

Журнал физической химии, 2019, T. 93, № 1, стр. 99-108
Структура, термодинамические свойства, ударное сжатие расплавов висмут–свинец и корреляционный эффект при диффузии. метод молекулярной динамикиД. К. Белащенко
a Национальный исследовательский технологический университет “Московский институт стали и сплавов”
Москва, Россия
* E-mail: dkbel@mail.ru
Поступила в редакцию 14.03.2018
Аннотация
Методом молекулярной динамики построены модели растворов Bi–Pb при температурах до 30000 К и давлениях до 282 ГПа. Применили потенциалы модели погруженного атома (ЕАМ) для чистых компонентов, а также потенциал Леннард–Джонса для пар 12. Изобара плотности при нормальном давлении и изотерма теплот образования растворов при 700 К хорошо согласуются с опытом. Рассчитаны термодинамические (плотность, энергия, теплоемкость, модуль всестороннего сжатия, скорость звука, коэффициенты Грюнайзена), структурные и диффузионные свойства эвтектического расплава Bi55Pb45 при нормальном давлении и температурах до 2900 К, а также в условиях ударного сжатия. С ростом давления отрицательные отклонения от идеальности изменяются на положительные. На моделях расплава проведен молекулярно-динамический расчет коэффициентов взаимной диффузии при 550–928 К. Коэффициенты взаимной диффузии, найденные методом молекулярной динамики, меньше измеренных экспериментально капиллярным методом в 1.25–1.64 раз. Подтверждено существование корреляционного эффекта при диффузии в расплавах, аналогичного катафоретическому эффекту в ионных растворах. В растворах Bi–Pb с отрицательными отклонениями от идеальности фактор корреляции существенно меньше единицы. Установлено, что в уравнении для коэффициента взаимной диффузии термодинамический множитель и фактор корреляции частично гасят друг друга.
Жидкие висмут, свинец и расплавы Bi–Pb являются перспективными объектами ядерной энергетики и технологии конверсии солнечной энергии. Опубликованы обзоры, посвященные анализу свойств этих металлических систем [1, 2] и их взаимодействию с конструкционными материалами. Особый интерес представляет эвтектика Bi55Pb45 из-за ее низкой температуры плавления (398 К). В связи с высокой летучестью компонентов данные о свойствах расплавов получены экспериментально в ограниченном диапазоне температур. Поэтому представляет интерес исследование свойств системы Bi–Pb методом компьютерного моделирования. В настоящей работе для этой цели применили метод молекулярной динамики (МД).
МЕТОДЫ РАСЧЕТА
Для описания межчастичного взаимодействия в расплавах Bi–Pb применили потенциалы модели погруженного атома (ЕАМ). Эти потенциалы для свинца и висмута были взяты из [3, 4] без изменений, и никакие преобразования исходных выражений для потенциалов не проводились. Это можно было сделать, так как значения средней эффективной электронной плотности в потенциалах ЕАМ чистых компонентов вблизи от температуры плавления были равны стандартному значению 1.000. Потенциал взаимодействия пар 12 (т.е. Bi–Pb) выбрали в форме Леннард–Джонса:
(1)
${{\varphi }_{{12}}}(r) = 4{{e}_{{12}}}[{{({{r}_{0}}{\text{/}}r)}^{{12}}} - {{({{r}_{0}}{\text{/}}r)}^{6}}].$Подбор параметров r0 и ε12 провели с учетом того, что при 700 К плотность эвтектического раствора Bi55Pb45 равна 10.16 г/см3 [1] и теплота смешения исходных чистых компонентов составляет –1070 Дж/моль [5]. Равновесную плотность растворов находили при расчетах в NpT-ансамбле при нулевом давлении. Наилучшее согласие с опытом по плотности и энергии получено при r0 = 3.005 Å и ε12 = 0.070 эВ. Дальнейшие молекулярно-динамические (МД) прогоны проводили в NpT- и NVT-ансамбле.
РЕЗУЛЬТАТЫ РАСЧЕТОВ
Свойства раствора Bi55Pb45. Некоторые расчетные свойства раствора приведены в табл. 1. Плотность моделей растворов определяли в режиме NpT-ансамбля при нулевом давлении. По данным [1], при 45% Pb плотность ρ(T), г/см3 = 11.065 – 1.293 × 10–3 T. Зависимость плотности моделей эвтектики Bi55Pb45 от температуры показана в табл.1 и на рис. 1. Расчетная плотность расплава очень хорошо согласуется с опытными данными [1] при всех температурах до 2000 К. Получить устойчивые состояния при нулевом давлении и температурах выше 2900 К не удалось, так как модели неограниченно расширялись, попадая в область пара на фазовой диаграмме.
Таблица 1.
Расчетные свойства раствора Bi55Pb45 с потенциалами ЕАМ
| T, K | d, г/см3 | CV, Дж/(моль К) | Cp, Дж/(моль К) | (∂p/∂T)V, MПа/K | γ | KT, ГПа | us, м/с | δ | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MД | [1] | MД | [1] | MД | [1] | по (3) | [7, 8] | по (4) | |||||
| 448 | 10.51 | 10.48 | 24.95 | 30.36 | 30.7 | 3.716 | 2.95 | 30.97 | 1894 | 1760 | 1.575 | 1.594 | – |
| 550 | 10.36 | 10.35 | 23.82 | 29.49 | 30.3 | 3.317 | 2.80 | 28.84 | 1856 | 1738 | 1.468 | 1.514 | – |
| 650 | 10.23 | 10.22 | 23.27 | 28.71 | 29.8 | 3.008 | 2.63 | 27.42 | 1818 | 1717 | 1.396 | 1.441 | – |
| 700 | 10.16 | 10.16 | 22.06 | 28.34 | 29.6 | 2.855 | 2.65 | 21.05 | 1631 | 1707 | 1.368 | 1.416 | 1.258 |
| 773 | 10.06 | 10.06 | 21.96 | 27.84 | 29.4 | 2.832 | 2.67 | 19.60 | 1572 | 1691 | 1.333 | 1.406 | – |
| 928 | 9.857 | 9.865 | 20.84 | 26.90 | 28.8 | 2.290 | 2.32 | 18.60 | 1560 | 1658 | 1.278 | 1.339 | – |
| 1000 | 9.759 | 9.772 | 20.03 | 26.51 | 28.6 | 2.355 | 2.41 | 16.64 | 1502 | 1643 | – | – | – |
| 1500 | 9.114 | 9.126 | 17.04 | 24.81 | 27.8 | 1.556 | 2.08 | 11.26 | 1367 | – | – | – | – |
| 2000 | 8.468 | 8.479* | 16.38 | 24.80 | – | 1.022 | 1.53 | 5.94 | 1030 | – | – | – | – |
| 2500 | 7.775 | – | 16.91 | 26.49 | – | 0.937 | 1.48 | 4.65 | 968 | – | – | – | – |
| 2850 | 7.286 | – | – | – | – | – | – | 4.05 | – | – | – | – | – |
| 2875 | 7.231 | – | 15.80 | 29.68 | – | 0.632 | 1.15 | – | – | – | – | – | – |
| 2900 | 7.170 | – | 17.68 | 29.20 | – | 0.389 | 0.639 | 4.09 | 985 | – | – | – | – |
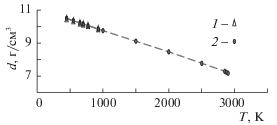
Объемы чистых компонентов при 700 К и нулевом давлении равны 21.16 см3/моль для висмута и 19.62 см3/моль для свинца. Объем растворов при 700 К изменяется с концентрацией практически аддитивно. Небольшие отклонения объема от аддитивных значений не превышают 0.036 см3/моль.
Значения энергии чистых компонентов при 700 К и нулевом давлении равны –178.51 кДж/моль для висмута и –173.07 кДж/моль для свинца. Теплота смешения реального расплава Bi55Pb45 при 700 К равна –1070 Дж/моль [5], то есть растворы образуются с небольшим выделением теплоты. Расчетная изотерма теплот образования ΔH растворов Bi–Pb из чистых компонентов при 700 К (при нулевом давлении ΔH = ΔU) показана на рис. 2. МД-расчеты в общем хорошо согласуются с опытом [5].
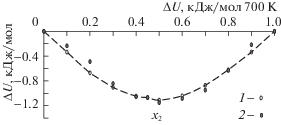
При нагревании теплота смешения убывает по абсолютной величине. С учетом данных [3, 4] для энергии чистых компонентов, теплота смешения расплава Bi55Pb45 равна –686 Дж/моль при 1000 К и –410 при 1500 К. Эта зависимость хорошо описывается выражением ΔH = –867640/T + 173.18 Дж/моль.
Расчетная теплоемкость Сp при нагревании постепенно отклоняется от фактической Сp в меньшую сторону (табл. 1). Модуль всестороннего сжатия KT при изобарном нагревании монотонно убывает. Расчетная скорость звука близка к данным эксперимента. Так, при 550 К она равна 1778 м/с при фактическом значении 1736 м/с [6]. Соответственно при 1000 К расчетная скорость равна 1504 м/с при фактическом значении 1616 м/с [6]. Коэффициент Грюнайзена γ = = (V/CV)(∂p/∂T)V убывает при нагревании до 2900 К примерно в 4 раза.
Термодинамический множитель. По своим свойствам растворы висмут–свинец близки к регулярным. Обозначим через Xi и ai мольную молю и термодинамическую активность i-го компонента. Теплота смешения регулярных растворов описывается выражением ΔH = hX1X2. При X1 = 0.55 и ΔH = –1070 Дж/моль коэффициент h равен ‒4323 Дж/моль. В случае регулярных растворов коэффициент активности первого компонента γ1 = a1/X1 описывается уравнением:
Отсюда следует, что термодинамический множитель δ = dln a1/dln X1 = dln a2/dln X2 = 1 + + dln γ1/dln X1 равен:
С учетом величины h = –4323 Дж/моль находим в приближении регулярных растворов, что δ = 1 + (8646 Дж)X1X2/(RT). При 700 К и X1 = 0.55 получается δ = 1.368. Значения δ, найденные по (3) при X1 = 0.55 и при различных температурах, приведены в табл. 1.
Активности компонентов реальной системы висмут–свинец были измерены при температурах до 1320 К [7–9]. Коэффициенты активности γi описываются формулой теории регулярных растворов ln γi = β(1 – Xi)2, так что δ = 1 – 2βX1X2. Для системы висмут–свинец β = –A – B/T, причем A = 0.2025 и B = 447 K [7, 8]. Значения δ, найденные в [7, 8], приведены в табл. 1. Они немного выше, чем найденные по (3).
Возможен и прямой расчет значений δ по МД-данным. Исходная модель при ε12 = 0.070 эВ имеет теплоту смешения –1070 Дж/моль и близкий к аддитивному объем. Изменяя в (1) величину ε по формуле ε = λε12, можно добиться обращения ΔH в нуль и перехода в состояние идеального (совершенного) раствора, в котором коэффициенты активности равны единице. Методом проб установлено, что условие ΔH = 0 достигается при значении λ = 0.961 и ε = 0.961ε12 = 0.06727 эВ. При таком значении ε величина ΔH обращается в нуль, а объем раствора равен 20.536 см3/моль. Аддитивный объем раствора Bi55Pb45 при 700 К равен 20.438 см3/моль, так что изменение объема при смешении равно всего ΔV = 0.10 см3/моль и достаточно мало, чтобы считать раствор близким к идеальности.
Далее можно рассчитать изменение энергии Гиббса раствора Bi55Pb45 при 700 К при изменении ε от 0.06727 до 0.070 эВ по формуле [10]:
(4)
$\Delta G = \mathop \smallint \limits_{_{{{{\lambda }_{1}}}}}^{_{{{{\lambda }_{2}}}}} (dG{\text{/}}d\lambda )d\lambda = \mathop \smallint \limits_{_{{{{\lambda }_{1}}}}}^{_{{{{\lambda }_{2}}}}} ({{U}_{{12}}}{\text{/}}\lambda )d\lambda ,$Свойства в сжатых состояниях. Было построено 100 моделей раствора Bi55Pb45 при температурах от 298 до 30000 К и степенях сжатия Y = V/V0 от 1 до 0.5 (V – мольный объем, V0 – нормальный объем при 298 К). Модели строили в режиме NVT. Было принято, что в исходном состоянии (при 298 К и нулевом давлении) объем V0 можно определить по правилу аддитивности, так что V0 = = 19.903 см3/моль (у твердого висмута реальный объем V0 = 21.246 и у свинца 18.262). Аддитивная энергия в этом состоянии равна U298 = = ‒189.5 кДж/моль (–191.6 кДж/моль у висмута и –186.9 для свинца). Однако из-за различия кристаллических решеток твердых висмута и свинца и недостаточной точности потенциалов ЕАМ из [4] для описания кристаллических фаз было довольно сложно моделировать поликристаллические состояния при невысоких температурах (в соответствии с фазовой диаграммой). Поэтому все модели были построены в жидком или аморфном состоянии. Условной границей между этими состояниями была величина среднего коэффициента самодиффузии 1.0 × 10–5 см2/с. В табл. 2 приведены значения энергии раствора Bi55Pb45 в зависимости от объема и температуры. Ячейки с аморфными состояниями раствора закрашены. К значениям энергии каждой модели были добавлены электронные вклады, рассчитанные по методу работы [4]. Среднее число электронов на атом было принято равным 4.55. Суммарные величины давления моделей (с учетом электронных вкладов) приведены в табл. 3. В исходном состоянии энергия аморфной модели (–188.4 кДж/моль) немного отличается от аддитивного значения (–189.5), а давление модели (–0.77 ГПа) не равно нулю, однако эти отклонения сравнительно невелики.
Таблица 2.
Энергия U раствора Bi55Pb45 с учетом электронных вкладов, кДж/моль
| T, K | Y = V/V0 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1.00 | 0.90 | 0.85 | 0.80 | 0.75 | 0.70 | 0.65 | 0.60 | 0.55 | 0.50 | |
| U | ||||||||||
| 298 | –188.4 | –188.9 | –184.2 | –174.2 | –157.5 | –132.5 | –95.5 | –44.3 | 19.7 | 116.2 |
| 500 | –183.0 | –183.1 | –178.3 | –168.4 | –151.9 | –127.1 | –90.1 | –38.9 | 25.2 | 121.8 |
| 1000 | –170.9 | –169.5 | –164.3 | –154.4 | –137.8 | –113.6 | –77.1 | –26.0 | 40.4 | 135.8 |
| 2000 | –148.8 | –145.4 | –139.2 | –127.9 | –110.2 | –84.8 | –48.9 | 1.9 | 69.1 | 162.1 |
| 5000 | –79.6 | –73.4 | –65.2 | –52.0 | –32.4 | –5.2 | 32.5 | 86.2 | 155.5 | 241.8 |
| 10 000 | 62.0 | 68.9 | 77.6 | 91.4 | 111.5 | 140.3 | 179.5 | 236.5 | 306.7 | 404.6 |
| 15 000 | 239.1 | 243.3 | 250.8 | 263.5 | 283.4 | 311.5 | 351.8 | 409.4 | 484.6 | 584.6 |
| 20 000 | 448.3 | 447.5 | 453.7 | 463.7 | 481.5 | 508.2 | 547.7 | 605.2 | 682.7 | 788.1 |
| 25 000 | 682.6 | 678.0 | 681.1 | 689.2 | 704.3 | 728.3 | 766.6 | 822.4 | 900.3 | 1004 |
| 30 000 | 937.0 | 927.9 | 928.6 | 934.7 | 946.7 | 969.0 | 1005 | 1059 | 1134 | 1240 |
Таблица 3.
Давление p раствора Bi55Pb45 с учетом электронных вкладов, ГПа
| T, K | Y = V/V0 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1.00 | 0.90 | 0.85 | 0.80 | 0.75 | 0.70 | 0.65 | 0.60 | 0.55 | 0.50 | |
| p | ||||||||||
| 298 | –0.77 | 3.85 | 8.07 | 14.03 | 21.88 | 30.93 | 43.45 | 62.65 | 73.09 | 110.7 |
| 500 | 0.09 | 4.79 | 8.95 | 14.96 | 22.60 | 31.66 | 44.13 | 63.61 | 75.05 | 112.6 |
| 1000 | 1.69 | 6.76 | 11.13 | 17.17 | 24.57 | 33.72 | 46.41 | 66.01 | 81.99 | 117.6 |
| 2000 | 4.11 | 9.70 | 14.46 | 28.39 | 32.05 | 38.00 | 51.62 | 71.37 | 90.82 | 124.2 |
| 5000 | 9.90 | 16.76 | 22.11 | 28.96 | 43.33 | 48.74 | 63.96 | 85.53 | 108.8 | 138.3 |
| 10 000 | 18.65 | 27.15 | 33.32 | 39.72 | 51.10 | 64.50 | 82.13 | 106.6 | 132.0 | 171.3 |
| 15 000 | 27.77 | 37.52 | 44.54 | 53.27 | 64.81 | 79.82 | 99.91 | 126.6 | 157.6 | 200.0 |
| 20 000 | 37.60 | 48.46 | 56.37 | 66.02 | 78.71 | 95.22 | 117.1 | 146.1 | 181.6 | 229.6 |
| 25 000 | 47.82 | 60.12 | 68.73 | 79.50 | 93.10 | 111.0 | 134.9 | 165.7 | 204.5 | 254.5 |
| 30 000 | 58.42 | 72.18 | 81.52 | 93.37 | 108.0 | 127.4 | 152.7 | 185.4 | 226.5 | 281.7 |
Представленная в табл. 2 и табл. 3 информация достаточна для расчета остальных термодинамических свойств, в частности, теплоемкостей CV и Cp, производной (∂p/∂T)V, модуля всестороннего сжатия, скорости звука и коэффициентов Грюнайзена γ = (V/CV) (∂p/∂T)V. Теплоемкость CV заметно растет с температурой благодаря электронному вкладу в энергию (от 20.7–27.9 при 298 К до 47.3–51.4 при 25000 К), а теплоемкость Cp растет от 22.1–29.0 при 298 К до 59.2–72.5 при 25000 К. Коэффициенты Грюнайзена убывают при нагревании до 25000 К в 2–3 раза (табл. 4).
Таблица 4.
Коэффициенты Грюнайзена γ моделей раствора Bi55Pb45
| T, K | Y = V/V0 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1.00 | 0.90 | 0.85 | 0.80 | 0.75 | 0.70 | 0.65 | 0.60 | 0.55 | 0.50 | |
| γ | ||||||||||
| 298 | 2.49 | 2.47 | 2.47 | 2.83 | 3.47 | 2.24 | 2.39 | 2.31 | 3.62 | 2.41 |
| 500 | 2.44 | 2.43 | 2.43 | 2.75 | 3.36 | 2.21 | 2.36 | 2.29 | 3.54 | 2.40 |
| 1000 | 2.30 | 2.31 | 2.31 | 2.56 | 3.11 | 2.15 | 2.29 | 2.24 | 3.35 | 2.36 |
| 2000 | 2.05 | 2.09 | 2.10 | 2.22 | 2.65 | 2.04 | 2.16 | 2.15 | 3.00 | 2.28 |
| 5000 | 1.50 | 1.58 | 1.60 | 1.45 | 1.62 | 1.73 | 1.83 | 1.88 | 2.18 | 2.07 |
| 10 000 | 1.08 | 1.13 | 1.16 | 0.98 | 0.93 | 1.35 | 1.43 | 1.51 | 1.52 | 1.77 |
| 15 000 | 0.95 | 0.98 | 1.01 | 1.01 | 0.96 | 1.15 | 1.22 | 1.27 | 1.35 | 1.50 |
| 20 000 | 0.91 | 0.94 | 0.97 | 1.10 | 1.10 | 1.04 | 1.10 | 1.12 | 1.31 | 1.29 |
| 25 000 | 0.87 | 0.90 | 0.92 | 1.01 | 1.04 | 0.98 | 1.01 | 1.03 | 1.16 | 1.15 |
Ударное сжатие раствора Bi55Pb45. Методом, описанным в [4], был проведен расчет ударной адиабаты раствора Bi55Pb45 по данным табл. 2 и табл. 3. Было принято, что в исходном состоянии (при 298 К и нулевом давлении) объем равен V0 = 19.903 см3/моль, а энергия U298 = –188.4 кДж/моль. Результаты приведены в табл. 5. Расчетная адиабата Гюгонио показана на рис. 3, где для сравнения приведены также ударные адиабаты чистых висмута и свинца, найденные в [4] тем же методом (и практически совпадающие с реальными). Адиабата раствора Bi55Pb45 располагается между адиабатами чистых компонентов, но гораздо ближе к адиабате свинца. На рис. 4 показаны температуры вдоль этих адиабат. Температура на адиабате раствора ближе к температуре на адиабате висмута, но при Y < 0.6 превышает последнюю на 2000–4000 К. В целом видно, что расчет адиабаты раствора по известным адиабатам чистых компонентов с помощью какого-либо простого правила (например, по аддитивности) был бы весьма неточным.
Таблица 5.
Ударное сжатие раствора Bi55Pb45V0 = = 19.903 см3/моль
| Y | p, ГПа | U, кДж/моль | T, K |
|---|---|---|---|
| 1 | 0 | –188.4 | 298 |
| 0.90 | 4.38 | –184.0 | 474 |
| 0.85 | 10.8 | –172.2 | 686 |
| 0.80 | 16.0 | –156.4 | 935 |
| 0.75 | 32.9 | –106.6 | 2127 |
| 0.70 | 44.5 | –55.6 | 3028 |
| 0.65 | 65.3 | 39.0 | 5219 |
| 0.60 | 103.8 | 224.6 | 9655 |
| 0.55 | 168.1 | 564.4 | 17 066 |
| 0.50 | 274.5 | 1177 | 28 725 |
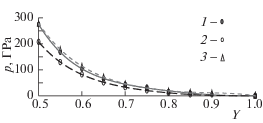
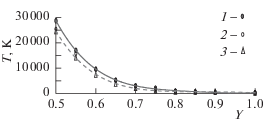
Структура растворов. Экспериментальные данные по структуре растворов системы Bi–Pb были получены в [11, 12] в серии дифракционных измерений при различных концентрациях, при перегревах на 50 К выше линии ликвидус. Парциальные структурные факторы растворов при 550 К (рис. 5) и 650 К были рассчитаны в предположении, что они не зависят от состава; поэтому они вряд ли очень точны.
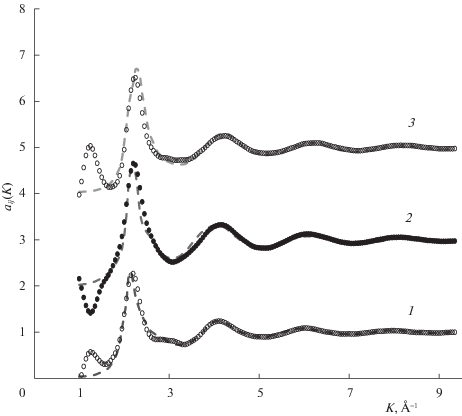
Парциальные парные корреляционные функции gij(r) (ППКФ) раствора Bi55Pb45 были рассчитаны по МД-данным при 550 К (рис. 6) и 650 К до расстояния 20 Å. Первый пик ППКФ для пар 12 при обеих температурах заметно выше, чем у пар 11 и 22. Это означает преимущественное окружение атомов раствора частицами противоположного сорта. Парциальные структурные факторы (Фабера–Займана) были рассчитаны по этим ППКФ с помощью обычного фурье-преобразования (рис. 5). На рис. 5 видны, в частности, ложные осцилляции в малоугловой области, которые обычно получаются при фурье-преобразовании из-за обрыва ППКФ на больших расстояниях. Некоторые характеристики структуры раствора Bi55Pb45 приведены в табл. 6. Через r1(ij) обозначены координаты первых пиков ППКФ пар i–j, а через К1(ij) – координаты первых пиков парциальных структурных факторов пар i–j. Согласие расчетных структурных факторов с дифракционными данными здесь вполне разумное.
Рис. 6.
Парциальные парные корреляционные функции gij(r) раствора Bi55Pb45 при 550 К: 1 – пары 11, 2 – пары 12, 3 – пары 22.
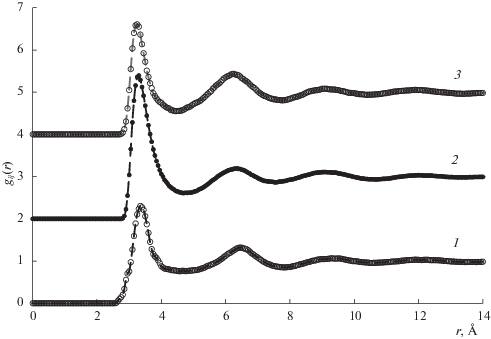
Таблица 6.
Структурные данные для эвтектики Bi55Pb45 при 550 К
| Т, К | 550 | 650 |
|---|---|---|
| Данные МД | ||
| r1(11) | 3.36 | 3.35 |
| r1(12) | 3.28 | 3.35 |
| r1(22) | 3.24 | 3.22 |
| g1(11) | 2.305 | 2.157 |
| g1(12) | 3.394 | 3.171 |
| g1(22) | 2.607 | 2.568 |
| К1(11) | 2.18 | 2.19 |
| К1(12) | 2.22 | 2.21 |
| К1(22) | 2.22 | 2.23 |
| Данные [12] | ||
| К1(11) | 2.13 | 2.13 |
| К1(12) | 2.18 | 2.18 |
| К1(22) | 2.27 | 2.27 |
При сильном сжатии раствора структура существенно изменяется. В случае эвтектического раствора в состоянии при 5000 К и давлении 137.5 ГПа (сжатие в 2 раза по объему) высота первого пика (6.47) и форма второго пика ППКФ для пар 11 характерны для аморфного сплава. Однако форма пиков ППКФ пар 22 (высота первого пика 3.55) имеет вид, отвечающий жидкости. Соответственно, коэффициент самодиффузии частиц свинца вдвое выше, чем у висмута. Первый пик ППКФ для пар 12 невысок (высота 1.63), гораздо ниже пиков пар 11 и 22 и расположен правее них, что указывает на пониженное число пар 12 и преимущественное окружение атомов частицами того же компонента. Это характерно для растворов с положительными отклонениями от идеальности. Следовательно, при высоких давлениях характер отклонений растворов Bi–Pb от идеальности меняется с отрицательного на положительный.
Самодиффузия и взаимная диффузия. Результаты, полученные в настоящей работе для диффузионной подвижности в расплавах Bi–Pb, дают дополнительный материал для решения общей проблемы взаимосвязи параметров самодиффузии и взаимной диффузии в простых жидкостях.
Коэффициент взаимной диффузии описывает величину потоков компонентов ji при наличии градиента концентрации Ci, если в системе координат, связанной с жидкостью, выполняется закон Фика:
где Di – парциальный коэффициент диффузии i-го компонента.С каждым потоком компонента связан соответствующий поток объема ${{J}_{i}}{{v}_{i}}$, где ${{v}_{i}}$ – парциальный объем атомов компонента. Если парциальные коэффициенты диффузии D1 и D2 в формуле (5) произвольны, то при диффузии может возникнуть поток жидкости, аналогичный смещению решетки при эффекте Киркендалла. Если капилляр, в котором идет диффузия, закрыт с одного конца, то суммарный объем частиц, проходящих в обоих направлениях через любое сечение образца, равен нулю. Это условие позволяет определить макроскопическую скорость w жидкости при диффузии.
При наличии потока жидкости потоки диффузии по отношению к капилляру равны
Кроме того,
Подставляя (6) в (7) и выражая концентрацию в атомных долях Xi, получаем:На следующем шаге следует учесть термодинамические свойства раствора. Если коэффициент самодиффузии компонента не зависит от наличия градиента концентрации (Предположение 1), то парциальные коэффициенты диффузии равны:
Здесь $D_{i}^{0}$ – коэффициенты самодиффузии, и δ = dln a1/dln X1 – введенный выше термодинамический множитель. Выражение для коэффициента взаимной диффузии принимает вид:(8)
$\tilde {D} = (D_{1}^{0}{{X}_{2}} + D_{2}^{0}{{X}_{1}}{)\;}\frac{{d\ln {{a}_{i}}}}{{d\ln {{X}_{i}}}}.$Таблица 7.
Коэффициенты самодиффузии D0, D* и взаимной диффузии $\tilde {D}$ в расплаве Bi55Pb45, см2/с
| № | T, K | 448 | 550 | 650 | 700 | 773 | 928 |
|---|---|---|---|---|---|---|---|
| 1 | 105$D_{{{\text{Bi}}}}^{0}$ | 1.333 | 2.108 | 2.941 | 3.584 | 4.048 | 5.666 |
| 2 | 105$D_{{{\text{Pb}}}}^{0}$ | 1.301 | 1.962 | 2.663 | 3.346 | 3.593 | 5.266 |
| 3 | 105${{\tilde {D}}_{{{\text{и д }}}}}$ по (9) | 1.315 | 2.028 | 2.788 | 3.453 | 3.798 | 5.446 |
| 4 | 105$D_{{{\text{Bi}}}}^{*}$ | – | – | – | 3.640 | 4.248 | 5.945 |
| 5 | 105$D_{{{\text{Pb}}}}^{*}$ | – | – | – | 3.091 | 3.926 | 5.238 |
| 6 | 105${{\tilde {D}}_{{{\text{М Д }}}}}$ | – | – | – | 3.07 | 3.18 | 4.57 |
| 7 | 105Dвз [13] | 1.24 | ∼2.40 | 3.19 | ∼3.8 | 4.82 | 7.5 |
| 8 | FМД | – | – | – | 0.65 | 0.63 | 0.66 |
| 9 | F по [7, 8, 13] | 0.59 | 0.78 | 0.79 | 0.78 | 0.90 | 1.03 |
| 10 | F по (14) | 0.686 | 0.722 | 0.837 | 0.838 | 0.708 | 0.826 |
При расчетах диффузионных явлений обычно применяется выражение (8). Однако в [14, 15] было показано, что в общем случае необходимо включить в него дополнительный фактор корреляции F, учитывающий увлечение атомов потоком частиц другого компонента, мигрирующих в противоположную сторону по отношению к потоку данного компонента (аналогично катафоретическому эффекту при переносе заряда в ионных растворах). В итоге коэффициент взаимной диффузии связан с коэффициентами самодиффузии компонентов уравнением:
(10)
$\tilde {D} = F(D_{1}^{0}{{X}_{2}} + D_{2}^{0}{{X}_{1}})d\ln {{a}_{1}}{\text{/}}d\ln {{X}_{1}}.$В настоящее время возможна независимая проверка уравнения (10) компьютерными методами. Для этого следует моделировать процесс взаимной диффузии в растворе. Ранее такие расчеты, видимо, не проводились. Вначале следует создать модель с заданным распределением концентрации. Исходная неоднородная (по оси x) концентрация C(x) в модели задавалась следующим образом. В однородной модели раствора Bi55Pb45 при 700 К проводили перебор всех атомов подряд. Для каждого атома с x – координатой xi находили равномерно распределенное случайное число 0 < A < 1 и с помощью метода Монте-Карло назначали сорт данного атома (1 или 2), сравнивая это число с величиной
(L – длина ребра основного куба). При A < B атому присваивали сорт 1, а при A ≥ B – сорт 2. В итоге в модели было задано распределение концентрации C(x) со средним значением C0 и косинусоидальной добавкой с амплитудой a0: На границах основного куба (при x = 0 и x = L) производная dC/dx = 0, так что в процессе взаимной диффузии должно происходить только выравнивание косинусоидальной компоненты концентрации и приближение ее к нулю.При исходном профиле концентрации в основном кубе вида (12) решение уравнения диффузии ∂С/∂t = D∂2С/∂x2 имеет точно такой же вид, но с коэффициентом, зависящим от времени:
(13)
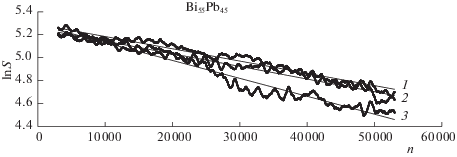
$С (x,t) = {{С }_{0}} + {{a}_{0}}\cos (2\pi {{x}_{i}}{\text{/}}L){{e}^{{ - 4{{\pi }^{2}}Dt/{{L}^{2}}}}}.$МД-опыты по измерению коэффициента взаимной диффузии в растворе Bi55Pb45 были проведены при 700, 773 и 928 К, при значениях C0 = 55 ат. % и a0 = 10 или 20%. Продолжительность диффузионных опытов составляла 50000 шагов по времени с длиной шага 0.01t0 (t0 = 1.472 × 10–13 с – внутренняя единица времени). Зависимости ln S от числа шагов n в МД-прогоне показаны на рис. 7 для прогонов, где a0 = 20%. Эти зависимости имеют довольно нерегулярную форму. При увеличении числа частиц в модели до 16000 они становятся более гладкими. Коэффициенты взаимной диффузии для случаев a0 = 20% были рассчитаны с учетом формулы (13); они обозначены как ${{\tilde {D}}_{{{\text{М Д }}}}}$ и приведены в табл. 7.
Рис. 7.
Зависимости lnS(t) для раствора Bi55Pb45 при C0 = 55 ат. % и a0 = 20 ат. %: 1 – 700 К, 2 – 773 К, 3 – 928 К.
Реальные коэффициенты взаимной диффузии в растворе Bi55Pb45 были измерены капиллярным методом в [13]. Эти данные обозначены в табл. 7 как Dвз. Они выше рассчитанных методом МД в 1.25–1.64 раза. Причины расхождений обсуждать сложно из-за обычной неясности в отношении возможной роли конвекции при измерениях скорости диффузии. Однако следует учитывать, что градиенты концентрации в реальных и компьютерных экспериментах по измерению коэффициента взаимной диффузии отличаются на много порядков. В реальных экспериментах величины dC/dx имеют обычно порядок величины 101 мол. доли/м, а в нашем компьютерном эксперименте ∼108 мол. доли/м. При различии масштабов на 7 порядков линейность уравнения диффузии (5) может нарушаться.
В строке 8 табл. 7 приведены коэффициенты корреляции в расплаве Bi55Pb45, найденные с учетом МД-данных по самодиффузии (строки 1, 2), взаимной диффузии (строка 6) и величине δ в приближении регулярных растворов (формула (3)). В строке 9 даны коэффициенты корреляции, найденные с учетом МД-данных по самодиффузии (строки 1, 2), экспериментальных данных по взаимной диффузии [13] и термодинамических данных [7, 8] (табл. 1). Расхождение результатов расчетов в строках 8 и 9 обусловлено различием коэффициентов взаимной диффузии, полученных методом МД и измеренных в [13].
Теоретическая оценка коэффициента корреляции F в формуле (10) была проведена в [14, 15] аналогично расчетам катафоретического эффекта в теории электролитов. Для частиц компонента 1 получено выражение:
(14)
$F = 1 + {{X}_{{1{\;}}}}{{r}_{1}}\frac{N}{V}\mathop \smallint \limits_0^\infty \,4\pi r[{{g}_{{11}}}(r) - {{g}_{{12}}}(r)]{\kern 1pt} {\kern 1pt} dr,$Отметим, что отклонения фактора F и термодинамического множителя δ от единицы противоположны по направлению и частично гасят друг друга, а значения F, рассчитанные по выражению (14), практически полностью гасят фактор δ, найденный методом МД по уравнению (4) (4 строка в табл. 1). Интересно было бы выяснить, в какой мере реализуется это гашение в системах с сильными отклонениями от идеальности.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Проведенные выше расчеты показывают, что свойства растворов Bi–Pb можно удовлетворительно описать с помощью потенциалов ЕАМ чистых компонентов, вводя в качестве подгоночной функции лишь парный потенциал пар 12 в форме Леннард–Джонса. Предсказаны свойства растворов в условиях ударного сжатия (до температур ∼30000 К и давлений ∼282 ГПа). При повышении давления отклонения термодинамических свойств от идеальности меняются с отрицательных на положительные. Ударная адиабата раствора Bi55Pb45 располагается вблизи адиабат чистых компонентов, но не совпадает с соответствующей средневзвешенной функцией.
При обсуждении диффузионных свойств можно исходить из того, что коэффициенты самодиффузии обычно рассчитываются методом МД довольно точно и неплохо согласуются с измеренным радиоизотопным методом, особенно в условиях микрогравитации [16]. Приведенные выше данные свидетельствуют, что корреляционный эффект при взаимной диффузии в двойных расплавах несомненно существует. Трудность его обнаружения связана с невысокой точностью измерения коэффициентов взаимной диффузии и с тем, что факторы корреляции рассмотренных систем в общем близки к единице. Кроме того, налицо компенсационный эффект, при котором термодинамический множитель δ и фактор F частично гасят друг друга.
Второй момент относится к результатам МД-расчета коэффициента взаимной диффузии ${{\tilde {D}}_{{{\text{М Д }}}}}$. Мы имеем здесь дилемму: либо экспериментальные данные [13] по взаимной диффузии завышены в 1.25–1.6 раза (что отнюдь не редкость в работах на эту тему), либо на моделях расплава размером ∼2000 частиц нельзя успешно моделировать макроскопическую взаимную диффузию. Быстрый монотонный рост коэффициента F в девятой строке табл. 7 от 0.59 до 1.03 (в отличие от умеренного роста в 10-й строке) однозначно указывает на увеличение конвективного вклада в диффузию при увеличении температуры реального эксперимента. С другой стороны, данные в шестой и восьмой строках выглядят немного заниженными, так что возникают вопросы относительно адекватности методики МД измерения коэффициента взаимной диффузии. Выше было уже отмечено, что градиенты концентрации в реальных и компьютерных экспериментах по измерению коэффициента взаимной диффузии отличаются на семь порядков, так что линейность уравнения диффузии (5) в этом интервале градиентов может нарушаться. Тем не менее, факторы корреляции в строках 9 и 10 табл. 7 довольно близки при температурах до 700 К, а расхождения при более высоких температурах могут быть вызваны небольшим конвективным вкладом в реальном эксперименте. Если это действительно так, то учет корреляции при диффузии вполне можно проводить по уравнению (14), возможно, с коррекцией коэффициента перед интегралом.
Список литературы
Handbook on Lead-bismuth Eutectic Alloy and Lead Properties, Materials Compatibility, Thermal-hydraulics and Technologies // 2015 Edition. © OECD 2015. NEA. № 7268 Nuclear Energy Agency Organisation For Economic Co-Operation And Development.
Popović M.P., Bolind A.M., Cionea C., Hosemann P. // Contemporary Materials. 2015. VI–1. P. 20.
Белащенко Д.К. // Журн. физ. химии. 2012. Т. 86. № 5. С. 872.
Белащенко Д.К. // ТВТ. 2017. Т. 55. № 3. С. 386.
Hultgren R., Desai P.D., Hawkins D.T., Gleiser M., Kelley K.K. Selected Values of the Thermodynamic Properties of Binary Alloys. ASM. Metal Park. 1973.
Borisenko A.V., Yagodin D.A., Filippov V.V. et al. // Russian Metallurgy (Metally). V. 2012. № 8. P. 659.
Moser Z. // Z. Metallkd. 1973. Bd. 64. S. 40.
Orlov Y.I. The Main Impurities and Their Condition in the Pb-Bi Coolant. Contract SSC RF IPPE/CEA-DRN-DER 5010 6 8 B049630, Substage 2.1. Obninsk. 1998.
Prasad, R., Venugopal V., Sood D.D. // J. Chem. Thermodyn. 1977. V. 9. P. 765.
Белащенко Д.К., Гопенгауз И.Е., Островский О.И. // Журн. физ. химии. 1992. Т. 66. № 7. С. 1753.
Waseda Y., Yokoyama K., Suzuki K. // Physics and Chemistry of Liquids. 1975. V. 4. № 4. P. 267.
Данные на сайте: http://res.tagen.tohoku.ac.jp/~waseda/scm/AXS/
Tanigaki M., Toyota Yu., Harada M., Eguchi W. // J. Chem. Engineering Japan. 1983. V. 16. № 2. P. 92.
Белащенко Д.К. Явления переноса в жидких металлах и полупроводниках. М.: Атомиздат. 1970. 400 с.
Белащенко Д.К., Митяев В.В. // ФММ. 1969. Т. 27. С. 81.
Frohberg G., Kraatz K.-H., Wever H. // Proc. 5th Europ. Symp. on Material Sciences under Microgravity. Schloss Elmau, 5–7 Nov. 1984. (ESA SP-222). P. 201.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии