35].

На спектре Co2p3/2 до травления (спектр 1 рис. 4б) можно увидеть сигнал, соответствующий Co(II): согласно литературе, линия при 781.3 эВ соответствует Co(OH)2 [36]. Линии CoO, Co3O4 и CoOOH обычно наблюдались при 780.0–780.2 эВ [36–38], и в спектре в нашей работе выражены значительно слабее, чем линия Co(OH)2. После травления ионами Ar+ (8 мин при энергии 4 кэВ) линия Co(II) исчезает (спектр 2 рис. 4б), и появляется четкая линия при 778.6 эВ, приписываемая в литературе металлическому Co [36, 39]. Известно, что травление Ar+ может вызывать восстановление оксидов некоторых металлов, однако Choudhury и соавторы показали, что травление при 5 кэВ в течение 10 мин не приводило к образованию металлического Co [1]. Появление линии металлического Со наблюдалось лишь спустя 30 мин после начала травления. Таким образом, можно предположить, что во внутренних слоях осадка Sm–Co Co находится в основном в металлическом состоянии, в то время как окисление Co на поверхности осадка, по-видимому, происходило в результате контакта осадка с воздухом.
На ЭДР-спектрах осадка при –1.445 В (т.е. вблизи максимума К3) и –1.595 В наблюдаются пики Sm и Co (рис. 6). Атомное соотношение Sm : Co, по данным ЭДРС, составляло 1 : 13 в обоих случаях. Наличие кислорода и углерода на ЭДР-спектрах подтверждает включение в осадок продуктов разложения ИЖ и окисления Sm и Co. Можно предположить, что образование оксидов/гидроксидов Sm вызвано химической реакцией с остаточной водой в растворе ИЖ, концентрация которой (~0.012 М) сравнима с концентрацией ионов Sm(III) (0.010 М).
Рис. 6.
ЭДР-спектры осадков Sm–Co, полученных на Pt-фольге в течение 3600 с при –1.445 В (спектр 1) и ‒1.595 В (спектр 2) в растворе [BMP][DCA] + 0.01 М Sm(OTf)3 + 0.01 М CoCl2.

Влияние воды на электроосаждение Sm и Co, а также на их соосаждение было исследовано в растворах с контролируемым содержанием воды. На рис. 7 показаны ЦВА Pt(111) в растворах ИЖ [BMP][DCA], содержащих ионы Sm(III) или Co(II), без добавки и с добавками воды. В первую очередь, отметим, что увеличение ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ приводит к существенному сдвигу начала катодного разложения ИЖ (волна К0) в сторону менее отрицательных потенциалов. По мере увеличения ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ пики К1 и К2 также смещаются в положительную сторону, что указывает на ускорение процессов осаждения Sm и Co соответственно. При этом при добавлении 3.092 М H2O сдвиг пика осаждения Sm составляет 0.78 В, что существенно больше, чем сдвиг пика осаждения Co, равный 0.15 В. Зависимости потенциалов пиков от ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ приведены на рис. 8. Интересно, что при ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ ≥ 1.552 М осаждение Sm происходит при потенциалах более положительных, чем осаждение Co. Отметим также, что интенсивность пика А2 (окисление осадка Co) при ~ –0.30 В снижается при добавлении воды, и при ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 3.092 М пик А2 практически отсутствует (рис. 7б). При этом существенно возрастают анодные токи при Е > 0.4 В. Можно предположить, что в присутствии значительного количества воды в ИЖ электроосаждение Co сопровождается его химическим окислением с образованием оксида/гидроксида (по крайней мере, части осадка), что приводит к торможению растворения осадка. В то же время, анодные токи при Е > 0.4 В могут быть связаны с электрокаталитическим окислением воды на поверхности оксидов/гидроксидов Co [40, 41]. Данный аспект не является предметом этой работы и требует дополнительных исследований. Предположение о формировании оксидов/гидроксидов железа в ходе его осаждения из [BMP][DCA] с добавками воды было также высказано ранее [4].
Рис. 7.
Первые циклы ЦВА Pt(111) в [BMP][DCA] + 0.01 М Sm(OTf)3 + H2O (а), [BMP][DCA] + 0.01 М CoCl2 + H2О (б). Концентрация воды: 0.012 (кривые 1, без добавки воды), 0.320 (кривые 2) и 3.092 М (кривые 3). Скорость развертки потенциала – 0.01 В/с.


Ускорение электроосаждения металлов из дицианамидных ИЖ в присутствии воды было также обнаружено ранее для Eu [5], Nd [4, 8], Fe [4], Zn [42]. Мы связываем это ускорение с эффектами на границе раздела ИЖ/электрод [4]. Увеличение концентрации воды может приводить к разрушению многослойной упорядоченной структуры ионов ИЖ, образующейся вблизи заряженной электродной поверхности [43, 44], способствуя нуклеации и росту осадка. Ранее мы также показали, что хотя добавление воды и ускоряет осаждение Nd и Fe в соответствующих однокомпонентных растворах [BMP][DCA], однако приводит к торможению соосаждения Nd–Fe [4]. На рис. 9 показаны первые циклы ЦВА Pt(111) в двухкомпонентном растворе [BMP][DCA] + + 0.01 М Sm(OTf)3 + 0.01 М CoCl2 с добавками воды (кривые 1). Как и в случае соосаждения Nd–Fe, добавление воды приводит к торможению соосаждения Sm–Co. С увеличением ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ пик К3 смещается в отрицательную сторону и при ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = = 1.552 М исчезает (рис. 9, см. также зависимость потенциала пика К3 от ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ на рис. 8). Кроме того, с увеличением ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ уменьшаются анодные токи процесса растворения осадка Sm–Co, обозначенного как А3 (рис. 9).
Рис. 9.
Первые циклы ЦВА Pt(111) в [BMP][DCA] + + 0.01 М Sm(OTf)3 + 0.01 М CoCl2 + H2O (кривые 1) и [BMP][DCA] + H2O (кривые 2). Концентрация воды указана рядом с кривыми. Скорость развертки потенциала – 0.01 В/с.

На рис. 9 также приведены ЦВА, полученные в растворах ИЖ без солей металлов, но с такими же добавками воды (кривые 2). Увеличение концентрации воды приводит к существенному сдвигу начала катодного разложения ИЖ (К0) в сторону менее отрицательных потенциалов. Эта тенденция наблюдается независимо от того, есть ли в растворе ионы металлов или нет (см. рис. 7 и 9). Таким образом, в присутствии воды катодное разложение ИЖ ускоряется как на Pt-электроде без осадка, так и на электроде, по крайней мере частично покрытом осадком. В ИЖ с высокой ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ можно было бы ожидать интенсивного выделения водорода. Подщелачивание приэлектродного слоя вследствие восстановления воды, в свою очередь, способствовало бы образованию гидроксидов Sm и/или Co на поверхности электрода в ИЖ, содержащей соответствующие соли металлов. Однако в растворах [BMP][DCA] + H2O даже при высоких ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ не наблюдается появления дополнительной катодной волны, которую можно было бы приписать реакции выделения водорода (рис. 9, кривые 2).
На рис. 10 показаны увеличенные фрагменты ЦВА, полученные в растворах ИЖ, содержащих один или оба компонента (Sm + Co). Помимо пика К3 (сосаждение Sm–Co), на ЦВА можно наблюдать катодную волну К1' при потенциалах, близких к осаждению Sm в ИЖ, содержащей только ионы Sm(III). Однако более выраженной она становится при ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.320 М и выше (рис. 10). Мы приписываем К1' осаждению Sm, которое не катализируется соосаждением Co, т.е. происходит вследствие достижения потенциала, достаточно отрицательного для осаждения Sm (как в случае раствора ИЖ, содержащего только ионы Sm(III)). Особенно это заметно при высоком содержании воды (3.092 М, рис. 10в), когда соосаждение Sm‒Co сильно заторможено и положение пиков К1 и К1' практически совпадает. Более того, хотя, как видно из рис. 7б и 8, осаждение Co ускоряется в присутствии воды в ИЖ, содержащей только ионы Co(II), в ИЖ с ионами обоих металлов Sm(III) и Co(II) в присутствии воды (≥0.320 М) осаждение Co сильно тормозится.
Рис. 10.
Фрагменты первых циклов ЦВА Pt(111) в [BMP][DCA] + 0.01 М Sm(OTf)3 + H2O (кривые 1), [BMP][DCA] + + 0.01 М CoCl2 + H2O (кривые 2) и [BMP][DCA] + 0.01 М Sm(OTf)3 + 0.01 М CoCl2 + H2O (кривые 3). Концентрация воды: (а) 0.012 (без добавки воды), (б) 0.320 и (в) 3.092 М. Скорость развертки потенциала – 0.01 В/с.

Как и при соосаждении Nd–Fe [4], торможение соосаждения Sm–Co можно объяснить на основе модели индуцированного соосаждения в ИЖ, недавно предложенной Xu и соавторами [13]. Исследуя соосаждение Nd–Fe из ИЖ [EMIm][DCA] (1-этил-3-метилимидазолий дицианамид), содержащей соли NdCl3 и FeCl2, авторы предположили, что сначала ион Fe(II) восстанавливается до частицы Fe* в переходном состоянии, которая, в свою очередь, катализирует восстановление Nd(III) до Nd(0). Хотя модель была предложена для соосаждения Nd–Fe, сходство между вольтамперометрическими данными для систем Nd–Fe [4] и Sm–Co (настоящая работа) в ИЖ с различным содержанием воды указывает на аналогичный механизм катализируемого соосаждения в обеих системах и подавления соосаждения в присутствии воды. По-видимому, добавление воды стимулирует формирование оксидов/гидроксидов как Co, так и Sm на поверхности электрода в процессе их соосаждения. Образующаяся оксидная пленка препятствует дальнейшему восстановлению ионов Co(II) и, следовательно, и образованию частиц Co* в переходном состоянии. Таким образом, в растворе ИЖ с ионами обоих металлов в присутствии воды тормозится как осаждение Co, так и катализируемое соосаждение Sm.
Другой механизм соосаждения Sm–Co был предложен Manjum и соавторами [18], которые исследовали соосаждение Sm–Co в [BMP][TFSI] (TFSI – бис-(трифторметансульфонил)имид) при комнатной температуре. При катодной поляризации авторы наблюдали формирование наночастиц SmCo7 как на поверхности электрода, так и в растворе вблизи поверхности электрода. Образование наночастиц SmCo7 было объяснено диспропорционированием ионов Sm(II), образующихся в ходе электровосстановления ионов Sm(III) в присутствии элементарного Co(0) [18]. При этом реакция диспропорционирования Sm(II) с образованием Sm(III) и Sm(0) в отсутствие ионов Co(II) была несущественной. Однако важно отметить, что в ИЖ [BMP][TFSI], содержащей только ионы Sm(III), авторы наблюдали четко выраженный пик восстановления Sm(III) только до Sm(II) и противопик на анодной развертке, соответствующий обратной электрохимической реакции. При этом процесс наработки ионов Sm(II) происходил при потенциалах осаждения Co. На ЦВА, полученных нами в растворах ИЖ с дицианамидным анионом ([BMP][DCA]), не наблюдается явно выраженной катодной волны перехода Sm(III) в Sm(II), т.е. ионы Sm(III) в основном восстанавливаются до Sm(0), что подтверждается данными АСМ и РФЭС. Кроме того, мы также не наблюдали образования наночастиц в растворе. Поэтому ускорение соосаждения Sm в присутствии ионов Co через реакцию диспропорционирования ионов Sm(II) в нашей системе представляется нам маловероятным. Различие в механизмах соосаждения может быть связано с разными анионами ИЖ ([TFSI]– в [18] и [DCA]– в нашей работе). Анионы ИЖ могут входить в состав внутренней координационной сферы иона металла и, таким образом, определять состав активной частицы, которая восстанавливается до металла. Благодаря этому, они могут значительно влиять как на потенциал восстановления ионов металлов, так и на стабилизацию ионов с промежуточной валентностью [6], например, Sm(II). Кроме того, в работе [18] в качестве рабочего электрода использовался стеклоуглерод, который, как правило, проявляет меньшую активность при электроосаждении металлов из ионных органических сред. Так, другим научным коллективом при исследовании соосаждения Sm–Co из [BMP][TFSI] на золотом электроде формирование осадка Sm–Co было приписано электрохимическому восстановлению Sm(III) и Co(II) [19], а не результату диспропорционирования Sm(II), что согласуется с нашими результатами.
ЗАКЛЮЧЕНИЕ
В работе изучено электроосаждение Sm и Co и их соосаждение в дицианамидной ИЖ, [BMP][DCA], с контролируемым содержанием воды. Показано, что как Co, так и Sm могут быть электрохимически осаждены из соответствующих однокомпонентных растворов в ИЖ, и потенциал их осаждения смещается в положительную сторону по мере увеличения концентрации воды. Особенно явно ускорение осаждения в присутствии воды выражено для Sm: сдвиг пика осаждения Sm составляет 0.78 В в присутствии 3.092 М H2O. В растворе ИЖ, содержащем ионы обоих металлов (Sm(III) и Co(II)), наблюдается соосаждение Sm–Co. При этом Sm соосаждается при потенциалах, существенно менее отрицательных (на 0.42 В), чем осаждение Sm из раствора ИЖ, содержащего только ионы Sm(III). Увеличение концентрации воды приводит к торможению процесса соосаждения Sm–Co, и при ${{c}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ ≥ 1.552 М катодный пик, приписываемый соосаждению, уже не наблюдается на ЦВА. Эти результаты сходны с полученными нами ранее данными по соосаждению Nd–Fe из той же ИЖ с добавками воды [4]. Торможение соосаждения Sm–Co можно объяснить, основываясь на предложенной ранее модели индуцированного соосаждения в ИЖ [13]. Согласно этой модели, при восстановлении Co(II) могут образовываться частицы Co* в переходном состоянии, которые катализируют восстановление ионов Sm(III) до Sm(0), смещая, таким образом, потенциал восстановления Sm(III) в положительную сторону. Добавление воды, однако, стимулирует реакцию окисления Co и Sm с образованием оксидов/гидроксидов, протекающую параллельно с электрохимическим осаждением. Такая оксидная/гидроксидная пленка препятствует восстановлению Co(II) и образованию интермедиата Co*, что приводит к заметному торможению как осаждения Co, так и соосаждения Sm–Co в растворе ИЖ, содержащем ионы обоих металлов.
Список литературы
Zhou, J., Meng, X., Zhang, R., Liu, H., and Liu, Z., Progress on Electrodeposition of Rare Earth Metals and Their Alloys, Electroanal., 2021, vol. 12, p. 628.
Gutfleisch, O., Willard, M.A., Brück, E., Chen, C.H., Sankar, S.G., and Liu, J.P., Magnetic Materials and Devices for the 21st Century: Stronger, Lighter, and More Energy Efficient, Adv. Mater., 2011, vol. 23, p. 821.
Zhu, H., Rare Earth Metal Production by Molten Salt Electrolysis, in Encyclopedia of Applied Electrochemistry, Kreysa, G., Ota, K.-i., and Savinell, R.F., Eds, N.Y.: Springer New York, 2014. P. 1765.
Molodkina, E.B., Ehrenburg, M.R., Arkhipushkin, I.A., and Rudnev, A.V., Interfacial effects in the electro(co)deposition of Nd, Fe, and Nd–Fe from an ionic liquid with controlled amount of water, Electrochim. Acta, 2021, vol. 398, p. 139342.
Ehrenburg, M.R., Molodkina, E.B., Mishchenko, A., and Rudnev, A.V., The promoting effect of water on the electrodeposition of Eu in a dicyanamide ionic liquid, Electrochim. Acta, 2021, vol. 379, p. 138169.
Rudnev, A.V., Electrodeposition of lanthanides from ionic liquids and deep eutectic solvents, Russ. Chem. Rev., 2020, vol. 89, p. 1463.
Glukhov, L.M., Greish, A.A., and Kustov, L.M., Electrodeposition of rare earth metals Y, Gd, Yb in ionic liquids, Russ. J. Phys. Chem. A, 2010, vol. 84, p. 104.
Periyapperuma, K., Pringle, J.M., Sanchez-Cupido, L., Forsyth, M., and Pozo-Gonzalo, C., Fluorine-free ionic liquid electrolytes for sustainable neodymium recovery using an electrochemical approach, Green Chem., 2021, vol. 23, p. 3410.
Ota, H., Matsumiya, M., Sasaya, N., Nishihata, K., and Tsunashima, K., Investigation of electrodeposition behavior for Nd(III) in [P2225][TFSA] ionic liquid by EQCM methods with elevated temperatures, Electrochim. Acta, 2016, vol. 222, p. 20.
Lebedeva, O., Kultin, D., and Kustov, L., Electrochemical Synthesis of Unique Nanomaterials in Ionic Liquids, Nanomaterials, 2021, vol. 11, p. 3270.
Endres, F., Abbott, A., and MacFarlane, D., Electrodeposition from Ionic Liquids, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2017.
Danilov, F.I. and Protsenko, V.S., Electrodeposition of composite coatings using electrolytes based on deep eutectic solvents: A mini-review, Voprosy khimii i khimicheskoi tekhnologii, 2018, p. 13.
Xu, X., Sturm, S., Zavasnik, J., and Rozman, K.Z., Electrodeposition of a Rare-Earth Iron Alloy from an Ionic-Liquid Electrolyte, ChemElectroChem, 2019, vol. 6, p. 2860.
Liu, P., Du, Y., Yang, Q., Tong, Y., and Hope, G.A., Induced Codeposition of Sm–Co Amorphous Films in Urea Melt and Their Magnetism, J. Electrochem. Soc., 2006, vol. 153, p. C57.
Gómez, E., Cojocaru, P., Magagnin, L., and Valles, E., Electrodeposition of Co, Sm and SmCo from a Deep Eutectic Solvent, J. Electroanal. Chem., 2011, vol. 658, p. 18.
Cojocaru, P., Magagnin, L., Gomez, E., and Vallés, E., Using deep eutectic solvents to electrodeposit CoSm films and nanowires, Mater. Lett., 2011, vol. 65, p. 3597.
Chen, Y., Wang, H., and Li, B., Electrodeposition of SmCo alloy nanowires with a large length-diameter ratio from SmCl3–CoCl2–1-ethyl-3-methylimidazolium chloride ionic liquid without template, RSC Adv., 2015, vol. 5, p. 39620.
Manjum, M., Serizawa, N., Ispas, A., Bund, A., and Katayama, Y., Electrochemical Preparation of Cobalt-Samarium Nanoparticles in an Aprotic Ionic Liquid, J. Electrochem. Soc., 2020, vol. 167, p. 042505.
Ispas, A., Buschbeck, M., Pitula, S., Mudring, A., Uhlemann, M., Bund, A., and Endres, F., Electrodeposition of Co, Sm and Co-Sm Thin Layers, ECS Trans., 2009, vol. 16, p. 119.
Panzeri, G., Tresoldi, M., Rinaldi, C., and Magagnin, L., Electrodeposition of Magnetic SmCo Films from Deep Eutectic Solvents and Choline Chloride-Ethylene Glycol Mixtures, J. Electrochem. Soc., 2017, vol. 164, p. D930.
Li, J., Lai, H., Fan, B., Zhuang, B., Guan, L., and Huang, Z., Electrodeposition of RE–TM (RE = La, Sm, Gd; TM = Fe, Co, Ni) films and magnetic properties in urea melt, J. Alloys Compd., 2009, vol. 477, p. 547.
Sanchez-Cupido, L., Pringle, J.M., Siriwardana, A.I., Hilder, M., Forsyth, M., and Pozo-Gonzalo, C., Correlating Electrochemical Behavior and Speciation in Neodymium Ionic Liquid Electrolyte Mixtures in the Presence of Water, ACS Sustain. Chem. Eng., 2020, vol. 8, p. 14047.
Sanchez-Cupido, L., Pringle, J.M., Siriwardana, A., Pozo-Gonzalo, C., and Forsyth, M., Electrochemistry of Neodymium in Phosphonium Ionic Liquids: The Influence of Cation, Water Content, and Mixed Anions, Aust. J. Chem., 2020, vol. 73, p. 1080.
Clavilier, J., Flame-annealing and cleaning technique, in Interfacial Electrochemistry: Theory, Experimental, and Applications, A. Wieckowski, Ed, New York: Marcel Dekker, 1999. P. 231.
McGrath, L.M. and Rohan, J.F., Pyrrolidinium Containing Ionic Liquid Electrolytes for Li-Based Batteries, Molecules, 2020, vol. 25, p. 6002.
Rudnev, A.V., Ehrenburg, M.R., Molodkina, E.B., Abdelrahman, A., Arenz, M., Broekmann, P., and Jacob, T., Structural Changes of Au(111) Single-Crystal Electrode Surface in Ionic Liquids, ChemElectroChem, 2020, vol. 7, p. 501.
Molodkina, E.B., Ehrenburg, M.R., Broekmann, P., and Rudnev, A.V., Initial stages of silver electrodeposition on single crystal electrodes from ionic liquids, Electrochim. Acta, 2019, vol. 299, p. 320.
Atkin, R., El Abedin, S.Z., Hayes, R., Gasparotto, L.H.S., Borisenko, N., and Endres, F., AFM and STM Studies on the Surface Interaction of [BMP]TFSA and [EMIm]TFSA Ionic Liquids with Au(111), J. Phys. Chem. C, 2009, vol. 113, p. 13266.
Andrew, C., Murugesan, C., and Jayakumar, M., Electrochemical Behavior of Sm(III) and Electrodeposition of Samarium from 1-Butyl-1-Methylpyrrolidinium Dicyanamide Ionic Liquid, J. Electrochem. Soc., 2022, vol. 169, p. 022503.
Pan, Y. and Hussey, C.L., Electrochemical and Spectroscopic Investigation of Ln3+ (Ln = Sm, Eu, and Yb) Solvation in Bis(trifluoromethylsulfonyl)imide-Based Ionic Liquids and Coordination by N,N,N′,N′-Tetraoctyl-3-oxa-pentane Diamide (TODGA) and Chloride, Inorg. Chem., 2013, vol. 52, p. 3241.
Chen, L., Li, Y., Shi, X., Wang, D., Wang, G., Jiao, C., and Zhang, M., Electrochemical properties of Ln(III) (Ln = Ce, Gd) in 1-butyl-3-methylimidazolium tetrafluoroborate ionic liquid, J. Radioanal. Nucl. Chem., 2021, vol. 329, p. 1269.
Rudnev, A.V. and Wandlowski, T., An influence of pretreatment conditions on surface structure and reactivity of Pt(100) towards CO oxidation reaction, Russ. J. Electrochem., 2012, vol. 48, p. 259.
Liu, D., Niu, F., Zhang, X., Meng, Y., and Yang, Y., Fabrication of SmCo5 alloy via cobalt-induced calciothermic reduction and magnetic properties of its ribbon, J. Rare Earths, 2021, vol. 39, p. 572.
Uwamino, Y., Ishizuka, T., and Yamatera, H., X-ray photoelectron spectroscopy of rare-earth compounds, J. Electron Spectrosc. Relat. Phenom., 1984, vol. 34, p. 67.
Xie, M., Zhu, L., Li, W., Liu, H., and Zhang, T., Electrodeposition of Sm–Co Alloy Films with Nanocrystalline/Amorphous Structures from a Sulphamate Aqueous Solution, Int. J. Electrochem. Sci., 2017, vol. 12, p. 11 330.
Choudhury, T., Saied, S.O., Sullivan, J.L., and Abbot, A.M., Reduction of oxides of iron, cobalt, titanium and niobium by low-energy ion bombardment, J. Phys. D: Appl. Phys., 1989, vol. 22, p. 1185.
Biesinger, M.C., Payne, B.P., Grosvenor, A.P., Lau, L.W.M., Gerson, A.R., and Smart, R.S.C., Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, vol. 257, p. 2717.
Schenck, C.V., Dillard, J.G., and Murray, J.W., Surface analysis and the adsorption of Co(II) on goethite, J. Colloid Interf. Sci., 1983, vol. 95, p. 398.
Lebugle, A., Axelsson, U., Nyholm, R., and Mårtensson, N., Experimental L and M Core Level Binding Energies for the Metals 22Ti to 30Zn, Phys. Scr., 1981, vol. 23, p. 825.
Hunter, B.M., Gray, H.B., and Müller, A.M., Earth-Abundant Heterogeneous Water Oxidation Catalysts, Chem. Rev., 2016, vol. 116, p. 14120.
Moysiadou, A., Lee, S., Hsu, C.-S., Chen, H.M., and Hu, X., Mechanism of Oxygen Evolution Catalyzed by Cobalt Oxyhydroxide: Cobalt Superoxide Species as a Key Intermediate and Dioxygen Release as a Rate-Determining Step, J. Amer. Chem. Soc., 2020, vol. 142, p. 11 901.
Periyapperuma, K., Pozo-Gonzalo, C., MacFarlane, D.R., Forsyth, M., and Howlett, P.C., High Zn Concentration Pyrrolidinium-Dicyanamide-Based Ionic Liquid Electrolytes for Zn2+/Zn0 Electrochemistry in a Flow Environment, ACS Appl. Energy Mater., 2018, vol. 1, p. 4580.
Cui, T., Lahiri, A., Carstens, T., Borisenko, N., Pulletikurthi, G., Kuhl, C., and Endres, F., Influence of Water on the Electrified Ionic Liquid/Solid Interface: A Direct Observation of the Transition from a Multilayered Structure to a Double-Layer Structure, J. Phys. Chem. C, 2016, vol. 120, p. 9341.
Kemna, A. and Braunschweig, B., Potential-Induced Adsorption and Structuring of Water at the Pt(111) Electrode Surface in Contact with an Ionic Liquid, J. Phys. Chem. Lett., 2020, vol. 11, p. 7116.
Дополнительные материалы отсутствуют.