

Журнал физической химии, 2019, T. 93, № 1, стр. 11-22
Кинетика и термодинамика образования соединений ионов кальция с аминокислотами, их строение и устойчивостьО. А. Голованова, И. А. Томашевский
О. А. Голованова a, *, И. А. Томашевский a
a Омский государственный университет им. Ф.М. Достоевского
Омск, Россия
* E-mail: golovanoa2000@mail.ru
Поступила в редакцию 22.03.2018
Аннотация
Определены термодинамические константы устойчивости координационных соединений и их термодинамические потенциалы комплексообразования в водных растворах при 298 К. Рассчитаны кинетические характеристики реакции комплексообразования ионов кальция с аминокислотами в водных растворах при 298 К. Показана способность ионов кальция как комплексообразователя координировать аминокислоты в качестве лигандов, установлена закономерность в комплексообразовании различные аминокислот различного строения. Координационные соединения изучены с помощью метода потенциометрического титрования с применением математических методов обработки данных.
Известно, что кальций в ионизированном виде играет важнейшую роль в осуществлении ряда самых важных клеточных и физиологических функций, таких как: формирование костной ткани, минерализация зубов; регуляция внутриклеточных процессов; участие в процессах свертывания крови; поддержание стабильной сердечной деятельности и др. [1].
В настоящее время единой теории, объясняющей природу взаимодействия минеральной (солей кальция) и органической составляющих (аминокислот) не существует. В основном рассматривается три варианта этого взаимодействия: прямое участие органической матрицы в построении биоминерала путем связывания компонентов минеральных фаз и инициирование собственной минерализации (кости, зубы) [2, 3]; ингибирование и/или промотирование кристаллизации минеральных фаз путем избирательного адсорбционного взаимодействия, образование координационных соединений ионов кальция с аминокислотами [4–6]; косвенное участие за счет патогенного взаимодействия в камнеобразующих растворах под действием внешних и внутренних факторов (инфекции, нарушение кальциевого, белкового обмена и т.д.) [7–10]. Например, нарушение процессов ремоделирования костной ткани негативно сказывается на ее структуре и свойствах, в результате чего развиваются различные костно-суставные патологии [3]. Несмотря на значительное число исследований, посвященных изучению костного матрикса, диагностика и коррекция подобных состояний относится к числу социально-значимых и не до конца решенных задач [2, 3].
В случае нарушения регуляции ионов кальция, в организме возникают патологические изменения, которые являются причиной почти 150 заболеваний. По статистике, за последние годы увеличился процент патогенного минералообразования в организме человека [4–9]. Проблема камнеобразования сегодня становится более ощутимой и реальной угрозой для современных жителей всех стран, в особенности – мегаполисов. На протяжении всего двадцатого века различные болезни, приводящие к образованию камней (органоминеральных агрегатов – ОМА) в органах человека, лишь увеличивали скорость их распространения [10–14]. Известно, что кристаллизация ОМА протекает в сложных по составу биологических жидкостях, как в физиогенных, так и в патогенных условиях [10–14]. Данное явление связано с целым рядом факторов экзогенного и эндогенного характера [2–4, 9].
К настоящему времени, сведения о закономерностях кристаллизации ОМА в многокомпонентных по составу физиологических растворах малочисленны и противоречивы. Очевидно, что сложность изучения кристаллизации из модельных растворов биологических жидкостей состоит в том, что в их состав входит большое число органических и неорганических соединений, а также существует множество факторов, влияющих на пусковые механизмы кристаллизации. Кроме того, образование ОМА чаще всего происходит в неравновесных условиях и за их возникновение отвечают кинетические факторы.
При этом, установление закономерностей кристаллизации из прототипов биожидкостей позволяет раскрыть механизмы образования и роста кристаллических фаз, что важно как для понимания природы этих процессов, так и для проведения профилактических мероприятий при борьбе с рядом социально-значимых заболеваний.
Таким образом, понимание природы и характеристик образующихся соединений ОМА поможет количественно охарактеризовать степень влияния разрушающих факторов, а также показать роль образования/разрушения соединений в патологических и физиологических процессах, происходящих в живых тканях. Кроме того, изучение процесса взаимодействия ионов кальция с АК послужит опорным теоретическим материалом при создании медицинских препаратов, направленных на точечную доставку ионов кальция по месту назначения. Решение подобной задачи возможно только путем систематического исследования взаимодействия ионов кальция с органическими лигандами.
Исследование в данной области усложняется тем, что в литературе приводится мало достоверных данных о взаимодействии между ионами кальция и АК. Практически во всех литературных источниках имеются сведения о том, что в водных растворах координируется комплекс состава 1 : 1 [1, 15, 16]. Это связано с тем, что ионы кальция, как и ионы всех металлов IIA группы, несмотря на координационное число, равное 6 [17], являются слабыми комплексообразователями как таковыми, к тому же, АК являются стерически объемными лигандами и их координирование по ионам кальция при подходе сразу множества молекул затруднен. В связи с этим, далее обсуждение результатов будет вестись с учетом состава комплексов 1 : 1.
Известно, что для изучения процессов кристаллизации из прототипов биожидкостей, как правило, используют спектрофотометрические, ионнообменные и полярографические методы [15–18]. На данный момент не представлено ни одного литературного источника, в котором были бы указаны все термодинамические и кинетические характеристики комплексов Ca2+ c АК. Поэтому, важным является разработка алгоритма, который предполагал бы чувствительное, точное, быстрое и селективное определение характеристик образования КС. Одним из способов такой оценки может стать использование заместительного потенциометрического титрования смесей Ca2+ и АК раствором гидроксида натрия NaOH с последующей расшифровкой экспериментальных кривых титрования с помощью компиляции нескольких математических методов, которые будут представлены ниже [19, 20].
Целью настоящей работы является изучение способности взаимодействовать АК с ионами кальция в водных растворах при T = 298 К по целому спектру параметров: характеристики устойчивости комплексов, термодинамические характеристики комплексообразования, кинетические характеристики комплексообразования, интерпретация полученных характеристик в рядах исследуемых систем в соответствии со строением изучаемых АК.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы и материалы
В качестве лигандов использовались АК в виде сухих веществ марки х.ч. Навески АК, взятые с точностью до 0.0001 г, растворяли в мерной колбе на 200.0 мл для создания конечной концентрации аминокислоты в мерной колбе, равной 10–2 моль/л. АК, исследуемые в составе комплексов, имеют разный состав и строение, их характеристики приведены в табл. 1 [21].
Таблица 1.
Характеристика аминокислот
| АК | Обозна- чение | Брутто-формула | Константы диссоциации, pK | pI | ||
|---|---|---|---|---|---|---|
| α-COOH | α-NH2 | пр. ион. группы | ||||
| Глицин | Gly | C2H5NO2 | 2.35 | 9.78 | 6.20 | |
| Аланин | Ala | C3H7NO2 | 2.35 | 9.78 | 6.11 | |
| Аспарагиновая кислота | Asp | C4H7NO4 | 1.99 | 9.90 | β-COOH-3.90 | 2.98 |
| Изолейцин | Ile | C6H13NO2 | 2.32 | 9.76 | 6.04 | |
| Аргинин | Arg | C6H15N4O2 | 1.82 | 8.99 | ε-NH2 12.48 | 10.76 |
| Гистидин | His | C6H9N3O2 | 1.80 | 9.33 | >NH-6.04 | 7.60 |
| Валин | Val | C5H11NO2 | 2.29 | 9.74 | 6.02 | |
| Серин | Ser | C3H7NO3 | 2.19 | 9.21 | 5.70 | |
| Глутаминовая кислота | Glu | C5H9NO4 | 2.10 | 9.47 | γ-COOH-4.07 | 3.09 |
| Метионин | Met | C5H11NO2S | 2.13 | 9.28 | 5.71 | |
| Пролин | Pro | C5H9NO2 | 1.95 | γ-NH-10.64 | 6.30 | |
| Лизин | Lys | C6H14N2O2 | 2.16 | 9.06 | δ-NH2-10.54 | 9.80 |
| Аспарагин | Asn | C4H8N2O3 | 2.14 | 8.72 | 5.43 | |
| Треонин | Thr | C4H9NO3 | 2.11 | 9.10 | 5.61 | |
В качестве источника ионов кальция использовалась соль нитрата кальция Ca(NO3)2 ⋅ 5H2O марки “х.ч.”. Навески нитрата кальция, взятые с точностью до 0.0001 г, растворяли в мерной колбе на 200.0 мл для создания конечной концентрации ионов кальция (II) в мерной колбе, равной 10–3 моль/л. Комплекс ионов кальция Ca2+ с каждой из АК (табл. 1) был получен путем смешения аликвот исходных растворов АК и нитрата кальция. После этого, перемешивали содержимое колбы в течение 30 минут.
Методы анализа
В работе использовался метод потенциометрического титрования смесей Ca2+ и АК раствором гидроксида натрия NaOH с последующей расшифровкой экспериментальных кривых титрования с помощью компиляции нескольких математических методов, которые будут представлены ниже. Исследование проводилось с применением следующего средства измерения: иономер И-160-МИ, точность измерения ЭДС прибора = ±0.1 мВ; – ионселективный электрод марки ЭЛИС-121Са (в соответствии с возможностями использования, указанными в работе [22], хлорсеребряный электрод сравнения. Точность метода потенциометрического титрования считается достаточной (общая погрешность определения – 0.5–1.0%) для установления характера взаимодействия аминокислот и ионов кальция) [23].
Первоначальное значение ЭДС раствора устанавливалось для всех титрований в пределах ±0.1 мВ в течение 3 мин. Титрование для всех АК проводилось с шагом в 0.5 мл из бюретки, в качестве титранта использовался свежеприготовленный раствор 0.10 М NaOH, перед титрованием каждой АК раствор титранта стандартизовали раствором 0.10 М HCl с кислотно-основным индикатором фенолфталеином.
Значение ЭДС перед добавлением каждой порции титранта устанавливалось в течение 45 с. Титрование продолжалось до момента, пока не начинал выпадать осадок Ca(OH)2. С целью улучшения характеристик повторяемости эксперимента и исключения влияния больших систематических погрешностей, для каждой аминокислоты проводилось три параллельных титрования. Поскольку предел повторяемости (r) для каждого измерения каждого параллельного испытания не превысил 5 мВ (в относительном значении для всех случаев и всех АК < 3%, в большинстве случаев < 1%), в качестве источника данных использовались интегральные кривые, полученные усреднением значений ЭДС для каждого шага титрования. Также, с помощью ионселективного электрода определяли значение pCa при каждом титровании [21].
Для установления кинетических характеристик образования комплекса Ca2+ и АК, был использован метод Грана. В данном методе точка эквивалентности определяется по графику в координатах: ∆V/∆E – V, где ∆V – шаг титрования, ∆E – разность ЭДС между двумя крайними значениями V – объем добавленного титранта. Перед точкой эквивалентности и после нее кривая Грана линейна. Точка эквивалентности находится как точка пересечения этих прямых. Достоинства и удобства метода Грана особенно заметны при анализе разбавленных растворов, позволяющих определить точку эквивалентности с достаточной точностью вследствие линейности графика, а также в тех случаях, когда кривая титрования выражена плохо [24].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Изменение распределения ионизированных форм аминокислот при различных pH
Для расшифровки и интерпретации результатов потенциометрического титрования, были построены ионные диаграммы, демонстрирующие формы нахождения АК (в долях от единицы) при определенных значениях pH. По виду ионных диаграмм, исследуемые АК можно разбить на три группы (рис. 1а–в):
Рис. 1.
Распределения ионизированных форм АК от pH: а – первая группа; б – вторая группа; в – третья группа.
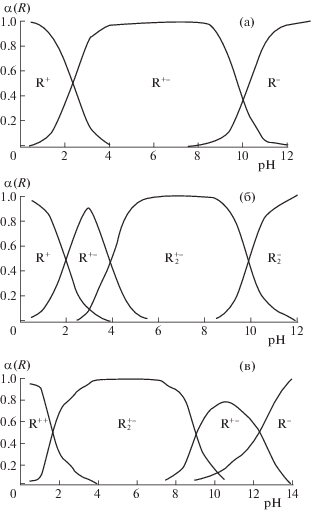
1. Большинство изучаемых АК не имеют ионизированных групп в радикале, их общий типичный вид диаграмм представлен на рис. 1а.
2. Аспарагиновая и глутаминовая кислоты имеют в своем составе две карбоксильные группы, поэтому они будут иметь ионную диаграмму, типичную для кислых АК (рис. 1б).
3. Три из исследуемых АК – аргинин, гистидин, лизин – помимо α-NH2 группы, имеют в своем составе ионизируемые основные группы, Гистидин имеет в своем составе имидазольную группу, именно благодаря имидазольной группе при физиологических колебаниях значений рН (от 6.9 до 7.4) гистидин заряжен либо нейтрально, либо положительно. Аргинин имеет в составе сильноосновную гуанидиниевую группировку, лизин содержит дополнительную аминогруппу на конце боковой цепи. Они будут иметь зависимость от pH, представленную на рис. 1в.
С варьированием pH, данные АК будут претерпевать следующие изменения:
Первая группа:
1. При pH < pK (α-COOH), за счет избытка протонов в водном растворе, будет преобладать протонированная форма АК
(1)
$\begin{gathered} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {COOH\;}{\kern 1pt} + {\kern 1pt} {{{\text{H}}}^{ + }} \rightleftarrows \\ \rightleftarrows {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{COOH}}{\text{.}} \\ \end{gathered} $2. При pK (α-COOH) < pH < pK (α-NH2), начинает диссоциировать α-карбоксильная группа с образованием цвиттер-иона
(2)
$\begin{gathered} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{COOH}} \rightleftarrows \\ \rightleftarrows {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }}{\kern 1pt} + {\kern 1pt} {{{\text{H}}}^{ + }}{\text{.}} \\ \end{gathered} $3. При pH > pK (α-NH2), депротонирует атом азота α-NH3 группы с образованием аниона АК
(3)
$\begin{gathered} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} \rightleftarrows \\ \rightleftarrows {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} + {{{\text{H}}}^{ + }}. \\ \end{gathered} $Пролин, фактически, отличается в данном случае от данной группы АК тем, что в процессах протонизации/депротонизации участвует не α-NH2, а γ-NH-группа.
Вторая группа:
1. При pH < pK (α-COOH), за счет избытка протонов в водном растворе, будет преобладать протонированная форма АК
(4)
$\begin{gathered} {\text{HOOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {COOH\;} + {{{\text{H}}}^{ + }} \rightleftarrows \\ \rightleftarrows {\text{HOOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{COOH}}{\text{.}} \\ \end{gathered} $2. При pK (α-COOH) < pH < pK (β-COOH или γ-COOH), начинает диссоциировать α-карбоксильная группа с образованием цвиттер-ион
(5)
$\begin{gathered} {\text{HOOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{COOH}} \rightleftarrows \\ \rightleftarrows {\text{HOOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} + {{{\text{H}}}^{ + }}{\text{.}} \\ \end{gathered} $3. При pK (β-COOH или γ-COOH) < pH < pK (α-NH2), диссоциирует вторая карбоксильная группа А
(6)
$\begin{gathered} {\text{HOOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} \rightleftarrows \\ \rightleftarrows {}^{ - }{\text{OOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} + {{{\text{H}}}^{ + }}{\text{.}} \\ \end{gathered} $4. При pH > pK (α-NH2), депротонирует атом азота $\alpha - {\text{NH}}_{3}^{ + }$-группы с образованием аниона АК
(7)
$\begin{gathered} {}^{ - }{\text{OOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} \rightleftarrows \\ \rightleftarrows {}^{ - }{\text{OOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} + {{{\text{H}}}^{ + }}{\text{.}} \\ \end{gathered} $Как видно, уже при pH 4 образуются анионы, что принципиально отличает эти две АК от других исследуемых α-аминокислот.
Третья группа:
1. При pH < pK (α-COOH), за счет избытка протонов в водном растворе и положительно заряженного радикала, будет преобладать наиболее протонированная форма АК
(8)
$\begin{gathered} {\text{H}}{{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {COOH\;} + {{{\text{H}}}^{ + }} \rightleftarrows \\ \rightleftarrows {\text{H}}{{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{COOH}}{\text{.}} \\ \end{gathered} $2. При pK (α-COOH) < pH < pK (α-NH2), начинает диссоциировать карбоксильная группа с образованием разнополярного иона
(9)
$\begin{gathered} {\text{H}}{{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{COOH}} \rightleftarrows \\ \rightleftarrows {\text{H}}{{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} + {{{\text{H}}}^{ + }}{\text{.}} \\ \end{gathered} $3. При pK (α-NH2) < pH < pK (ε, δ –NH2 или >NH), диссоциирует вторая карбоксильная группа АК
(10)
$\begin{gathered} {\text{H}}{{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} \rightleftarrows \\ \rightleftarrows {\text{H}}{{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} + {{{\text{H}}}^{ + }}{\text{.}} \\ \end{gathered} $4. При pH > pK (ε, δ –NH2 или >NH), депротонирует атом второй аминогруппы с образованием аниона АК
(11)
$\begin{gathered} {\text{H}}{{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} \rightleftarrows \\ \rightleftarrows {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} + {{{\text{H}}}^{ + }}{\text{.}} \\ \end{gathered} $Данные АК несут положительный заряд в наибольшем интервале значений pH. Стоит подчеркнуть, что указанные зависимости на рис. 1а–в являются типичными, а распределение ионизированных форм каждой отдельной АК изменяется от pK группировок, присутствующих в ней [21].
Таким образом, зная зависимости распределения ионизированных форм при pH 3 для каждой из исследуемых АК, в начале титрования, а также, получив экспериментальным путем интегральные кривые потенциометрического титрования, представляется возможным охарактеризовать процесс образования/разрушения комплексов ионов кальция (комплексообразователь) с АК (органические лиганды) на всех стадиях комплексообразования. На рис. 2 изображены интегральные кривые заместительного потенциометрического титрования изучаемых КС. Изучаемые АК разбиты на два ряда по семь АК на каждом, интегральные кривые АК первого ряда (рис. 2а) имеют скачок титрования ближе к началу координат, чем кривые АК второй группы (рис. 2б).
Рис. 2.
Интегральные кривые потенциометрического титрования: а – первого ряда АК: – изолейцин;
+ аргинин; х‑метионин; - пролин;  лизин;
лизин;  аспарагин; $\vartriangle $ треонин; б – второго ряда АК: × – аланин; + гистидин; $\blacksquare $ валин; * серин; $\vartriangle $ глицин;
аспарагин; $\vartriangle $ треонин; б – второго ряда АК: × – аланин; + гистидин; $\blacksquare $ валин; * серин; $\vartriangle $ глицин;  аспарагиновая кислота;
аспарагиновая кислота;  глутаминовая кислота.
глутаминовая кислота.
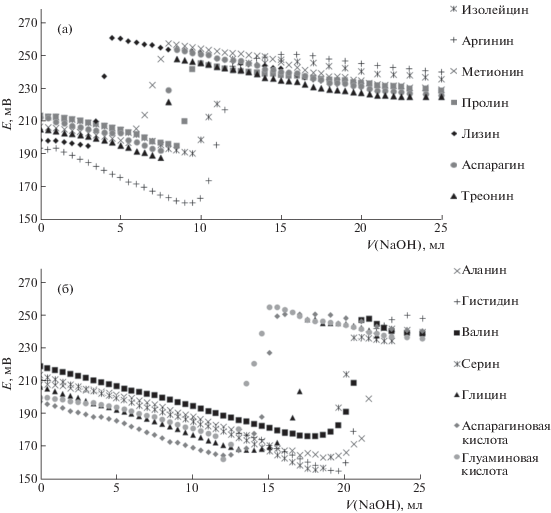
Видно, что для всех КС кривые имеют, в целом, аутентичную форму. Это дает основание предполагать, что во всех случаях процессы образования и разрушения комплексов при данном титровании имеют некий общий характер. При этом, различное расположение скачков титрования относительно оси абсцисс координат и различная величина скачка титрования указывает на разные количественные характеристики образования/разрушения каждого из комплексов.
Исходя из данных (рис. 1–2, табл. 1), возможно предположить, что в результате потенциометрического титрования происходят следующие процессы:
1. Первоначально, смесь ионов кальция и АК объемом 20.0 мл подкислялась 6 М раствором HCl до pH 3.
При этих значениях pH (сильнокислая среда), согласно ионным диаграммам, 11 АК (изолейцин, аланин, глицин, валин, серин, метионин, пролин, аспарагин, треонин, аспарагиновая и глутаминовая кислоты) в начале титрования будут находиться преимущественно в форме цвиттер-иона
(12)
$\begin{gathered} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{COOH}} + {\;}{{{\text{H}}}^{ + }} \rightleftarrows \\ \rightleftarrows {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }}{\text{.}} \\ \end{gathered} $В составе аспарагиновой и глутаминовой кислот имеется вторая карбоксильная группа, которая также может частично принимать участие в перераспределении электронного заряда, также, есть некоторая доля катионной формы АК, однако, в целом, молекулы аспарагиновой и глутаминовой кислот пребывают в состоянии, аналогичном цвиттер-ионам вышеперечисленных АК
(13)
$\begin{gathered} {\text{HOOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{COOH}} + {{{\text{H}}}^{ + }} \rightleftarrows \\ \rightleftarrows {\text{HOOC}}{\kern 1pt} - {\kern 1pt} {\text{R}}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} + {{{\text{H}}}^{ + }}{\text{.}} \\ \end{gathered} $Аргинин, гистидин и лизин, как указано выше, имеют в своем составе основные группы, поэтому на данном этапе АК заряжены положительно
(14)
$\begin{gathered} {{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{N}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{COOH}} \rightleftarrows \\ \rightleftarrows {{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }}{\text{.}} \\ \end{gathered} $Для удобства, примем здесь и далее следующее обозначение:
Ионы Ca2+ выступают в смеси в качестве комплексообразователя, поэтому, АК координируется по ним
(15)
${\text{C}}{{{\text{a}}}^{{2 + }}} + {{{\text{L}}}^{ \pm }} \rightleftarrows {{{\text{[С a}}{{{\text{L}}}^{ \pm }}{\text{]}}}^{{2 + }}}.$Для аргинина, гистидина, лизина координация происходит по уравнению
(16)
$\begin{gathered} {\;}{{{\text{R}}}^{ + }}{\kern 1pt} - {\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{NH}}_{3}^{ + }{\kern 1pt} - {\kern 1pt} {\text{CO}}{{{\text{O}}}^{ - }} = {{{\text{L}}}^{{ \pm + }}}, \\ {\text{C}}{{{\text{a}}}^{{2 + }}} + {{{\text{L}}}^{{ \pm + }}} \rightleftarrows {{[{\text{С a}}{{{\text{L}}}^{{ \pm + }}}]}^{{2 + }}}{\text{.}} \\ \end{gathered} $2. В начале титрования с добавлением титранта ЭДС электрохимической цепи медленно уменьшается, это связано с тем, что ионная сила раствора возрастает, снижая активность свободных ионов кальция. При этом, комплексы ${{{\text{[С a}}{{{\text{L}}}^{ \pm }}{\text{]}}}^{{2 + }}}$ и ${{[{\text{С a}}{{{\text{L}}}^{{ \pm + }}}]}^{{2 + }}}$ остаются устойчивыми.
3. В определенный момент, после добавления очередной порции титранта, для всех АК наблюдается резкое увеличение (скачок) значения ЭДС. Данное изменение связано с увеличением концентрации несвязанных ионов кальция, которые появляются в растворе с разрушением комплекса
(17)
${{{\text{[С a}}{{{\text{L}}}^{ \pm }}{\text{]}}}^{{2 + }}} \to {\text{C}}{{{\text{a}}}^{{2 + }}} + {{{\text{L}}}^{ \pm }}$(18)
${{[{\text{С a}}{{{\text{L}}}^{{ \pm + }}}]}^{{2 + }}} \to {\text{C}}{{{\text{a}}}^{{2 + }}} + {{{\text{L}}}^{ \pm }}.$Это подтверждается соответствующими измерениями pCa при добавлении каждой порции титранта. В качестве примера, на рис. 3 приведено изменение активности ионов кальция при добавлении 0.10 М гидроксида кальция (шаг = 0.5 мл) к смеси ионов кальция с серином.
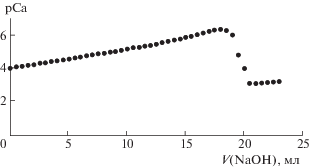
Как видно, при добавлении достаточного объема титранта, при котором возникает скачок ЭДС происходит резкое снижение pCa, что и связано с разрушением комплекса кальций-аминокислотного комплекса. Увеличение показателя pCa связано со снижением активности ионов кальция.
По нашему мнению, в точке эквивалентности (т. экв.) 50% молекул комплексного соединения устойчивы, 50% – разрушены, т.е. имеют функцию образования $\bar {n}$ = 0.5 [25].
4) После завершения разрушения комплекса, ЭДС также начинает монотонно падать по мере увеличения ионной силы и соответственно снижения активности ионов кальция.
5) При добавлении около 25.0 мл, в растворе достигается pH 10–10.5, который приводит к образованию малорастворимого осадка гидроксида кальция (рПР = 5.26)
(19)
${\text{C}}{{{\text{a}}}^{{2 + }}} + 2{\text{O}}{{{\text{H}}}^{ - }} \to {\text{С a}}{{({\text{OH}})}_{{2{\;}}}}.$Поскольку в т.экв. разрушается 50% молекул комплексных соединений от общего числа изначально образованных, объем раствора NaOH, затраченный на титрование для достижения точки эквивалентности кривой титрования, может служить полуколичественной характеристикой величины взаимодействия ионов кальция и каждой из аминокислот. Соответственно, чем больше объем затраченного титранта, тем больше его необходимо на разрушение комплекса и тем получившийся комплекс устойчивее. Обозначим эту характеристику как Vn = 0.5.
Данная характеристика рассчитана по потенциометрическим кривым для комплексов всех АК с ионами кальция (табл. 2).
Таблица 2.
Значения условного критерия Vn = 0.5 для комплексов аминокислот с Са2+
| АК | Vn = 0.5 | АК | Vn = 0.5 |
|---|---|---|---|
| Lys | 3.6 | Glu | 13.5 |
| Met | 7.6 | Asp | 14.7 |
| Asn | 7.8 | Gly | 17.3 |
| Thr | 7.8 | Ser | 19.5 |
| Pro | 9.2 | Val | 20.3 |
| Ile | 10.5 | His | 20.8 |
| Arg | 11.2 | Ala | 21.5 |
На основании полученных данных, все АК можно ранжировать по возрастающей устойчивости комплекса Ca2+ с АК
(20)
$\begin{gathered} {\text{Lys}} \to {\text{Met}} \to {\text{Asn}} \to {\text{Thr}} \to {\text{Pro}} \to {\text{Ile}} \to \\ \to {\text{Arg}} \to {\text{Glu}} \to {\text{Asp}} \to {\text{Gly}} \to \\ \to {\text{Ser}} \to {\text{Val}} \to {\text{His}} \to {\text{Ala}}{\text{.}} \\ \end{gathered} $Графически, с указанием структурных формул АК и их функциональных групп, данный ряд можно представить следующим образом (рис. 4).
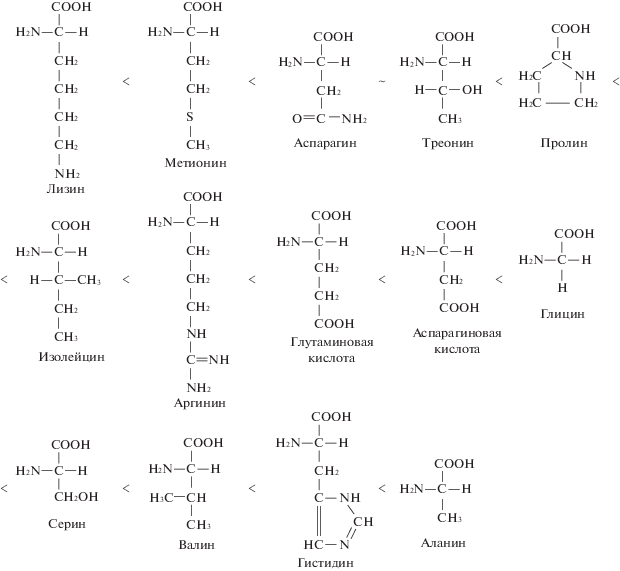
Видно, что структурные формулы трех вышеперечисленных АК различаются между собой а) по количеству атомов углерода в структурной формуле; б) по природе функциональных групп и их количеству; в) по расположению функциональных групп.
Для удобства объяснения увеличения устойчивости комплексов в предложенном ряду, будем принимать во внимание три данных фактора: а) большое количество атомов углерода будут создавать препятствие при координации лигандов АК к ионам кальция; б) ион кальция относится к первой группе катионов, для которых комплексообразование осуществляется, главным образом, за счет атомов кислорода – –COOH-группы АК. Влияние донорных атомов азота к образованию координационных связей с кальцием менее значительно, но возможно. Также, в комплексообразовании/разрушении комплексов могут принимать участие и другие функциональные группы, являющиеся донором или акцептором электронной плотности. Соответственно, увеличение количества однотипных функциональных групп усиливает их эффект; в) расположение функциональных групп относительно друг друга и относительно α-атома углерода носит ключевой характер в комплексообразовании [21].
Разделим условно весь ряд на три группы:
А) Неустойчивые. Как видно, самым неустойчивым комплексом является комплекс ионов кальция с лизином. Это связано с тем, что молекула данной АК имеет большое количество атомов углерода в радикале, связанном с α-атомом углерода. Этим самым, создается стерическое препятствие при подходе молекулы АК к ионам кальция, соответственно, данный комплекс будет разрушаться проще всего, несмотря на NH2 на конце углеродного радикала, которая также может служить источником электронной плотности, но гораздо хуже, поскольку стерический фактор в данном случае будет преобладать над электронным. Метионин имеет в своем составе боковой радикал, имеющий на одну CH2-группу меньше, чем в лизине, и содержит после γ-атома углерода –S-группу, которая может принимать участие в комплексообразовании. Соответственно, комплекс Ca2+-Met чуть более устойчив, чем Ca2+–Lys. Устойчивость систем Ca2+–Asn и Ca2+–Thr начинает повышаться, поскольку они содержат в своем составе амидную и гидроксильную группировки, которые принимают участие в создании дополнительных связей с ионами кальция. Пролин, при этом, подобных группировок не имеет, но, за счет образования циклического радикала, имеет выигрыш к компактности, соответственно, данная АК имеет чуть более устойчивый комплекс с ионами кальция.
Б) Слабоустойчивые. Молекула изолейцина имеет разветвленный боковой радикал, соответственно, это позволяет подходить большему числу молекул лиганда к комплексообразователю, –NH2- и –COOH-группы ориентируются по комплексообразователю. Молекула аргинина имеет аналогичное изолейцину строение, но вместо метилового заместителя на противоположном от карбоксильной группы конце молекулы расположена гуанидиновая группа NH2–C(NH)–NH2. Большое количество аминогрупп создает большую электронную плотность и, как следствие, дополнительные связи с комплексообразователями.
Что касается аминокислот с двумя карбоксильными группами, то, как уже сказано выше, координация жестких ионов металлов в реакциях комплексообразования с аминокислотами осуществляется преимущественно за счет атомов кислорода карбоксильных групп. При этом, ‒COOH-группа координируется бидентатно с образованием циклической структуры либо мостиковой структуры [26]
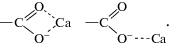
Поскольку этих групп в молекуле две, он считается более сильным лигандом, чем предыдущие АК, и комплексы Ca2+ c аспарагиновой и глутаминовой кислотами более устойчивы, чем с предыдущими аминокислотами, что хорошо согласуется с полученными результатами. Разница в комплексообразовании между этими двумя АК исключительно в том, что глутаминовая кислота содержит на один атом углерода больше, что сильнее делокализует электронную плотность, создаваемую двумя карбоксильными группами, делая комплекс с Са2+ чуть менее устойчивым, чем с аспарагиновой кислотой. Замена карбоксильной группы на амидную у аспарагина наглядно показывает падение устойчивости комплекса с ионами кальция.
В) Относительно устойчивые. В отличие от двух предыдущих групп, молекулы данных АК, за исключением гистидина, состоят из меньшего числа атомов углерода, и не имеют объемных CH3-заместителей, которые могут являться стерическим препятствием при координировании свободных s-орбиталей ионов комплексообразователя Ca2+ и электронных пар лигандов.
Именно поэтому комплексы Ca2+ с АК данной группы более устойчивы, чем те, которые имеют в своем составе объемные заместители и большее число атомов углерода в основной цепи.
Большая устойчивость гистидина объясняется тем, что боковой радикал, помимо атомов азота, имеет в своем составе двойную связь –С=С–, и вместо того, чтобы делокализовывать электронную плотность, атомы углерода способны ее создавать. Таким образом, боковой радикал может являться активным участником комплексообразования.
Обобщая, можно отметить, что все перечисленные факторы активно влияют на комплексообразование и ранжируют устойчивость комплексов с Са2+ в определенной последовательности.
Как видно, устойчивость комплексов исследуемых АК с Са2+ установлена на качественном и полуколичественном уровне за счет ввода условной характеристики Vn = 0.5. С помощью математических приемов, возможно и количественное описание данных систем.
Общеизвестно, что главная количественная характеристика любого процесса комплексообразования – это общая константа устойчивости, для данных компонентов она выражается следующей формулой
(21)
${M\;} + {\;}n{L\;} \rightleftarrows {\;}[{\text{M}}{{{\text{L}}}_{n}}],\quad {{{\beta }}_{n}} = \frac{{\left[ {{\text{M}}{{{\text{L}}}_{n}}} \right]}}{{\left[ {\text{M}} \right]{{{\left[ {\text{L}} \right]}}^{n}}}},$Как было отмечено ранее, данная система является очень сложной для изучения. Это объясняет тот факт, что значения констант устойчивости КС встречаются в литературных источниках крайне редко, а сами исследования не являются комплексными, в лучшем случае, содержат данные по комплексам ионов кальция с одной или двумя АК. Кроме того, такие исследования проведены достаточно давно, а новых исследований по данной тематике не проводилось [27–30].
Однако, в случае, если данные будут достоверными, их, возможно, использовать для того, чтобы вычислить константы устойчивости исследуемых комплексов в данной статье с учетом полученных экспериментальных данных и их математической обработки.
Для этого, необходимо знать значения хотя бы двух (достоверно установленных) констант устойчивости КС, которые будут приняты в качестве опорных. Сложность, помимо вышеуказанных факторов, состоит в том, что в литературных источниках найденные константы устойчивости вычислялись, как правило, при иных значениях ионной силы водных растворов, нежели в данной статье, и выбор ионной силы для одной кислоты резко отличался от других. Чтобы получить опорные значения констант при значении ионной силы I = 0.5, был использован метод экстраполяции констант устойчивости при ионных силах I = 0.1 и I = 3.0 комплекса Ca2+ с серином и при ионных силах I = 0.1 и I = 1.0 комплекса Ca2+ с глутаминовой кислотой на значение ионной силы I = 0.5 при Т = 298 К. Данный выбор неслучаен и связан с тем, что в литературе для этих двух АК имеются значения констант системы Ca2+–АК для двух разных значений ионной силы [31–34]. Для всех остальных АК встречаются лишь значения констант устойчивости при I = 0.1. Также, метод исследования для нахождения констант – потенциометрический во всех четырех случаях, что дает возможность сопоставлять результаты данных литературных источников (табл. 3).
Экстраполяция на ионную силу I = 0.5 дает следующие значения lg K°:
Для системы Ca2+–Ser lg K°=3.68, для системы Ca2+–Glu lg K° = 2.14. Данные значения констант здесь далее используются как опорные для нахождения констант устойчивости при одинаковых условиях проведения испытаний для всех исследуемых систем. Стоит отметить, что в этом случае lg K°(Ca2+–Ser) > lg K° (Ca2+–Glu), тогда как Vn = 0.5 (Ca2+–Ser) > Vn = 0.5 (Ca2+–Glu), что говорит о хорошем согласовании экспериментальных и расчетных данных по данным комплексам с другими литературными источниками [18].
Связав значения lg K° для систем Ca2+–Ser и Ca2+–Glu с соответствующими им полуколичественными условными характеристиками Vn = 0.5 и путем применения дальнейшей экстраполяции, представляется возможным найти значения lg K° для всех исследуемых систем. Произведя соответствующие преобразования, был получен ряд значений логарифмов констант устойчивости КС (табл. 4).
Таблица 4.
Значения lg K° для комплексов аминокислот с Са2+, найденных методом экстраполяции значений Vn = 0.5 и lg K°
| АК | lg K° | АК | lg K° |
|---|---|---|---|
| Lys | 0.39 | Glu | 2.14 |
| Met | 0.63 | Asp | 2.45 |
| Asn | 0.68 | Gly | 3.11 |
| Thr | 0.68 | Ser | 3.68 |
| Pro | 1.04 | Val | 3.88 |
| Ile | 1.37 | His | 4.01 |
| Arg | 1.55 | Ala | 4.19 |
Полученные значения констант позволяют рассчитать термодинамические характеристики образования КС, в частности, энергию Гиббса по формуле
где T – температура проведения испытания = = 298 К; R – универсальная газовая постоянная = = 8.314 Дж/(моль К).Далее, если принять, что энтропия самой системы Са2+ – АК $S_{{{\text{298}}}}^{^\circ }$ стремится к 0, то становится возможным расчет изменения энтропии $\Delta S_{{{\text{298}}}}^{^\circ }$ в ходе реакции комплексообразования, используя $S_{{{\text{298}}}}^{^\circ }$ ионов Са2+ (в H2O) из справочника = = +56.5 Дж/моль и рассчитав $S_{{{\text{298}}}}^{^\circ }$ для каждой из АК, используя следующий упрощенный способ. Зависимость между молекулярной теплоемкостью и молекулярной энтропией вещества выражается уравнением
где K – константа (для жидких тел при 298 К равна 1.4); Ср – общая теплоемкость органических соединений (в жидком состоянии), которая рассчитывается по уравнению(24)
${{С }_{р }} = {\text{2}}.{\text{5}}({\text{С }}) + {\text{2}}.{\text{3}}({\text{Н }}) + {\text{6}}.{\text{5}}({\text{О }}) + {\text{7}}.0({\text{N}}) + {\text{8}}.{\text{5}}({\text{S}}).$Все коэффициенты в данном уравнении являются атомными теплоемкостями, предназначенными для приближенного вычисления. Каждый коэффициент необходимо сначала умножить на число атомов данного элемента, входящего в соединение, и уже затем суммировать.
В соответствии с уравнениями (23), (24) и данных табл. 1 и преобразований согласно закону Гесса, были рассчитаны значения $\Delta S_{{298}}^{^\circ }$ образования всех исследуемых систем.
Далее, исходя из уравнения расчета энергии Гиббса, которая является разностью энтальпийного и энтропийного термодинамических факторов по уравнению
становится возможным расчет энтальпии комплексообразования КС при Т = 298К путем подстановки найденных ранее значений $\Delta G_{{298}}^{^\circ }$ и $\Delta S_{{298}}^{^\circ }$.Для удобства, значения термодинамических потенциалов для исследуемых систем сведены в табл. 5.
Таблица 5.
Значения термодинамических потенциалов для процессов комплексообразования аминокислот с Са2+
| АК | $ - \Delta H_{{298}}^{^\circ }$, кДж/моль | $ - \Delta S_{{298}}^{^\circ }$, Дж/(моль К) | $ - \Delta G_{{298}}^{^\circ }$, кДж/моль |
|---|---|---|---|
| Lys | 41.2 | 130.7 | 2.23 |
| Met | 40.3 | 123.3 | 3.60 |
| Asn | 39.3 | 118.9 | 3.89 |
| Thr | 38.1 | 114.8 | 3.89 |
| Pro | 38.8 | 110.2 | 5.93 |
| Ile | 44.2 | 121.9 | 7.83 |
| Arg | 52.4 | 146.0 | 8.85 |
| Glu | 49.2 | 124.2 | 12.20 |
| Asp | 48.8 | 117.1 | 13.95 |
| Gly | 45.6 | 93.5 | –17.74 |
| Ser | 53.0 | 107.6 | 20.95 |
| Val | 56.3 | 114.8 | 22.12 |
| His | 60.3 | 125.7 | –22.84 |
| Ala | 53.8 | 100.6 | 23.86 |
Видно, что во всех случаях процесс комплексообразования протекает самопроизвольно, при этом, значения энергии Гиббса $\Delta G_{{298}}^{^\circ }$ сравнительно невелики, что говорит об образовании малоустойчивых комплексов, реакция образования комплексов является обратимой. Тем не менее, реакция при условиях эксперимента протекает самопроизвольно. Кроме того, как энтальпийная (экзотермический тепловой эффект реакции), так и энтропийная составляющая (упорядочивание в системе, что характерно для подобных процессов) реакций комплексообразования являются отрицательными. При этом, стоит отметить, что энтальпиийная составляющая, в целом, вносит больший вклад в значение энергии Гиббса, однако и размер энтропийной составляющей в данном случае тоже довольно важен, и ее влиянием в процессе комплексообразования пренебречь нельзя.
Помимо всего вышеперечисленного, в данном исследовании стояла задача установления скорости образования/разрушения системы “Ca2+–АК” для всех изучаемых систем. Поэтому, для решения данной задачи, была проведена математическая обработка всех интегральных кривых потенциометрического титрования, изображенных на рис. 6 , согласно которым, получены первая и вторая производные кривых титрования, а также кривые титрования, построенные по методу Грана, который указан в методах анализа.
Вследствие того, что данные кривых титрования в нашем эксперименте отличаются от классических, получаемых при потенциометрическом титровании, для оценки лабильности кальций-аминокислотных комплексов был использован метод Грана, который был модифицирован* тем, что по оси ординат вместо разностей ЭДС и объемов между двумя крайними значениями откладывались разности между текущим значением ЭДС и объема затраченного титранта и их значениями перед началом титрования соответственно. Данная обработка кривых позволяет избежать искажения кривых от различных побочных процессов и более удобна для анализа. В качестве примера, на рис. 5 приведены дифференцированные кривые титрования смесей Ca(NO3)2 с изолейцином и аргинином.
Рис. 5.
Дифференцированные кривые потенциометрического титрования по методу Грана смесей Ca(NO3)2) и изолейцина и аргинина раствором гидроксида натрия $\bigcirc $ изолейцин;  аргинин.
аргинин.

Полученные кривые как таковые могут использоваться для полуколичественного описания того, насколько тот или иной комплекс Ca2+ с АК лабилен. Для того, чтобы определить степень лабильности образовавшихся комплексов, на преобразованных кривых, предлагается ввести полуколичественную характеристику, описывающую поведение разрушающегося комплекса – δ, которая представляет собой следующее выражение
(26)
${\delta } = \frac{{\Delta V{\;}}}{{\Delta E}}\quad {\text{т }}.\,{\text{п о с л е }}\;{\text{т }}.\,{\text{э к в }}.{\;} - {\;}\frac{{\Delta V{\;}}}{{\Delta E}}\quad {\text{т }}.\,{\text{д о }}\;{\text{т }}.\,{\text{э к в }}{\kern 1pt} .{\kern 1pt} {\text{,}}$Для каждого комплекса Ca2+ – АК по ее кривой было найдено значение δ и сравнено с другими АК (табл. 6).
Таблица 6.
δ-Характеристики для процессов комплексообразования аминокислот с Са2+
| АК | δ | АК | δ |
|---|---|---|---|
| Lys | 1.1 | Glu | 2.2 |
| Met | 2.1 | Asp | 2.1 |
| Asn | 0.9 | Gly | 6.3 |
| Thr | 0.9 | Ser | 1.9 |
| Pro | 3.1 | Val | 2.8 |
| Ile | 2.2 | His | 1.4 |
| Arg | 5.3 | Ala | 3.1 |
Как видно, по своей лабильности, комплексы Ca2+ с данными АК располагаются в следующий ряд: δ(Ca2+–Asn) = δ(Ca2+–Thr) < δ(Ca2+– Lys) < < δ(Ca2+–His) < δ(Ca2+–Ser) < δ(Ca2+–Asp) = = δ(Ca2+–Met) < δ(Ca2+–Ile) = δ(Ca2+–Glu) < < δ(Ca2+–Val) < δ(Ca2+–Ala) = δ(Ca2+–Pro) < < δ(Ca2+–Arg) < δ(Ca2+Gly), т.е., самые лабильные комплексы у ионов кальция с аспарагином и треонином, а самые стабильные – с глицином.
Полученные экспериментальные и расчетные данные представляют собой научный интерес и могут служить теоретической базой и опорным материалом при диагностике и лечении болезней, связанных с патогенным минералообразованием в организме человека, и служить руководством при создании и/или разработке медицинских препаратов узкого спектра действия.
Таким образом, предложены три группы образующихся комплексов аминокислот с ионами кальция согласно их устойчивости; доказано увеличение констант устойчивости комплексов в ряду исследуемых систем “ионы кальция – аминокислота” в водном растворе при T = 298 K; рассчитаны основные термодинамические потенциалы изучаемых систем в водных растворах при Т = 298 К: энтальпия образования $\Delta H_{{298}}^{^\circ }$, энтальпия образования $\Delta S_{{298}}^{^\circ }$, энергия Гиббса $\Delta G_{{298}}^{^\circ }$; установлено различие в скорости взаимодействия ионов кальция и ряда изучаемых аминокислот в водных растворах при Т = 298 К с помощью ввода полуколичественного критерия δ.
Список литературы
Добрынина Н.А. Бионеорганическая химия. М.: МГУ, 2007. 36 с.
Лунева С.Н. Дис. Биохимические изменения в тканях суставов при дегенеративно-дистрофических заболеваниях и способы биологической коррекции докт. биол. наук. Тюмен. гос. унив. 2003. 297 с.
Golovanova O.A., Gerk S.A., Mylnikova T.S. // Biogenic-Abiogenic Interactions in Natural and Anthropogenic Systems Lecture Notes in Earth System Sciences. 2016. P. 443.
Голованова О.А. // Патогенные минералы в организме человека. Омск. 2007. 395 с.
Голованова О.А., Ачкасова Е.Ю., Пунин Ю.О. и др. // Кристаллография. 2006. Т. 51. № 2. С. 376.
Голованова О.А., Пунин Ю.О., Изатулина А.Р. и др. // Журн. структур. химии. 2014. Т. 55. С. 178.
Голованова О.А., Пятанова П.А., Россеева Е.В. // Докл. Академии наук. 2004. Т. 395. № 5. С. 1.
Голованова О.А., Россеева Е.В., Франк-Каменецкая О.В. // Вестн. СПбГУ. 2006. Сер. 4. № 2. С. 123.
Ламанова Л.М. // Вестник ТГУ. 2010. № 337. С. 194.
Golovanova O.A., Solodyankina A.A. // J. Crystallography Reports. 2017. T. 62. № 2. C. 342.
Golovanova O.A., Korol’kov V.V., Kuimova M.V. // IOP Conf. Series: Materials Science and Engineering 168. 2017. 012066 doi: 10.1088/1757-899X/168/1/ 012066
Golovanova O.A., Gerk S.A., Mylnikova T.S. // Biogenic-Abiogenic Interactions in Natural and Anthropogenic Systems Lecture Notes in Earth System Sciences. 2016. P. 443.
Korolkov V.V., Golovanova O.A., Kuimova M.V. // Ibid. 2016. P. 485.
Chikanova E.S., Golovanova O.A., Kuimova M.V. // Ibid. Switzerland. 2016. P. 501.
Курочкин В.Ю. // Дисс. “Термодинамика процессов комплексообразования ионов кальция с аминокислотами в водном растворе” канд. хим. наук. М.: Иванов. государственный химико-технологический ун-т. 2011. 121 с.
Амерханова Ш.К., Голованова О.А., Шляпов Р.М. // Вестн. Омского ун-та. 2015. № 2. С. 45.
Кожомуратова Ж.С., Миронов Ю.В., Шестопалов М.А. и др. // Координац. химия. 2007. Т. 33. № 1. С. 3.
Гусев А.Н., Шульгин В.Ф., Мешкова С.Б. и др. // Ученые записки Крымского федерального университета имени В.И. Вернадского. Биология. Химия. 2009. Т. 22. № 1. С. 154.
Томашевский И.А., Спиридонова К.С. // Матер. XIX международной экологической студенческой конференции “Экология России и сопредельных территорий”. 2014. С. 242.
Чернова С.П., Трубачева Л.В. // Аналитика и контроль. 2006. С. 336.
Селифонова Е.И., Чернова Р.К. // Изв. Саратов. ун-та. 2012. Т. 12. В. 3. С. 25.
Никольский Б.П., Матерова Е.А. Ионоселективные электроды. Л.: Химия, 1980. С. 240.
Агасян П.К., Николаева Е.Р. Основы электрохимических методов анализа (потенциометрический метод). М.: МГУ, 1986. 198 с.
Посыпайко В.И., Козырева Н.А., Логачева Ю.П. Химические методы анализа. М.: Выс. школа. 1989. С. 42.
Дятлова Н.М., Темкина В.Я., Попов К.И. Комплексоны и комплексонаты металлов. М.: Химия, 1988. С. 48.
Болотин С.Н., Буков Н.Н., Волынкин В.А. и др. Координационная химия природных аминокислот. М.: Издательство ЛКИ, 2008. С. 240.
Bottari E., Festa M. // J. Ann. Chim. 1996. V. 86. P. 133.
De Robertis A., De Stefano C., Gianguzza A. // J. Thermochim. Acta. 1991. V. 177. P. 39.
Daniele P., De Robertis A., De Stefano C. // Dalton Trans. 1985. Is. 11. P. 2353.
Khan M., Satyanarayana S. // J. Indian Chem. 1983. V. 22A. P. 584.
Rey F., Antelo J., Arce F. et al. // J. Polyhedron. 1990. V. 9. Is. 5. P. 665.
Bottari E., Porto R. // J. Ann. Chim. 1985. V. 75. P. 393.
Khalil M., Attia A. // J. Chem. Eng. Data. 2000. V. 45. Is. 45. P. 1108.
Burger K., Sipos P., Veber M. // J. Inorg. Chim. Acta. 1988. V. 152. Is. 4. P. 233.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии