

Журнал физической химии, 2019, T. 93, № 1, стр. 74-81
Термодинамика кислотно-основных равновесий глицил-глицил-глицина и его комплексообразования с ионом меди(II) в водно-органических растворителяхТ. Р. Усачева, Фам Тхи Лан, В. А. Шарнин
Т. Р. Усачева a, *, Фам Тхи Лан b, **, В. А. Шарнин a, ***
a Ивановский государственный химико-технологический университет
Иваново, Россия
b Вьетнамская академия наук и технологий, Институт тропической технологии
Ханой, Социалистическая Республика Вьетнам
* E-mail:
** E-mail:
*** E-mail:
Поступила в редакцию 07.03.2018
Аннотация
Проведено обобщение собственных данных по термодинамике комплексообразования иона меди (II) с глицил-глицил-глицином, кислотно-основных равновесий глицил-глицил-глицина в водно-органических растворителях, а также литературных источников, отражающих влияние концентрации органического сорастворителя на устойчивость координационных соединений аминокислот и пептидов с ионами d-металлов и энтальпию реакций их образования. Анализ сольватационных характеристик реагентов показал, что при переходе от воды к водно-этанольным и водно-диметилсульфоксидным растворителям упрочнение комплексов меди(II) с цвиттерионом глицил-глицил-глицина и глицинат-ионом происходит, в основном, за счет пересольватации лигандов, что характерно для реакций образования ионных комплексов d-металлов в водно-органических растворителях.
Изучение равновесий в растворах, содержащих аминокислоты и низкомолекулярные пептиды, представляет научный и практический интерес, так как эти молекулы принимают активное участие во многих процессах жизнедеятельности и могут служить в качестве моделей более сложных биосистем. В частности, их комплексообразование с ионами металлов может рассматриваться как модель активных центров ферментов [1].
К настоящему времени накоплен большой экспериментальный материал по термодинамике комплексообразования аминокислот и пептидов с ионами биометаллов, в том числе, с ионом меди(II) в водном растворе [2–13]. Комплексообразование ионов Ni(II), Ag(I) и Cu(II) с глицином и диглицином и кислотно-основные равновесия лигандов были изучены в водных растворах этанола, диметилсульфоксида и ацетона [14–25]. На примере этих взаимодействий было показано, что при переходе от воды к водно-органическим растворителям, изменяя сольватное состояние реагентов, можно целенаправленно изменять устойчивость координационных соединений ионов d-металлов с глицином и диглицином и энтальпии реакций их образования.
В данной работе проведено обобщение имеющихся экспериментальных данных по термодинамике комплексообразования иона меди(II) с глицил-глицил-глицином (триглицин, HL±) и кислотно-основным равновесиям лиганда с использованием сольватационного подхода, основанного на термодинамической характеристике сольватации реагентов [26–31].
Протолитические равновесия цвиттериона триглицина (NH$_{3}^{ + }$–CH–CO–NH–CH–CO–NH–CH–COO–, HL±) могут быть представлены уравнениями:
(1)
${{{\text{H}}}_{2}}{{{\text{L}}}^{ + }} \leftrightarrow {\text{H}}{{{\text{L}}}^{ \pm }} + {{{\text{H}}}^{ + }},\quad {{K}_{1}} = \frac{{[{\text{H}}{{{\text{L}}}^{ \pm }}][{{{\text{H}}}^{ + }}]}}{{[{{{\text{H}}}_{2}}{{{\text{L}}}^{ + }}]}},$(2)
${\text{H}}{{{\text{L}}}^{ \pm }} \leftrightarrow {{{\text{H}}}^{ + }} + {{{\text{L}}}^{ - }},\quad {{K}_{2}} = \frac{{[{{{\text{L}}}^{ - }}][{{{\text{H}}}^{ + }}]}}{{[{\text{H}}{{{\text{L}}}^{ \pm }}]}},$Константы диссоциации триглицина, состав и константы устойчивости триглицианатов меди в водно-этанольных растворителях были определены в [28, 29, 31]. Математическая обработка экспериментальных данных по программе “PHMETR” [32] показала, что в исследуемом интервале рН 2.0–7.5 в воде при соотношении металла и лиганда 1 : 1 система Cu(II)-триглицин наилучшим образом описывается с учетом равновесий (3–10), если предположить существование четырех комплексных частиц [CuHL]2+, [CuL]+, [CuH–1L] (координация Cu(II) с замещением протона одной амидной группы), [CuH–2L]– (координация Cu(II) с замещением протона обеих амидных групп). Данная модель процесса подтверждается методами спектрофотометрии и ЯМР [11].
(4)
$2{{{\text{H}}}^{ + }} + {{{\text{L}}}^{ - }} \rightleftarrows {{{\text{H}}}_{2}}{{{\text{L}}}^{ + }},$(5)
${\text{C}}{{{\text{u}}}^{{2 + }}} + {{{\text{L}}}^{ - }} + {{{\text{H}}}^{ + }} \rightleftarrows {{[{\text{CuHL}}]}^{{2 + }}},$(6)
${\text{C}}{{{\text{u}}}^{{2 + }}} + {{{\text{L}}}^{ - }} \rightleftarrows {{[{\text{CuL}}]}^{ + }},$(7)
${\text{C}}{{{\text{u}}}^{{2 + }}} + {{{\text{L}}}^{ - }} \rightleftarrows [{\text{Cu}}{{{\text{H}}}_{{ - 1}}}{\text{L}}] + {{{\text{H}}}^{ + }},$(8)
${\text{C}}{{{\text{u}}}^{{2 + }}} + {{{\text{L}}}^{ - }} \rightleftarrows {{[{\text{Cu}}{{{\text{H}}}_{{ - 2}}}{\text{L}}]}^{--}} + 2{{{\text{H}}}^{ + }},$(9)
${\text{C}}{{{\text{u}}}^{{2 + }}} + {\text{HOH}} \rightleftarrows {\text{CuO}}{{{\text{H}}}^{ + }} + {{{\text{H}}}^{ + }},$(10)
${{{\text{H}}}_{2}}{\text{O}} \rightleftarrows {{{\text{H}}}^{ + }} + {\text{O}}{{{\text{H}}}^{--}}.$ТЕРМОДИНАМИКА КИСЛОТНО-ОСНОВНЫХ РАВНОВЕСИЙ ТРИГЛИЦИНА В ВОДНО-ОРГАНИЧЕСКИХ РАСТВОРИТЕЛЯХ
Рост концентрации органического сорастворителя сопровождается увеличением pK1 как в водном этаноле, так и в водном DMSO. Ранее было отмечено аналогичное влияние водно-этанольного растворителя на процессы диссоциации глицил-глицина [14] и глицина [16] (рис. 1а). В водном диметилсульфоксиде для процессов диссоциации аминогруппы глицина и глицил-глицина характерна экстремальная зависимость pK2 = f(X2) с минимумом при XDMSO ≈ 0.2 мол. доли. Можно предположить аналогичное влияние водно-диметилсульфоксидного растворителя на процесс диссоциации глицил-глицил-глицина (рис. 1б). В водно-этанольном растворителе наблюдается монотонное уменьшение pK2 диссоциации глицина, глицил-глицина и глицил-глицил-глицина.
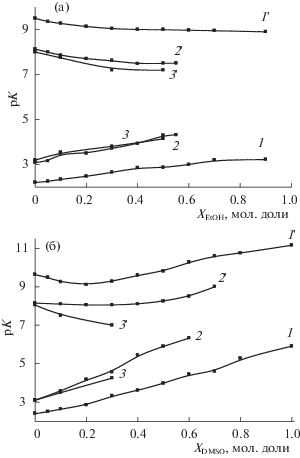
Увеличение длины цепи между карбоксильной и аминогруппой в структуре молекулы пептидов в ряду глицин, диглицин, триглицин и тетраглицин вызывает увеличение констант диссоциации карбоксильной группы и уменьшение основности аминогруппы в воде [33]. Такая закономерность объясняется тем, что при образовании пептидов электронная плотность на концевой аминогруппе понижается, а на карбоксильной группе, наоборот, увеличивается.
С использованием значений ∆rH1 и ∆rH2, полученных в [30], а также величин ∆rG1 и ∆rG2 соответствующих процессов [29], были рассчитаны изменения энтропии реакций (1) и (2) в водно-этанольном растворителе. Процессы диссоциации триглициний-иона и триглицина в растворителе вода-этанол во всей исследуемой области составов растворителя Н2О-EtOH эндотермичны. Эндотермичность диссоциации триглицина по карбоксильной группе (∆rH1) монотонно увеличивается с увеличением концентрации этанола в растворителе. Зависимость ∆rH2 = F(XEtOH) имеет незначительный эндотермический максимум в области XEtOH = 0.1 мол. доли. Дальнейший рост содержания этанола в растворителе приводит к уменьшению эндотермичности диссоциации триглицина по аминогруппе (рис. 2). Уменьшение эндотермичности диссоциации глицина по карбоксильной группе было установлено ранее в водно-этанольных [34] и водно-изопропанольных [35] средах.
Анализ соотношений энтальпийной и энтропийной составляющих энергии Гиббса диссоциации триглициний-иона и триглицина в растворителе Н2О–EtOH выявил преимущество энтальпийного вклада над энтропийным в изменении ΔG, причем данное преимущество выражено значительнее в случае диссоциации триглицина по аминогруппе (рис. 2). Аналогичная тенденция в соотношении энергетической и структурной составляющих энергии Гиббса характерна для реакций диссоциации глицина в растворителях H2O–EtOH [34]. Можно отметить, что при диссоциации по аминогруппе для триглицина, вклад TΔSr2 в ΔGr2 менее значим, чем при диссоциации глицина.
С целью выявления причин изменения энергий Гиббса и энтальпий реакций (1) и (2) в водно-этанольном растворителе был использован сольватационно-термодинамический подход основанный на термодинамической характеристике сольватации реагентов [36]. Анализ сольватационных вкладов реагентов в изменение энергии Гиббса реакций (1) и (2) при переходе от воды к водно-этанольным смесям проведен с использованием сольватационно-термодинамического подхода по уравнениям:
(11)
${{\Delta }_{{{\text{tr}}}}}{{G}_{1}} = {{\Delta }_{{{\text{tr}}}}}G({{{\text{H}}}^{ + }}) + {{\Delta }_{{{\text{tr}}}}}G({\text{H}}{{{\text{L}}}^{ \pm }})--{{\Delta }_{{{\text{tr}}}}}G({{{\text{H}}}_{2}}{{{\text{L}}}^{ + }}),$(12)
${{\Delta }_{{{\text{tr}}}}}{{G}_{2}} = {{\Delta }_{{{\text{tr}}}}}G({{{\text{H}}}^{ + }}) + {{\Delta }_{{{\text{tr}}}}}G({{{\text{L}}}^{ - }})--{{\Delta }_{{{\text{tr}}}}}G({\text{H}}{{{\text{L}}}^{ \pm }}).$Значения ΔtrG1 и ΔtrG2 для процессов (1) и (2) рассчитаны с использованием значений констант диссоциации триглицина [27, 29] в соответствии с уравнением:
(13)
${{\Delta }_{{{\text{tr}}}}}G = --2.303RT({\text{p}}{{K}_{{{\text{solv}}}}}--{\text{p}}{{K}_{{{{{\text{H}}}_{2}}{\text{O}}}}}),$Это позволило применить уравнения (11) и (12) для расчета величин ΔtrG(H2L+) и ΔtrG(L–) с привлечением литературных данных по изменениям энергии Гиббса переноса протона [37, 38] и триглицина [39, 40] из воды в растворители вода−этанол и вода–диметилсульфоксид.
Влияние состава водно-органического растворителя на изменение энергии Гиббса реакций диссоциации триглицина и пересольватацию реагентов представлено на рис. 3.
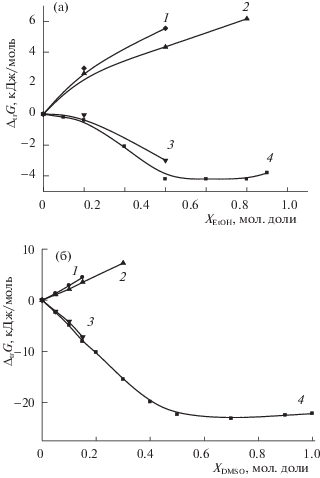
Ослабление кислотных свойств триглициний-иона (H2L+) при возрастании содержания этанола или диметилсульфоксида в растворителе определяется ослаблением сольватации цвиттерионной формы трипептида и усилением сольватации триглициний-иона. Согласно [14] ослабление кислотных свойств глицил-глициний-иона при переходе от воды к водному этанолу или к водному DMSO происходит, главным образом, из-за возрастающего различия в пересольватации нейтральной и протонированной форм дипептида, что не противоречит соотношениям аналогичных сольватационных характеристик реагентов в реакции кислотной диссоциации трипептида по карбоксильной группе.
В случае диссоциации триглицина наблюдается противоположная тенденция: кислотная сила триглицина возрастает с ростом содержания EtOH и DMSO в растворителе за счет ослабления сольватации цвиттериона трипептида и усиления сольватации протона. Изменение энергии Гиббса сольватации протона (ΔtrG(H+)) преобладает над разницей ΔG пересольватации глицил-глицил-глицинат-иона (ΔtrG(L–)) и цвиттериона триглицина (ΔtrG(HL±)), что аналогично для кислотной диссоциации глицина [16] и глицил-глицина [14]. Таким образом, можно предположить сходное влияние водно-органического растворителя на смещение кислотно-основных равновесий глициновых пептидов с различной длиной полипептидной цепи.
В соответствии с [36] энтальпии переноса реакций (1) и (2) из воды в растворитель вода−этанол можно представить в виде:
(14)
${{\Delta }_{{{\text{tr}}}}}{{H}_{{{\text{r1}}}}} = {{\Delta }_{{{\text{tr}}}}}H({\text{H}}{{{\text{L}}}^{ \pm }}) + {{\Delta }_{{{\text{tr}}}}}H({{{\text{H}}}^{ + }})--{{\Delta }_{{{\text{tr}}}}}H({{{\text{H}}}_{2}}{{{\text{L}}}^{ + }}),$(15)
${{\Delta }_{{{\text{tr}}}}}{{H}_{{{\text{r2}}}}} = {{\Delta }_{{{\text{tr}}}}}H({{{\text{L}}}^{--}}) + {{\Delta }_{{{\text{tr}}}}}H({{{\text{H}}}^{ + }})--{{\Delta }_{{{\text{tr}}}}}H({\text{H}}{{{\text{L}}}^{ \pm }})$(16)
${{\Delta }_{{{\text{tr}}}}}{{Н }_{{\text{r}}}} = {{\Delta }_{{\text{r}}}}{{Н }_{{{\text{S}}1 + {\text{S}}2}}}--{{\Delta }_{{\text{r}}}}{{Н }_{{{\text{S}}1}}},$Анализ соотношения сольватационных вкладов реагентов в изменение энергетики реакции (1) показал, что рост положительных значений энтальпии диссоциации при переносе триглициний-иона из воды в водно-этанольные растворители состава 0.20 мол. доли этанола определяется изменением сольватного состояния HL± при взаимной компенсации сольватационных вкладов протона и триглийциний-иона (рис. 4). Для реакции диссоциации триглицина различия в сольватном состоянии протона и триглицина {ΔtrH(H+) – ΔtrH(HL±)} определяют рост экзотермичности переноса реакции (2) из воды в водный этанол в области составов растворителя Х = 0.0–0.2 мол. доли EtOH. В растворителях Н2О-EtOH с большим содержанием EtOH, сольватационный вклад протона является основным фактором роста экзотермичности ΔtrHr2.
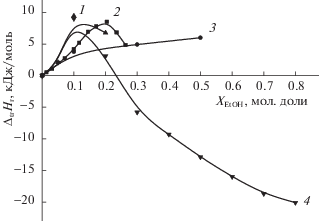
Сольватационные характеристики реагентов на рис. 5 имеют эндотермические максимумы в области малых концентраций этанола в растворителе, которые обычно связывают с упрочнением структуры воды в области малых добавок EtOH [43, 44]. Проводя сравнение энтальпийных характеристик реакции диссоциации триглицина (2) с энтальпийными характеристиками процессов кислотной диссоциации родственных соединений можно отметить, что сольватационный вклад протона также является определяющим фактором в смещении кислотно-основных равновесий в водно-спиртовых растворах глицина [45] и протонированных аминов [46–49].
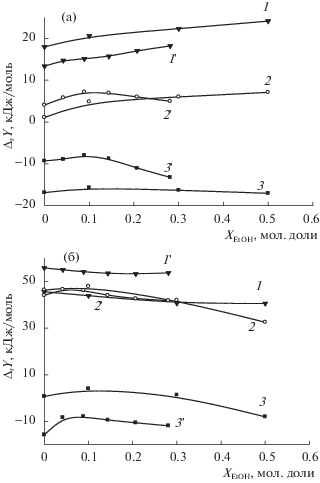
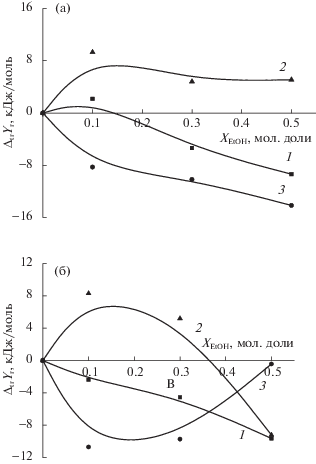
Рис. 5.
Термодинамические характеристики реакции комплексообразования иона меди(II) с триглицином (а) и триглицинат-ионом (б) в растворителе вода–этанол: 1 – ΔtrGr, 2 – (–TΔtrSr), 3 – ΔtrHr, [26].
ТЕРМОДИНАМИКА КОМПЛЕКСООБРАЗОВАНИЯ ТРИГЛИЦИНА С ИОНОМ МЕДИ(II) В ВОДНО-ОРГАНИЧЕСКИХ РАСТВОРИТЕЛЯХ
Значения констант устойчивости комплексов триглицианатов меди(II) в водно-этанольном и водно-диметилсульфоксидном растворителях приведены в табл. 1. Полученные нами константы устойчивости комплексов [CuL]+ и [CuHL]2+ в водном растворе при ионной силе 0.1, создаваемой различными фоновыми электролитами NaClO4 [27, 28] и NaCl [28] согласуются между собой и с данными литературы [7, 8].
Таблица 1.
Значения логарифмов констант устойчивости комплексов меди(II) с триглицином и триглицинат-ионом в водно-органических растворителях при I = 0.1 (NaCl или NaClO4) и температуре T = 298.15 K [27, 28]
| X2, мол. доли | I | Фоновый электролит | lg β[CuHL]2+ | lg β[CuL]+ |
|---|---|---|---|---|
| 0 | 0 | NaCl | 2.24 ± 0.03 | 5.25 ± 0.14 |
| 0.1 | NaCl | 2.39 ± 0.04 [28] | 5.39 ± 0.05 [28] | |
| 2.36 [8] | 5.25 [7] | |||
| NaClO4 [28] | 2.23 ± 0.06 | 5.26 ± 0.05 | ||
| 0.5 | NaCl | 2.90 ± 0.01 | 5.92 ± 0.01 | |
| 1.0 | NaCl | 3.61 ± 0.04 | 6.62 ± 0.04 | |
| H2O–EtOH [28] | ||||
| 0.1 | 0.1 | NaClO4 | 2.04 ± 0.06 | 5.68 ± 0.06 |
| 0.3 | 3.17 ± 0.06 | 6.06 ± 0.06 | ||
| 0.5 | 3.87 ± 0.06 | 6.95 ± 0.06 | ||
| H2O–DMSO [27] | ||||
| 0.1 | 0.1 | NaClO4 | 2.66 ± 0.13 | 5.59 ± 0.08 |
| 0.3 | 3.03 ± 0.08 | 5.88 ± 0.09 | ||
Значения lg β[CuL]+ и lg[CuHL]2+, полученные в воде при ионной силе 0.5 и 1.0, создаваемой NaCl, приводятся в данной работе впервые. Установлено, что с ростом ионной силы раствора устойчивость комплексов [CuL]+ и [CuHL]2+ монотонно увеличивается. Методики потенциометрических экспериментов и расчетов аналогичны приведенным нами ранее в работе [50].
При переходе от воды к водно-этанольным и водно-диметилсульфоксидным растворителям наблюдается рост устойчивости комплексов [CuHL]2+ и [CuL]+. Во всех составах водно-органических растворителей значения lg β[CuL]+ примерно на 3 порядка больше, чем значения lg β[CuHL]2+. Такое различие в устойчивости комплексов объясняется тем, что при образовании [CuL]+ связывание иона металла с пептидом осуществляется через аминогруппу с участием атома кислорода пептидной связи. В результате комплекс [CuL]+ имеет структуру пятичленного хелатного кольца [11].
Термодинамические параметры реакций комплексообразования триглицина и триглицинат-иона с ионом меди (II) в водно-этанольном растворителе представлены в табл. 2.
Таблица 2.
Термодинамические параметры реакций комплексообразования триглицина и триглицинат-иона с ионом меди(II) в водно-этанольном растворителе при Т = 298.15 K (I = 0.10, NaClO4) [26, 28]
| XEtOH, мол. доли | –∆r1H, кДж/моль | –Δr1G, кДж/моль | TΔr1S, кДж/моль | –∆r2H, кДж/моль | –Δr2G, кДж/моль | TΔr2S, кДж/моль |
|---|---|---|---|---|---|---|
| Cu2+ + HL± ↔ [CuHL]2+ (4) | Cu2+ + L– ↔ [CuL]+ (14) | |||||
| 0 | –6.62 ± 0.15 [26] | 2.72 ± 0.34 [28] | 19.3 ± 0.4 [26] | 18.03 ± 0.24 [26] | 30.01 ± 0.29 [28] | 12.0 ± 0.6 [26] |
| 26.36 ± 0.84 [3] | 28.74 ± 0.04 [3] | 2.38 ± 0.84 [3] | ||||
| 0.1 | 1.65 ± 0.20 [26] | 11.64 ± 0.34 [28] | 10.0 ± 0.4 [26] | 28.73 ± 0.20 [26] | 32.41 ± 0.34 [28] | 3.7 ± 0.4 [26] |
| 0.3 | 3.56 ± 0.20 [26] | 18.09 ± 0.34 [28] | 14.5 ± 0.4 [26] | 27.75 ± 0.25 [26] | 34.58 ± 0.34 [28] | 6.8 ± 0.4 [26] |
| 0.5 | 7.89 ± 0.20 [26] | 22.08 ± 0.34 [28] | 14.2 ± 0.4 [26] | 18.46 ± 0.20 [26] | 39.66 ± 0.34 [28] | 21.2 ± 0.4 [26] |
С ростом концентрации этанола в растворителе происходит монотонное увеличение экзотермичности комплексообразования [CuHL]2+. Энтальпии реакции образования комплекса [CuL]+ изменяются экстремально с максимумом экзотермичности при 0.1–0.3 мол. доли. EtOH. Упрочнение комплекса [CuHL]± определяется ростом экзотермичности комплексообразования. Изменения энтропийной составляющей энергии Гиббса происходят, в основном, в области составов растворителя от 0 до 0.1 мол. доли. Дальнейший рост содержания этанола в растворителе не приводит к значительным изменениям величины TΔr1S (рис. 5а). В случае [CuL]+ наблюдается типичный компенсационный эффект с нарастающим доминированием энтропийного вклада в Δr2G (рис. 5б).
Уравнения, аналогичные уравнениям (11)–(16), были использованы для расчета термодинамических функций переноса реакций (5) и (6) и сольватации комплексных частиц [CuHL]2+ и [CuL]+ с привлечением литературных данных по изменениям термодинамических параметров сольватации триглицина [39–41], триглицинат-иона [27, 29, 30] и иона Cu2+ [51–53] при переносе из воды в растворители вода–этанол и вода–диметилсульфоксид.
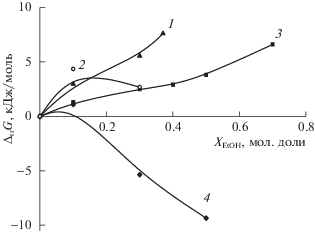
Анализ сольватационных вкладов реагентов в изменение устойчивости комплексов [CuHL]2+ и [CuL]+ показал, что при переходе от воды к водному этанолу упрочнение обоих комплексов происходит, в основном, за счет пересольватации лигандов при благоприятном сольватационном вкладе иона-комплексообразователя (рис. 6).
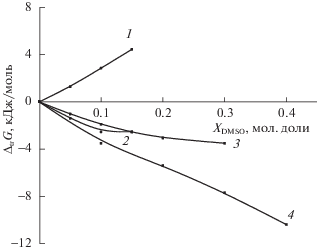
В отличии от водных растворов этанола, в водных растворах диметилсульфоксида усиление сольватации иона меди(II) при переходе от воды к ее смесям с DMSO не способствует упрочнению комплексов [CuHL]2+ и [CuL]+ (рис. 7). Однако, благодаря взаимной компенсации сольватационных вкладов комплексных частиц и иона меди(II), также как и в водно-этанольных растворителях, ослабление сольватации лигандов определяет изменение устойчивости триглицинатных комплексов.
Рис. 7.
Влияние водно-диметилсульфоксидного растворителя на изменение энергии Гиббса реакции комплексообразования меди(II) c триглицином и сольватации реагентов при переносе из воды в водно-диметилсульфоксидный растворитель: 1 – ΔtrG(HL±) [40], 2 – ΔtrG([CuHL]2+), 3 – ΔtrGr1 [27], 4 – ΔtrG(Cu2+) [52].
Динамика сольватационных вкладов реагентов в изменение энтальпии реакции образования комплекса [CuHL]2+ представлена на рис. 8.
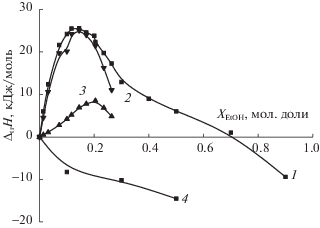
Увеличение экзотермичности реакции образования [CuHL]2+ при переходе от воды к водно-этанольным растворителям определяется эффектом лиганда (ΔtrH(HL±)) при взаимной компенсации сольватационных вкладов ионов (Cu2+ и [CuHL]2+). Близкие значения ΔtrH([CuHL]2+) и ΔtrH(Cu2+) свидетельствует об определяющей роли центрального иона в энергетике образования сольватной оболочки комплексной частицы [CuHL]2+. Соотношение энтальпийных характеристик реакции образования [CuL]+ аналогично соотношениям сольватационных вкладов реагентов в изменение энтальпии реакции образования [CuHL]2+.
Таким образом, анализ влияния растворителей Н2O–EtOH и Н2O–DMSO на изменение термодинамических параметров образования комплексов меди(II) с цвиттер-ионом глицил-глицил-глицина и триглицинат-ионом и кислотно-основные равновесия лигандов показал, что:
– изменения кислотно-основных свойств глицил-глицил-глицина обусловлены изменениями в сольватном состоянии протона;
– изменения термодинамических параметров комплексообразования определяются, в основном, пересольватацией лигандов.
Подобные соотношения сольватационных вкладов реагентов были отмечены ранее для реакций образования комплексов глицинат-иона и диглицинат-иона с медью(II) [19], никелем(II) [24] и серебром(I) [25] в водно-спиртовых растворителях и в водном DMSO. Обобщение результатов, представленных в данной работе, подтверждает установленную ранее определяющую роль сольватационного вклада лиганда для реакций образования ионных комплексов d-металлов в водно-органических растворителях [54].
Работа выполнена в Институте термодинамики и кинетики химических процессов ИГХТУ с использованием оборудования Центра коллективного пользования ИГХТУ.
Список литературы
Духович Ф.С., Дарховский М.Б., Горбатова Е.Н., Курочкин В.К. Молекулярное узнавание: фармакологические аспекты. М.: Медицина, 2004. 224 с.
Koval C., Margerum D. // Inorg. Chem. Acta. 1981. V. 20. № 7. P. 2311.
Brunetti A., Lim M., Nancollas G. // J. Am. Chem. Soc. 1968. V. 206. № 90. P. 5120.
Kim M., Martell A. // Ibid. 1966. V. 88. № 5. P. 914.
Brookes G., Pettit L. // J. Chem. Soc., Dalton Trans. 1975. Iss. 20. P. 2106.
Sigel H., Griesser R., Prijs B. // Naturforsch. 1972. V. 27. P. 353.
Yamauchi O., Nakao Y., Nakahara A. // Bull. Chem. Soc. Jpn. 1973. V. 46. № 7. P. 2119.
Kaneda A., Martell A. // J. Coord. Chem. 1975. V. 4. Iss. 3. P. 137.
Чернявская Н.В., Гридчин С.Н., Бычкова С.А. // Журн. неорган. химии. 2015. Т. 60. № 9. С. 1276.
Горболетова Г.Г., Метлин А.А., Бычкова С.А. // Журн. физ. хим. 2015. Т. 9. № 5. С. 787.
Илакин В.С., Штырлин В.Г., Захаров А.В., Конькин А.Л. // Журн. общ. химии. 2002. Т. 72. Вып. 3. С. 377.
Shtyrlin V.G., Gogolashvili E.L., Zakharov A.V. // J. Chem. Soc., Dalton Trans. 1989. № 7. P. 1293.
Hadweh S., Huet J., Jouini M., Lapluye G. // J. Chim. Phys. 1992. V. 89. № 10. P. 1973.
Наумов В.В., Исаева В.А., Шарнин В.А. // Журн. физ. химии. 2011. Т. 56. № 7. С. 1208.
Исаева В.А., Леденков С.Ф., Шарнин В.А., Шорманов В.А. // Там же. 1993. Т. 67. № 11. С. 2202.
Исаева В.А., Шарнин В.А., Шорманов В.А., Баранова И.А. // Там же. 1996. Т. 70. № 8. С. 1421.
Наумов В.В., Исаева В.А., Шарнин В.А. // Там же. 2011. Т. 85. № 10. С. 1881.
Исаева В.А., Наумов В.В., Шарнин В.А., Гессе Ж.Ф. // Там же. 2009. Т. 83. Вып. 3. С. 477.
Фадеев Ю.Ю., Шарнин В.А., Шорманов В.А. // Журн. неорган. химии. 1997. Т. 42. № 7. С. 1220.
Леденков С.Ф., Шорманов В.А., Шарнин В.А. // Журн. физ. хим. 1996. Т. 70. № 10. С. 1769.
Михеев С.В., Шарнин В.А. // Журн. физ. химии. 2010. Т. 84. № 2. С. 205.
Isaeva V.A., Naumov V.V., Sharnin V.A. // Russ. J. Coord. Chem. 2009. V. 35. № 11. P. 868.
Naumov V.V., Isaeva V.A., Sharnin V.A., Kuzina E.N. // Russ. J. Phys. Chem. A. 2011. V. 85. № 10. P. 1752.
Исаева В.А., Шарнин В.А., Шорманов В.А. // Коордирац. химия. 1999. Т. 25. № 12. С. 912.
Исаева В.А., Наумов В.В., Гессе Ж.Ф., Шарнин В.А. // Там же. 2008. Т. 34. № 8. С. 631.
Фам Тхи Л., Усачева Т.Р., Хренова Т.М., Шарнин В.А. // Журн. физ. химии. 2017. Т. 91. № 7. С. 1161.
Usacheva T.R., Pham Thi L., Kuzmina K.I., Sharnin V.A. // J. Therm. Anal. Calorim. 2017. V. 130. P. 471.
Фам Тхи Л., Усачева Т.Р., Тукумова Н.В. и др. // Журн. физ. химии. 2016. Т. 90. № 10. С. 1479.
Фам Тхи Л., Усачева Т.Р., Тукумова Н.В. и др. // Там же. 2016. Т. 90. № 2. С. 216.
Фам Тхи Л., Усачева Т.Р., Шарнин В.А. // Там же. 2016. Т. 90 № 12. С. 1815.
Badelin V.G., Barannikov V.P., Tarasova G.N. et al. // Russ. J. Phys. Chem. A. 2012. V. 86. № 1. P. 40.
Бородин В.А., Козловский Е.В., Васильев В.П. // Журн. неорган. химии. 1986. Т. 31. № 1. С. 10.
Sigel H., Martin R.B. // Chem. Rev. 1982. V. 82. P. 385.
Gao H., Hu X., Lin R. // Thermochim. Acta. 2000. V. 346. P. 1.
Tsurko E.N., Shihova T.M., Bondarev N.V. // J. Mol. Liq. 2002. V. 96–97. P. 425.
Крестов Г.А. Термодинамика ионных процессов в растворах. Л.: Химия, 1984. 272 с.
Marcus. Y. // Pure Appl. Chem. 1983. V. 55. Iss. 6. P. 977.
Kalidas C., Hefter G., Marcus Y. // Chem. Rev. 2000. V. 100. № 3. P. 819.
Nozaki Y., Tanford C. // J. Biol. Chem. 1971. V. 246. Iss. 10. P. 2211.
Усачева Т.Р., Кузьмина К.И., Фам Тхи Л. и др. // Журн. физ. химии. 2014. Т. 88. № 7–8. С. 1176.
Smirnov V.I., Badelin V.G. // Thermochim. Acta. 2008. V. 471. P. 97.
Невский А.В., Шорманов В.А., Крестов Г.А., Пирогова Е.С. // Изв. вузов. Хим. и хим. технология. 1984. Т. 2. № 6. С. 730.
Arnett E.M., Bentrude W.G., Burke J.J., Duggleby P. McC. // J. Amer. Chem. Soc. 1965. V. 87. Iss. 7. P. 1541.
Крестов Г.А., Никифоров М.Ю., Альпер Г.А. и др. Растворы неэлектролитов. М.: Наука, 1989. 263 с.
Гессе Ж.Ф., Исаева В.А., Репкин Г.И., Шарнин ВА. // Журн. физ. химии. 2012. Т. 86. № 1. С. 59.
Шорманов В.А., Репкин Г.И., Крестов Г.А. // Изв. вузов. хим. и хим. техн. 1983. Т. 36. № 5. С. 561.
Афанасьев В.Н., Шорманов В.А., Крестов Г.А. // Тр. Ивановского хим-тех. инст. 1972. Т. 13. С. 36.
Корякин Ю.С., Шорманов В.А., Крестов Г.А. // Изв. вузов. Хим. и хим. техн. 1979. Т. 22. № 4. С. 500.
Невский А.В., Шорманов В.А., Крестов Г.А. // Там же. 1984. Т. 27. № 2. С. 156.
Badelin V.G., Barannikov V.P., Tarasova G.N. et al. // Russ. J. Phys. Chem. A. 2012. V. 86. № 1. P. 40.
Lewandowski A. // Electrochim. Acta. 1985. V. 30. Iss. 3. P. 311.
Lewandowski A. // Ibid. 1986 V. 31. Iss. 1. P. 59.
Михеев С.В., Шарнин В.А., Шорманов В.А. // Журн. физ. химии. 1997. Т. 71. № 1. С. 91.
Шарнин В.А. // Изв. вузов. Хим. и хим. техн. 2005. Т. 48. № 7. С. 44.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии