

Журнал физической химии, 2019, T. 93, № 1, стр. 28-34
Деструкция лигнина при озонировании древесины сосныН. А. Мамлеева, Н. А. Бабаева, А. Н. Харланов, В. В. Лунин
Н. А. Мамлеева a, *, Н. А. Бабаева b, А. Н. Харланов a, В. В. Лунин a
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Филиал МГУ имени Ломоносова в г.
Баку, Азербайджан
* E-mail: mamleevana@bk.ru
Поступила в редакцию 21.02.2018
Аннотация
Изучено поглощение озона древесиной сосны с содержанием воды от 7 до 85%. Определено удельное поглощение озона, скорость поглощения и степень превращения озона при озонировании древесины с различным содержанием воды. Проанализированы ИК-спектры поглощения озонированного лигноцеллюлозного материала (ЛЦМ), полученного из древесины. Отмечена деструкция ароматических структур лигнина, образование карбонил- и карбоксил-содержащих соединений. С увеличением удельного поглощения озона содержание целлюлозы в образце озонированного ЛЦМ возрастает. Методом ВЭЖХ установлено, что при озонировании древесины сосны образуются продукты озонолиза лигнина-глиоксалевая, муравьиная и щавелевая кислоты. Показано, что в водной фазе образца протекают реакции окисления кислот озоном. На основании данных по деструкции лигнина и результатов изучения процесса озонирования древесины сделан вывод, что оптимальным для процесса обработки древесины сосны является содержание воды 60–63%. Отмечено, что деструкция лигнина при озонировании древесины осуществляется вследствие реакции озонолиза, а также при участии радикалов, образующихся в реакциях озона с водой.
Древесина является одним из наиболее перспективных материалов биологического происхождения, относящимся к высокопродуктивным возобновляемым ресурсам. С химической точки зрения древесина представляет собой композитный материал, в состав которого входят целлюлоза, гемицеллюлозы и лигнин, а также небольшие количества экстрактивных веществ [1, 2]. Высокое содержание целлюлозы в древесине определило ее широкое использование в качестве сырья для получения целлюлозы с помощью технологий, позволяющих разрушить лигнин и гемицеллюлозы.
Озон служит окислителем ароматических соединений, и в этой связи он используется для деструкции остаточного лигнина в процессах получения бумажной массы. Реакции озона с лигнином (ЛГ) достаточно глубоко изучены [3, 4].
Исследования превращений древесины осины под действием озона показали возможность проведения глубокой делигнификации материала, которая сопровождается деструкцией гемицеллюлоз [5–7].
Древесина хвойных пород отличается от лиственной древесины клеточным строением, структурой лигнина и гемицеллюлоз, а также степенью полимеризации целлюлозы и ее более высоким содержанием. [1, 2, 8, 9]. С практической точки зрения интерес к изучению превращений хвойной древесины связан с огромным количеством производственных отходов, которые могут служить сырьем для дальнейшей химической переработки.
Установлено [5, 6, 10–15], что одним из необходимых условий деструкции ЛГ биомассы под действием озона является присутствие воды в структуре образца, оптимальное количество которой различно для разных типов растительных субстратов.
Данная работа посвящена изучению процесса озонирования и делигнификации древесины сосны с различным содержанием воды. Целью исследования является определение оптимальных условий деструкции ЛГ древесины сосны под действием озона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исследуемого материала использовали опилки древесины сосны (Pinus silvestris) с размером частиц 0.315–0.63 мм и содержанием воды (moisture content – МС) от 7 до 85 ± 1% относительно массы абсолютно-сухой древесины (а.с.д.).
${\text{М С }} = ({{m}_{{{{{\text{Н }}}_{{\text{2}}}}{\text{О }}}}}{\text{/}}{{m}_{{{\text{а }}{\text{.с }}{\text{.д }}}}})$ × 100%. Навески воздушно-сухой древесины, содержащие известное количество воды, инкубировали при 20°C в герметичных сосудах для достижения набухания древесины, согласно [14]. Масса образцов составляла 0.42–0.45 г с объемом 0.75 ± 0.02 мл. Постоянство объема образцов позволяло проводить эксперимент при постоянном времени контакта реагента в реакционной зоне [15].
Озонирование проводили в проточной установке в термостатированном (25°С) реакторе с неподвижным слоем. Озон − кислородную смесь с концентрацией озона 55 ± 5 мг/л пропускали через реактор при объемной скорости потока 1 × × 10–3 л/с [14]. Чтобы оценить потери озона проводили “холостой” опыт. В качестве образца для этого опыта использовали образец озонированной древесины того же объема, что и исследуемый образец. Количество поглощенного озона в момент времени t рассчитывали из кинетических кривых зависимости концентрации озона, согласно уравнению
где U – скорость потока газовой смеси (л/с), Ct и $C_{t}^{{\text{*}}}$ – текущие значения концентрации озона (моль/л) на выходе из реактора с исследуемым образцом, и в холостом опыте, соответственно, mа.с.д – масса а.с.д. образца. Ошибка определения Qr(t) – 10%.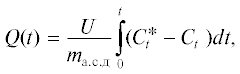
Степень превращения озона рассчитывали, согласно [14], как долю поглощенного озона (в %) относительно количества озона, прошедшего через реактор. Скорость поглощения озона определяли из наклона начального линейного участка кривых поглощения озона [6].
Сразу после обработки озоном образцы промывали водой, контактный раствор анализировали методом ВЭЖХ. Твердый остаток озонированного лигноцеллюлозного материала (ЛЦМ) отфильтровывали и высушивали на воздухе.
ИК-спектральные исследования проводили на приборе Bruker Equinox 55/S в режиме пропускания. Для этого просушенные образцы древесины дополнительно измельчали, смешивали с КВr и прессовали при давлении 4500 кгс/см2 в таблетки массой 300 мг. Содержание образца в КВr составляло 0.125 мас. %. Экспериментальные ИК-спектры корректировались вычитанием базовой линии в программе OPUS 6.0 (Bruker).
Для изучения продуктов, нерастворимых в воде, после проведения 10 опытов по озонированию древесины реактор промывали этанолом, затем раствор этанола наносили на поверхность оптического окошка из КВг и выпаривали. Полученная пленка растворившихся в этаноле продуктов озонирования исследовалась методом ИКС.
Анализ водорастворимых продуктов озонирования выполняли на жидкостном хроматографе Agilent 1100 c УФ-детектором (195 нм) и колонкой Rezex ROA. Подвижная фаза – 0.005 М H2SO4, температура – 65°С; скорость потока – 0.5 мл/мин.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Поглощение озона древесиной
На рис. 1 представлены кинетические кривые зависимости удельного поглощения и степени превращения озона для образцов древесины сосны с различным содержанием воды. Как видно из рис. 1а, с увеличением содержания воды в образце в ходе озонирования достигаются более высокие значения удельного поглощения озона.
Рис. 1.
Кинетические кривые зависимости удельного поглощения (а) и степени превращения озона (б) для образцов древесины сосны с различным содержанием воды; МС, %: 7(1), 25(2), 35 (3), 45 (4), 60 (5), 80 (6).
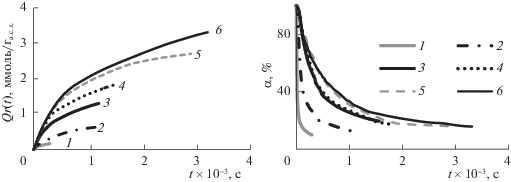
Зависимость удельного поглощения озона, соответствущее завершению озонирования (Qr), от содержания воды носит линейный характер (рис. 2).
Рис. 2.
Зависимости удельного поглощения (1) и скорости поглощения озона (2) от содержания воды в исходном образце древесины сосны; y = 0.0412x − 0.1361.
Степень превращения озона (α) в ходе озонирования падает для всех образцов от 100% в начале обработки до 14–16% при завершении опыта (рис. 1б). Уменьшение значений α объясняется тем, что в ходе обработки озоном в реакцию постепенно вступают менее реакционноспособные и менее доступные функциональные группы образца, и это наблюдается при всех значениях МС. Из рис. 1б видно, что с увеличением МС степень превращения озона снижается медленнее, что свидетельствует о возрастании эффективности поглощения озона. Как видно из рисунка 1б, процесс поглощения озона образцами с содержанием воды 60 и 80% характеризуется наибольшей эффективностью.
На рис. 2 приведены значения скорости поглощения озона для образцов с различным содержанием воды. Видно, что по мере увеличения МС скорость поглощения озона древесиной возрастает, а при МС выше 60–63% наблюдается снижение скорости.
Влияние воды на процесс поглощения озона и деструкцию растительной биомассы озоном обсуждается подробно многими исследователями [5–7, 10, 11, 14, 15]. В данной работе отметим, что влияние содержания воды на процесс поглощения озона, в первую очередь, обусловлено набуханием древесины. В точке набухания волокна (ТНВ) на поверхности древесины формируется слой “связанной воды” [16], многократно возрастает число доступных реагенту функциональных групп. Это объясняет увеличение удельного поглощения, начальной скорости поглощения озона и степени превращения озона при увеличении содержания воды до 28%, что соответствует ТНВ древесины сосны [17].
При содержании воды выше ТНВ, на поверхности растительного субстрата присутствует водная фаза, которую называют “свободной” водой [16]. Предполагается [5, 10, 14, 15], что в этих условиях процесс поглощения озона растительным субстратом состоит из нескольких стадий. Из газовой фазы озон переходит в воду, растворенный озон диффундирует к поверхности субстрата через слой свободной в слой связанной воды, где озон взаимодействует с функциональными группами ЛЦМ. Продукты деструкции ЛЦМ переходят в водную фазу, так как тепловое движение молекул и диффузия обеспечивают обмен между молекулами свободной и связанной воды. В свою очередь, в водной фазе молекулы реагента взаимодействуют с продуктами реакций, растворенными в воде [14]. Оптимум содержания воды, при котором процессы, протекающие в связанной и в свободной воде, составляют баланс, соответствующий наибольшей скорости поглощения озона, для древесины сосны соответствует МС 60–63%.
ИК-спектры поглощения озонированного лигноцеллюлозного материала
На рис. 3 приведены ИК-спектры поглощения образцов ЛЦМ, полученных при озонировании древесины с различным содержанием воды, а на рис. 4 приведены значения оптических плотностей (D) полос поглощения. Данные рисунков позволяют проследить основные тенденции изменения оптической плотности полос поглощения при увеличении удельного поглощения озона.
Рис. 3.
ИК-спектры поглощения озонированной древесины сосны; Qr, ммоль/га.с.д: 0 (1), 0.3 (2), 0.7 (3), 1.9 (4), 2.2 (5), 3.3(6); МС, %: 7 (2), 25 (3), 45 (4), 60 (5), 80 (6).
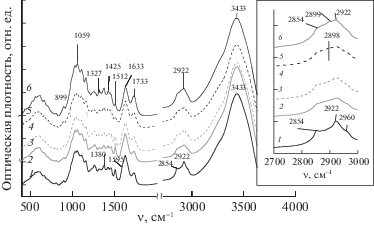
Рис. 4.
Оптическая плотность ИК-полос поглощения древесины сосны при различных значениях удельного поглощения озона: МС, %: 7 (2), 25 (3), 45 (4), 60 (5), 80 (6).
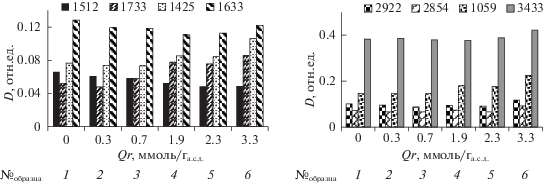
Валентные С–С-колебания в ароматических (гваяцильных) структурах ЛГ древесины сосны наблюдаются при 1595, 1512 и 1425 см–1 [18]. Оптическая плотность полосы 1512 см–1 уменьшается по мере увеличения расхода озона (рис. 4). Это особенно заметно для образцов № 4 (МС = 45%) и № 5 (Qr = 2.3 ммоль/га.с.д, МС = 60%). Увеличение Qr до 3.3 ммоль/га.с.д для образца № 6 (содержание воды 80%) не влияет на значение D1512.
Полоса поглощения 1633 см–1 представляет собой суперпозицию полос валентных С–С-колебаний ароматического кольца (1595 см–1) и деформационных О–Н-колебаний кристаллизационной воды [19]. Симбатный характер изменения D1633 и D1512 в интервале Qr 0.3–2.3 ммоль/га.с.д (образцы № 2 – № 5) объясняется разрушением лигнина. Полоса 1425 см–1 также относится к валентным колебаниям ароматических структур. В этой области спектра наблюдаются и деформационные С–Н-колебания лигнина и полисахаридов [19], поэтому увеличение D1425 может быть связано с их присутствием в составе озонированного ЛЦМ. Изменение D1425 согласуется с изменениями полосы 1733 см–1 С=О-валентных колебаний карбоксильных групп кислот и сложных эфиров [18]. Значение D1733 и D1425 возрастает при увеличении удельного поглощения озона в интервале Qr 1.9–3.3 ммоль/га.с.д (образцы № 4, № 5, № 6).
Контур полосы валентных С–Н-колебаний обусловлен суперпозицией полос поглощения, относящихся к νС–Н колебаниям алифатических групп. В этой области наблюдаются С–Н-колебания в метильных и метиленовых группах ЛГ, а также в метиленовых и метиновых группировках целлюлозы (2899 см–1) и гемицеллюлоз (2835, 2935 см–1 [18]).
С увеличением удельного поглощения озона контур спектра изменяется, возрастает оптическая плотность при 2899 см–1 (вставка на рис. 3, спектры 4, 5, 6), что свидетельствует о возрастающем содержании целлюлозы в образцах озонированных ЛЦМ. Это также подтверждается возрастанием оптической плотности полосы 1059 см–1 валентных С–О-колебаний целлюлозы. При Qr = = 3.3 ммоль/га.с.д значение D1059 почти в 1.5 раза выше, чем в спектре исходной древесины (рис. 4). Валентные С–Н-колебания в спектре исходной древесины характеризуются максимумами при 2922 и 2854 см–1. Для образцов, обработанных озоном, значения D2922 и D2854 повторяют изменения полосы 1059 см–1 по мере увеличения Qr (рис. 4).
Полоса 3433 см–1 относится к валентным колебаниям О–Н-групп, связанных водородной связью. Положение максимума этой полосы и ее интенсивность обусловлены взаимодействиями гидроксилов с ОН-, С=О- и СООН-группами образца. Небольшое увеличение оптической плотности этой полосы, а также полосы деформационных О–Н-колебаний при 1633 см–1 для образца № 6, по-видимому, объясняется отмеченным увеличением содержания полярных групп в составе образца.
Результаты изучения образцов озонированных ЛЦМ методом ИКС показывают, что обработка озоном приводит к разрушению ароматических структур древесины сосны, причем для глубокой деструкции ЛГ достаточно озона в количестве 2.3 ± 0.2 ммоль/га.с.д, при озонировании субстрата с содержанием воды 60%.
Продукты озонирования
Методом ВЭЖХ в контактных растворах озонированной древесины сосны обнаружены глиоксалевая, муравьиная и щавелевая кислоты, известными как продукты озонолиза ЛГ [3]. На рис. 5 данные по составу кислот при различных значениях удельного поглощения озона приведены в процентах на 1 г а.с.д. древесины.
Рис. 5.
Массовый, молярный состав продуктов и рН контактных растворов в зависимости от удельного поглощения озона; МС, %: 7 (1), 25 (2), 35 (3), 45 (4), 60 (5), 80 (6); I – щавелевая кислота, II – глиоксалевая кислота, III – муравьиная кислота.
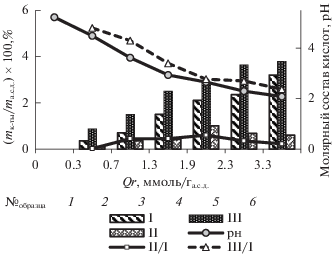
Видно, что по мере увеличения удельного поглощения озона количество кислот возрастает, а рН контактного раствора снижается от 5.7 для исходного образца до 2.2 при Qr = 3.3 ммоль/га.с.д. Данные по содержанию кислот в контактном растворе также представлены как зависимость молярного содержания муравьиной и глиоксалевой кислот относительно молярного содержания щавелевой кислоты от удельного поглощения озона. Изменение молярного состава кислот объясняется сочетанием двух процессов − образованием кислот при окислении ЛГ озоном и их последующим окислением озоном. Глиоксалевая кислота окисляется в щавелевую кислоту, а муравьиная и щавелевая кислоты окисляются до СО2 и Н2О [20].
Наиболее устойчивой к действию озона является щавелевая кислота (константа скорости реакции с озоном k < 0.01 л/(моль с) [4]), поэтому значение молярного содержания двух других кислот относительно щавелевой с ростом удельного поглощения озона уменьшается. Видно, что снижение относительного содержания муравьиной кислоты (III/I) наблюдается при более низких значениях Qr по сравнению со значением II/I для глиоксалевой кислоты. Для муравьиной кислоты константа скорости реакции с озоном k = = 30 л/моль с [4], а для глиоксалевой k = = 0.2 л/моль с [20], так что наблюдаемая закономерность, в принципе, согласуется со значениями констант скорости реакций этих соединений с озоном.
Данные по составу кислот в контактном растворе позволяют оценить их концентрацию в слое воды на поверхности ЛЦМ при озонировании. Так, например, для образца № 6 (МС = 80%, Qr = = 3.3 ммоль/га.с.д) концентрация щавелевой кислоты составляет ~0.4 моль/л, а муравьиной кислоты ~1.0 моль/л. Полученный результат важен для понимания процесса озонирования биомассы. При озонировании древесины “целевым” процессом является окислительная деструкция ЛГ. Реакции озона с ароматическими структурами лигнина характеризуются высокими значениями констант скорости (для гваякола k > 5 × 105 л/моль × с [21]). Из-за высокой концентрации кислот в водной фазе образца часть озона расходуется на реакции с продуктами окисления, реакции которых с озоном характеризуются значительно более низкими значениями констант скорости. Этот процесс становится лимитирующим, поэтому в области МС ~ 60% процесс поглощения озона замедляется, а при МС 65–80% скорость поглощения озона уменьшается (рис. 2). Участие озона в процессах окисления продуктов окисления ЛГ приводит также к увеличению степени превращения озона (рис. 1б).
Помимо водорастворимых соединений образуются бесцветные воскообразные продукты, растворимые в этаноле. Метод ИК-спектроскопии позволяет качественно охарактеризовать состав растворимых в этаноле продуктов озонирования растительного субстрата (рис. 6).
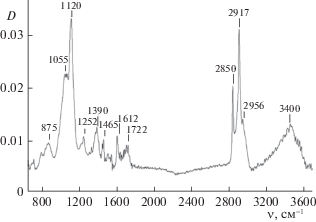
Интенсивные полосы при 2850 и 2917 см–1 относятся к симметричным и асимметричным С–Н-валентным колебаниям в СН2-группах. Валентные С–Н-колебания в СН3-группах характеризуются низкой интенсивностью и проявляются в спектре в виде плеча при 2956 см–1 [19]. Полосы поглощения 1120 и 1055 см–1 отвечают валентным С–О-колебаниям в алифатических простых эфирах и первичных спиртах соответственно. Полосы при 1465 и 1390 см–1 относятся к симметричным и асимметричным деформационным колебаниям С–Н-связей. В небольших количествах присутствуют альдегиды, сложные эфиры (возможно, формиаты) и карбоновые кислоты, определяемые по присутствию в спектре полос поглощения С=О и С–О валентных колебаний при 1722 и 1252 см–1 [19]). Широкая полоса поглощения при 3400 см–1 относится к валентным колебаниям ОН групп, связанных водородной связью. Приведенные данные показывают, что продукты озонирования, нерастворимые в воде, представляют собой смесь полимерных соединений, имеющих, в основном, структуру алифатических простых и сложных эфиров.
Реакции озона с лигнином в древесине
Результаты работы показывают, что после обработки озоном содержание ЛГ в ЛЦМ снижается, причем эти превращения обусловлены реакциями с озоном, растворенным в воде. Среди продуктов озонирования присутствуют карбоновые кислоты, которые, согласно современным представлениям, образуются из лигнина в результате озонолиза – реакции электрофильного цикло-присоединения озона к ароматическому кольцу с последующим раскрытием кольца по механизму Криге [3, 22]. Предполагается [3, 21, 22], что этот процесс осуществляется при участии молекулярной формы озона. Как отмечено в [23], β–O–4-связи ЛГ также окисляются молекулярным озоном по механизму.
Известно [24], что в водном растворе озон разлагается с образованием реакционноспособных ${\text{O}}{{{\text{H}}}^{ \bullet }}$-радикалов. Авторами работы [25] рассмотрены реакции ${\text{O}}{{{\text{H}}}^{ \bullet }}$-радикалов с ЛГ и его модельными соединениями. Отмечено, что радикал ${\text{O}}{{{\text{H}}}^{ \bullet }}$ обуславливает разрыв связей С–С боковой цепи ЛГ, расщепление связей алкил–арил и деметоксилирование ЛГ. Таким образом, помимо озонолиза, при обработке древесины озоном деструкция ЛГ может протекать и по радикальному механизму. Это соображение подкрепляется и отмеченным фактом присутствия нерастворимых в воде соединений, по-видимому, образовавшихся вследствие конденсации фрагментов деструкции ЛГ и других компонентов древесины.
Соотношение между реакциями молекулярного озона и радикальными процессами регулируется величиной рН среды [24, 26]. Согласно [26], генерация ${\text{O}}{{{\text{H}}}^{ \bullet }}$-радикалов в воде усиливается при рН > 3. На основании полученных данных по изменению рН среды можно предположить, что ${\text{O}}{{{\text{H}}}^{ \bullet }}$-радикалы генерируются на начальных этапах озонирования, и их роль особенно существенна при обработке озоном образцов с содержанием воды ниже 30%. В ходе озонирования при низких значениях рН возрастает роль процессов с участием молекулярного озона, что способствует образованию продуктов озонолиза. Таким образом, варьирование содержания воды позволяет проводить делигнификацию древесины по различным направлениям. Содержание воды 60–63% соответствует наиболее глубокой деструкции ЛГ древесины сосны в условиях эффективного, с точки зрения степени превращения озона, процесса озонирования.
Авторы выражают благодарность профессору Алтайского государственного университета Н.Г. Базарновой за предоставленный образец древесины сосны.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта № 16-08-00876).
Список литературы
Fengel D., Wegener G. Wood: chemistry, ultrastructure, reactions. Ed.W.de Gruyter. 1984. 613 p.
Азаров В.И. Химия древесины и синтетических полимеров. 1999. СПб., 629 с.
Matsumoto J. et al. // Int. Wood and Pulping Chem. 1983. V. 4. P. 68.
Разумовский С.Д., Заиков Г.Е. Озон и его реакции с органическими соединениями (кинетика и механизм). М.: Наука, 1974. 322 с.
Mamleeva N.A., Autlov S.A., Bazarnova N.G., Lunin V.V. // Int. J. Current Research. 2016. V. 8. № 11. P. 41714.
Мамлеева Н.А., Аутлов С.А., Базарнова Н.Г., Лунин В.В. // Химия растительного сырья. 2015. № 4. С. 5.
Mamleeva N.A., Autlov S.A., Bazarnova N.G., Lunin V.V. // Pure Appl. Chem. 2009. V. 81. № 11. P. 2081.
Papadopoulos A.N., Hill C.A.S., Gkaraveli A. // Holz als Roh- und Werlag 2003. V. 61. P. 453.
http://lesoved.pro/stanki/kletka_drevesiny_osobennosti_ obrazovaniya_i_rosta_kletok_drevesiny.php
Travaini R., Martín-Juárez J., Lorenzo-Hernando A., Bolado-Rodriges S. // Biores. Technol. 2016. V. 199. № 1. P. 2.
Li C., Wang L., Chen Z., Li Y. et al. // Biores. Technol. 2015. V. 183. P. 240.
Souza-Corrêa J.A., Ridenti M.A. et al. // J. Phys. Chem. B. 2013. V. 117. № 11. P. 3110.
Бенько Е.М., Манисова О.Р., Лунин В.В. // Журн. физ. химии. 2017. Т. 91. № 7. С. 1117.
Мамлеева Н.А., Кустов А.Л., Лунин В.В. // Там же. 2018. Т. 92. № 9. С. 1402.
Мамлеева Н.А., Лунин В.В. // Там же. 2016. Т. 90. № 11. С. 1626.
Walker J.C.F. Water in wood / Primary Wood Processing. 2010. 606. The Netherlands.
Мамлеева Н.А., Лунин В.В. // Журн. физ. химии. 2016. Т. 90. № 3. С. 456.
Базарнова Н.Г., Карпова Е.В., Катраков И.В. и др. / Методы исследования древесины и ее производных. ред. Н.Г. Базарновой. Изд-во Алт. гос. ун-та, Барнаул. 160 с. 2002.
Смит А. Прикладная ИК-спектроскопия. 1982. М.: Мир, 327 с.
Schöne L., Herrmann H. // Atmos. Chem. Phys. 2014. V. 14. P. 4503.
Худошин А.Г., Митрофанова А.Н., Лунин В.В. // Изв. РАН. Сер. химия. 2008. № 2. С. 276.
Bailey P.S. Ozonation in Organic Chemistry. V. 2. Acad. Press. New York. 1982. P. 31.
Bertaud F., Croué J.P., Legube B. // Ozone: Science & Engineering. 2001. V. 23. № 1. P. 139.
Staehelin J., Hoigné // Environ. Science Technol. 1982. V. 16. № 12. P. 666.
Демин В.А., Шерешовец В.В., Монаков Ю.Б. // Успехи химии. 1999. Т. 68. № 11. С. 1029.
Ragnar M., Eriksson T., Reitberger T. // Holzforschung. 1999. V. 53. P. 292.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии