

Журнал физической химии, 2019, T. 93, № 1, стр. 40-45
Окислительное дегидрирование этилбензола в стирол на смешанном оксидном катализаторе Mo–V–Te–NbОх в условиях термического и СВЧ-нагреваА. Л. Тарасов, Е. Д. Финашина
А. Л. Тарасов a, *, Е. Д. Финашина a
a Российская академия наук, Институт органической химии им. Н.Д. Зелинского
Москва, Россия
* E-mail:
Поступила в редакцию 13.03.2018
Аннотация
Проведено сравнительное исследование активности смешанного оксидного катализатора в реакции дегидрирования этилбензола (ЭБ) в стирол при атмосферном давлении с использованием в качестве окислителей О2 и СО2 в условиях традиционного термического и СВЧ-нагрева. Установлены зависимости конверсии и селективности от состава реакционных смесей. Показано, что замена кислорода на СО2 приводит к существенному уменьшению конверсии ЭБ при одновременном увеличении селективности процесса. Установлено, что СВЧ-нагрев при использовании О2 в качестве окислителя способствует увеличению эффективности процесса в диапазоне умеренных (до 420°С) температур. Показано, что при более высоких температурах происходит изменение фазового состояния смешанного оксидного катализатора. Сделано предположение, что это явление обусловлено появлением “горячих” точек в местах локализации активных центров реакции.
Основным промышленным способом производства стирола является в настоящее время дегидрирование этилбензола. Данная реакция является эндотермической и протекает с увеличением объема. Соответственно с повышением температуры и снижением парциального давления углеводорода увеличивается степень превращения этилбензола в стирол [1]. При давлении 0.1 МПа эта зависимость выглядит так:
В промышленности реакцию проводят при разбавлении водяным паром (соотношение пар : газ = (15–20) : 1) и при температуре 830–900 K. Используются катализаторы на основе оксида железа с добавками K и Cr. Селективность по стиролу составляет около 98%. Водяной пар, подаваемый на разбавление, не только сдвигает равновесие, но и газифицирует углистые отложения, образующиеся на поверхности катализатора.
Другим способом получения стирола является окислительное дегидрирование этилбензола, в котором при более низких температурах возможно получить более высокие выходы целевых продуктов с селективностью до 80–90% [2, 3]. В этой реакции широко исследуются различные оксидные системы, в том числе оксид алюминия и модифицированный фосфором оксид кремния [4]. Однако эффективность таких простых катализаторов невысока. Так при температуре 450°С выход стирола не превышает 29% при селективности около 80%. Кроме того, для Al2O3 отмечается быстрая дезактивация катализаторов вследствие образования коксовых отложений, количество которых может достигать 40% к массе катализатора [5].
В работе [6] при окислительном дегидрировании (ОД) этилбензола (ЭБ) на катализаторе Mg3(VO4)2–MgO селективность по стиролу достигает 96%, при этом носитель MgO не полностью и неравномерно покрыт активной фазой ортованадата Mg, а максимальная селективность достигается при концентрации ванадия выше 10 мас. %.
В последнее время возрос интерес к проведению дегидрирования алкилбензолов в присутствии более мягких, чем кислород, акцепторов водорода (CO2, NOx и др.), что позволяет заметно повысить селективность процесса за счет подавления реакций глубокого окисления углеводородов. В работе [7] в качестве катализатора неокислительного дегидрирования этилбензола при атмосферном давлении и температуре 560°С авторы использовали CeO2. Было установлено, что катализатор подвергается быстрой дезактивации в течение 0.5 ч, при этом в присутствии СО2 скорость дезактивации существенно уменьшается, однако конверсия ЭБ через несколько часов также не превышает 10%.
Известно [8], что оксиднованадиевые системы, в том числе и нанесенные [9], могут рассматриваться как весьма эффективные катализаторы для реакций окислительного дегидрирования C4-углеводородов и этилбензола в стирол. В работе [10] исследовано дегидрирование этилбензола в стирол в присутствии диоксида углерода на серии нанесенных алюмованадиевых катализаторов (VOx/Al2O3).
По данным [8] в ходе окислительного дегидрирования этилбензола в присутствии CO2 на катализаторе V/TiO2 при T = 550°С, P = 0.1 МПа и мольном отношении CO2/этилбензол = 11 : 1 конверсия этилбензола составляет 11.6% при 100%-й селективности по стиролу. Известны примеры успешного применения в окислительном дегидрировании этилбензола оксида ванадия, нанесенного на более сложные по составу носители, такие как CexZr1 – xO2/SiO2 [11]. Данный катализатор способен эффективно работать при атмосферном давлении, причем CexZr1 – xO2 заметно подавляет дезактивацию нанесенного V2O5, что обеспечивает стабильную работу катализатора и его высокую селективность по стиролу при проведении реакции в проточных условиях.
Ранее в наших работах [12] было показано, что сложный по составу ванадий содержащий смешанный оксидный катализатор Mo1.0V0.37Te0.2Nb0.12Ох, полученный в гидротермальных условиях из смеси водных растворов прекурсоров – солей металлов, достаточно эффективно работает в реакции окислительного дегидрирования этана в этилен при пониженных 380–420°С температурах. В работе [13] предложен способ данного катализатора в условиях СВЧ-активации реакционной массы, позволяющий получать более мелкие (300 нм) и однородные по размеру кристаллиты. Ранее было показано, что такие смешанные оксидные катализаторы могут быть нагреты с помощью микроволновой энергии, что позволяет изучать различные реакции, протекающие с их участием, в условиях микроволнового нагрева [14]. СВЧ-облучение поглощающих материалов (катализаторов, носителей и реакционной среды) способен вызывать быстрый локальный нагрев, модифицировать свойства поверхности и может быть использовано для снижения температуры реакции, повышения активности катализатора путем перевода процесса в мягкий режим.
Целью настоящей работы являлось сравнительное исследование каталитических свойств смешанного оксидного катализатора Mo1.0V0.37Te0.2Nb0.12Ох в реакции окислительного дегидрирования этилбензола в условиях традиционного термического и микроволнового нагрева.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализатора
Катализатор состава Mo1.0V0.37Te0.17Nb0.15Ох был получен методом гидротермального синтеза по описанной в литературе методике [15–17]. Гептамолибдат аммония тетрагидрат (26.5 г) и 5.75 г теллуровой кислоты растворяют в 195 мл дистиллированной воды при 80°C. К полученному раствору по каплям добавляют водный раствор гидроксида аммония (25%) до тех пор, пока величина рН не достигнет значения 7.5. Воду из раствора удаляют при постоянном перемешивании и температуре 80°C; твердый остаток высушивают при 90°C; 6.4 г полученного в результате белого порошка молибдотеллурата аммония растворяют в 20 мл воды при 80°C. К полученному раствору добавляют раствор 2.4 г сульфата ванадила в 10 мл воды. Смесь перемешивают в течение 5 минут затем к полученной смеси прибавляют суспензию 2.3 г оксалата ниобия в 10 мл воды. Смесь перемешивают в течение 10 мин и переносят в автоклав из нержавеющей стали с внутренним вкладышем из Teflon® (тетрафторэтилен). Воздух в автоклаве замещают инертным газом, автоклав герметизируют и нагревают до 175°С. Систему выдерживают при заданной температуре в течение 48 ч. После этого автоклав охлаждают до комнатной температуры, образовавшийся в результате гидротермального синтеза твердый осадок отфильтровывают, промывают дистиллированной водой до обесцвечивания промывной воды и сушат при 80°C. Полученный прекурсор прокаливают 2 ч в токе инертного газа при 600°C. Скорость нагрева составляла 1.7 K/мин. Полученный порошок прессуют, полученные таблетки дробят и просеивают через сито, отбирая фракцию с нужным размером частиц.
Методика каталитического эксперимента
Катализатор в виде фракции 0.8–1.0 мм в количестве 1 мл (0.75 г) загружали в проточный реактор, представляющий собой кварцевую трубку с внутренним диаметром 7 мм. Реакцию проводили в двух режимах: в условиях традиционного термического и в условиях микроволнового нагрева катализатора. Для этого реактор помещали в печь с электрообогревом или в камеру бытовой СВЧ-печи “Vigor” с переменной мощностью 100–1000 Ватт, работающей на частоте 2.45 ГГц, и при атмосферном давлении нагревали катализатор до минимальной реакционной температуры (380°С) в токе окислителя – кислорода или СО2. Далее включали шприцевой насос-дозатор для подачи этилбензола. Подача ЭБ составляла 1.5 и 3 ч–1. Реакцию проводили при мольном соотношении окислитель/ЕБ = 0.5 и 2. При объемной скорости подачи ЭБ = 1.5 ч–1 подача окислителя соответствовала 150 и 600 мл/ч.
Температуру катализатора в реакторе измеряли с помощью термопары, помещенной непосредственно в середину слоя катализатора и задавали при работе в режиме термического нагрева терморегулятором “Термодат-17”. При работе в режиме СВЧ-нагрева температуру регулировали изменением мощности СВЧ-излучения, а также вертикальным перемещением реактора с загруженным катализатором в камере СВЧ-печи, имеющей по высоте градиент электромагнитного поля.
Реакцию исследовали в диапазоне температур 380–500°С. Жидкие продукты накапливали в охлаждаемой до 0°С ловушке-приемнике в течение 1 ч реакции, а далее следовало повышение температуры на 40°С в течение 15 мин.
Анализ жидких продуктов осуществляли хроматографическим методом на хроматографе марки “3700”, производства НПО “Гранат”, с использованием капиллярной колонки SE-54 (30 м) и пламенно-ионизационного (ПИД) детектора в программируемом режиме 60°С (4 мин) далее 6 К/мин до 180°С. Газ носитель – гелий.
Конверсию этилбензола рассчитывали, как разницу между количеством подаваемого ЭБ и его остатком, идентифицированном в жидком катализате (г). Селективность по стиролу рассчитывали, как отношение его количества в жидком катализате к количеству превращенного ЭБ (ммоль).
Кристалличность и фазовый состав исходных и отработанных катализаторов определяли с помощью рентгенофазового анализа на установке ДРОН-2 (навеска образца 80 мг; 2θ от 5 до 40°).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Анализ жидких продуктов реакции показал, что независимо от условий ее проведения (температура, состав сырьевой смеси, термический или СВЧ-нагрев) помимо стирола образуются также бензол, толуол, кумол и в следовых количествах более тяжелые ароматические углеводороды. Таким образом, помимо целевой реакции дегидрирования в указанных условиях протекают также реакции крекинга, деалкилирования и диспропорционирования ЭБ. Однако, суммарная селективность образования этих продуктов не превышает 5% даже при максимальной температуре (500°С). Согласно проведенному нами термодинамическому расчету с использованием компьютерной программы HSC-4 (Haldor Topsøe A/S), вероятность протекания этих реакций в исследованном нами температурном диапазоне достаточно высока. Как пример, на рис. 1 представлен термодинамический расчет равновесных концентраций реагентов и продуктов в реакции деалкилирования ЭБ с образованием бензола и этилена.
Рис. 1.
Результаты термодинамического расчета равновесных концентраций продуктов и реагента в реакции деалкилирования ЭБ при давлении 1 атм.
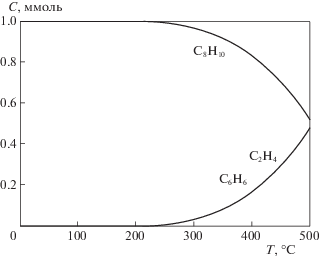
Из рис. 1 видно, что при 500°С конверсия ЭБ может достигать 50%, однако, согласно [18, 19] эту и другие вышеупомянутые реакции катализируют кислотные катализаторы. Таким образом, обнаруженная нами низкая селективность по ароматическим у/в (углеводородам) свидетельствует о том, что исследуемый нами Mo1.0V0.37Te0.17Nb0.12Ох катализатор не имеет сильных кислотных каталитических центров, а с учетом его высокой эффективности в реакции ОД этилена [12] позволяет предположить, что он может быть эффективен также и в реакции ОД этилбензола.
Результаты испытаний катализатора представлены на рис. 1–3.
Рис. 2.
Зависимости конверсии ЭБ (1, 2) и селективности (3, 4) образования стирола от температуры реакции при атмосферном давлении в смесях с различным соотношением О2/ЭБ: 0.5 (1, 3) и 2 (2, 4).
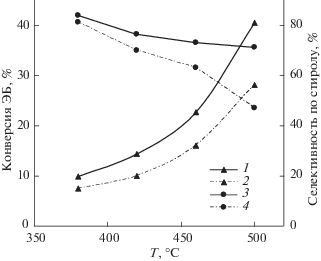
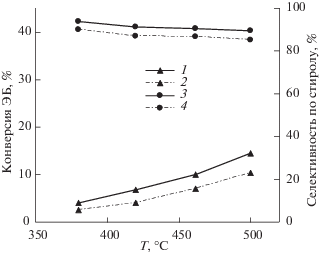
Рис. 3.
Зависимости конверсии ЭБ (1, 2) и селективности (2, 3) образования стирола от температуры реакции при атмосферном давлении в смесях с различным соотношением СО2/ЭБ: 0.5 (1, 3) и 2 (2, 4).
Из рис. 2 видно, что при проведении реакции в газовой смеси со стехиометрическим мольным соотношением реагентов (О2/ЭБ = 0.5) конверсия ЭБ во всем исследованном температурном диапазоне несколько превышает значения, полученные в смеси с избытком кислорода (О2/ЭБ = 2), что, по видимому, связано с уменьшением парциального давления ЭБ в разбавленной кислородом сырьевой смеси и, соответственно, большей линейной скоростью подачи реагентов. Конверсия ЭБ в обеих газовых сырьевых смесях экспоненциально растет с ростом температуры от 380 до 500°С, при этом селективность по стиролу уменьшается. Следует отметить, что в смеси с избытком О2 наблюдается резкое падение селективности при температуре выше 460°С. При 500°С она уже не превышает 50%, при этом в газообразных продуктах реакции наблюдается существенное увеличение концентрации СО2, что свидетельствует об увеличении вклада реакции полного окисления. В смеси же со стехиометрическим соотношением О2/ЭБ = 0.5 этого не происходит, и даже при повышенной температуре (500°С) селективность по стиролу остается выше 70%.
Замена кислорода на СО2 приводит к существенному уменьшению конверсии ЭБ. Максимально зафиксированное ее значение при 500°С не превышает 15% (рис. 3), при этом, независимо от состава сырьевой смеси, селективность образования стирола (85–90%) превышает значения, полученные при использовании кислорода в качестве окислителя. Таким образом, с учетом возможности рециркуляции непрореагировавшего ЭБ, а также актуальностью проблемы утилизации СО2, такой способ получения стирола также может рассматриваться как достаточно перспективный для дальнейшего исследования.
Поскольку, наилучшие показатели (конверсия ЭБ выше 40% и селективность по стиролу выше 40 и 70%, соответственно) в исследованном температурном диапазоне были получены нами для стехиометрической смеси реагентов, сравнительное исследование активности катализатора в условиях термического и микроволнового нагрева было проведено в этой сырьевой смеси состава О2/ЭБ = 0.5. Полученные результаты представлены на рис. 4.
Рис. 4.
Сравнительные показатели реакции окислительного дегидрирования ЭБ при атмосферном давлении в смеси с соотношением О2/ЭБ = 0.5 в зависимости от температуры катализатора в условиях его термического (1, 3) и СВЧ-нагрева (2, 4).
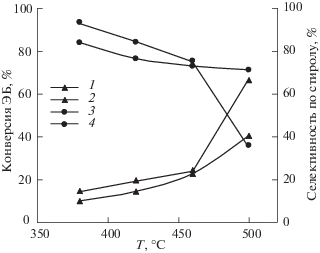
Из рис. 4 видно, что преимущество СВЧ-нагрева проявляется только в области умеренных температур (до 420°С), где наблюдается небольшое (около 10%) превышение конверсии ЭБ, достигнутой в условиях СВЧ-нагрева над конверсией, полученной при термическом нагреве. Это согласуется с данными работы [20], в которой авторы сравнивали каталитические показатели этой реакции в условиях термического и СВЧ-нагрева с использованием в качестве катализаторов оксида железа, нанесенного на углеродный носитель. По нашему мнению, объяснением этому эффекту может служить появление “горячих” точек в местах дислокации активных центров реакции, при этом термопара фиксирует среднюю температуру в слое катализатора [21]. Однако, с этой точки зрения трудно объяснить даже небольшое увеличение селективности процесса наблюдаемое (около 7–10%) в условиях СВЧ-нагрева (рис. 4).
Следует отметить, что с ростом температуры от 460 до 500°С в условиях СВЧ-нагрева наблюдается резкое увеличение конверсии ЭБ, сопровождающееся таким же резким падением селективности по стиролу (до 35%, рис. 4), при этом в газообразных продуктах реакции наблюдается существенный рост концентрации СО2. Нами сделано предположение, что это может быть связано с изменением фазового состояния оксидов в смешанном оксидном катализаторе.
На рис. 5 представлены рентгенограммы свежеприготовленного смешанного оксидного катализатора и катализатора после реакции окислительного дегидрирования ЭБ в условиях СВЧ-нагрева до 500°С. Рентгеновский дифракционный анализ полученного катализатора указывает на наличие хорошо кристаллизованной фазы M1, соответствующей литературным данным [12, 15]. На рентгенограмме образца 1 присутствует более 10 интенсивных рефлексов, наиболее интенсивные линии характеризуются следующими величинами дифракционных углов: 2Θ 7.7° ± 0.4°, 8.9° ± 0.4°, 22.1° + 0.4°, 26.6° ± 0.4°, 26.9° ± 0.4°, 27.1° ± 0.4°, 28.1° ± 0.4°, 31.2° ± 0.4°, 35.0° ± 0.4° и 45.06° ± 0.
Рис. 5.
Рентгенограммы смешанного оксидного катализатора: 1 – свежеприготовленный катализатор, прокаленный при 600°С в атмосфере N2; 2 – катализатор, после реакции в условиях СВЧ-нагрева до 500°С.
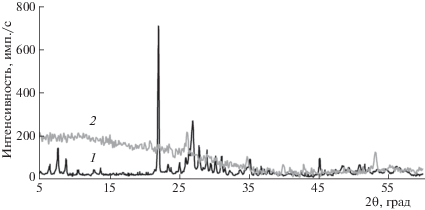
Рентгенограмма катализатора, полученная после проведения реакции ОД ЭБ в условиях СВЧ-нагрева до 500°С (образец 2), практически не содержит интенсивных рефлексов, что указывает на практически полное разрушение активной фазы катализатора, что является возможной причиной возникновения локальных перегревов, так называемых “горячих” точек, возникающих под действием СВЧ-излучения.
Таким образом, данные РФА подтверждают наше предположение об изменении фазового состояния катализатора, приводящего к резкому ухудшению каталитических свойств в реакции получения стирола.
Разработка нанесенных или гибридных наноразмерных катализаторов [22–28], возможно, позволит решить эту проблему благодаря снижению концентрации активной фазы в катализаторе и пространственному рассредоточению горячих точек в массе катализатора.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 14-50-00126).
Список литературы
http://newchemistry.ru/letter.php?n_id=6801&cat_id =5&page_id=1
Алхазов Т.Г., Лисовский А.Е. // Окислительное дегидрирование углеводородов. М.: Химия, 1980. 240 с.
Исагулянц Г.В., Беломестных И.П., Форбек Г., Перрегаард Й. // Рос. хим. журн. 2000. № 3. С. 69.
Nederlof C. et al. // Appl. Catal. A: General. 2014. V. 476. P. 204.
Zarubina V. et al. // J. Molec. Catal. A: Chemical. 2014. V. 381. P. 179.
Oganowski W., Hanuza J., Kepiński L. // Appl. Catal. A: General. 1998. V. 171. № 1. P. 145.
Kovacevic M. et al. // Appl. Catal. A: General. 2015. V. 505. P. 354.
Ren J., Li W.Y., Xie K.C. // Catal. Lett. 2004. V. 93. № 1–2. P. 31.
Hongxia F., Jie F., Xiaohong L. et al. // Chem. Eng. Sci. 2015. V. 135. P. 403.
Chen S., Qin Z., Xu X., Wang J. // Appl. Catal. A: General. 2006. V. 302. № 2. P. 185.
Reddy B.M., Lakshmanan P., Loridant S. et al. // J. Phys. Chem. B. 2006. V. 110. № 18. P. 9140.
Finashina E.D., Kucherov A.V., Kustov L.M. et al. // J. Adv. Oxid. Techn. 2016. V. 20. Is. 2. P. 165.
Кустов Л.М., Финашина Е.Д., Тарасов А.Л. // Заявка на патент РФ № 2017141368 от 28.11.2017.
Тарасов А.Л., Финашина Е.Д., Кустов Л.М. // Патент РФ № 2523801. опубл. 29.05.2014.
Lopez Nieto J.M., Botella P., Vazquez M., Garsia A. // US Patent № 7319179. Опyбл. 20.03.2008.
Кустов Л.М., Кучеров А.В., Кучерова Т.Н. и др. // Патент РФ № 2358958. Опубл. 20.06.2009.
Финашина Е.Д., Кучеров А.В., Кустов Л.М. // Журн. физ. химии. 2013. Т. 87. № 12. С. 2016.
Kustov L.M., Sachtler W.M.H. // J. Molec. Catal. 1992. V. 71. P. 233.
Khodakov A.Yu., Williams C., Kustov L.M., Kazansky V.B. // J. Chem. Soc., Faraday Trans. 1993. V. 89. P. 1393.
Nigrovski B., Zavyalova U., Scholz P. et al. // Carbon. 2008. V. 46. P. 1678.
Zhang X., Hayward D.O., Lee C., Mingos D.M.P. // Appl. Catal. 2001. V. 33. № 2. P. 137.
Vorob’eva M.P., Greish A.A., Ivanov A.V., Kustov L.M. // Appl. Catal.: General. 2000. V. 199. P. 257.
Ananikov V.P., Gordeev E.G., Egorov M.P. et al. // Mendeleev Commun, 2016. V. 26. P. 365.
Redina E.A., Greish A.A., Mishin I.V. et al. // Catal. Today. 2015. V. 241. P. 246.
Kustov L.M., Al-Abed S.R., Virkutyte J. et al. // Pure and Appl. Chem. 2014. V. 86. № 7. P. 1141.
Kirichenko O.A., Davshan N.A., Redina E.A. et al. // Chem. Eng. J. 2016. V. 292. P. 62.
Isaeva V.I., Belyaeva E.V., Fitch A.N. et al. // Crystal Growth and Design. 2013. V. 13. № 12. P. 5305.
Isaeva V.I., Barkova M.I., Kustov L.M. et al. // J. Mater. Chem. A. 2015. V. 3. P. 7469.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии