

Журнал физической химии, 2019, T. 93, № 10, стр. 1506-1516
Функциональные группы оксида графита: Экспериментальные данные и неэмпирическое моделирование
Е. А. Шиляева a, Ю. В. Новаковская a, *
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: jvnovakovskaya@gmail.com
Поступила в редакцию 15.03.2019
После доработки 15.03.2019
Принята к публикации 09.04.2019
Аннотация
Сопоставление экспериментального инфракрасного спектра поглощения синтезированного образца оксида графита и рассчитанных с использованием неэмпирических подходов спектров модельных систем CnOmHk, включающих базовый фрагмент графитового слоя С54 и различные модифицирующие его функциональные группы (гидроксильные, эпоксидные, карбоксильные и кето-группы) в разных количествах и сочетаниях в присутствии небольшого числа сольватирующих молекул воды, позволило прояснить природу ряда сигналов в средней и характеристичной области ИК-спектра оксида графита и выявить роль водородного связывания между различными функциональными группами и молекулами воды.
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ И МОДЕЛИ СТРОЕНИЯ ОКСИДА ГРАФИТА
В настоящее время развитие как экспериментальных методов исследования строения веществ, так и расчетных схем, позволяющих в рамках неэмпирического квантово-механического подхода моделировать не только достаточно большие отдельные молекулы или молекулярные кластеры и агломераты, но и стационарные фрагменты сложных макросистем нерегулярного строения, явилось стимулом более детального изучения практически важных химических систем. Сочетание эксперимента и теории оказывается весьма продуктивным в тех случаях, когда, будучи использованы независимо, эти два подхода в силу внутренних ограничений не дают возможность построить обоснованную корректную модель изучаемой системы или процесса. Яркий пример, иллюстрирующий справедливость сформулированного утверждения, – проблема строения оксида графита.
Оксид графита, впервые синтезированный в 1855 году [1], за более чем полуторавековую историю своего получения и применения прошел не один цикл всплеска и последующего затухания интереса к его структурным особенностям и возможным применениям. В последние десятилетия периодический рост числа исследований и выявление новых особенностей, как правило, были инициированы возникновением новых или существенным повышением точности существующих физических методов исследования. Их результаты служили отправной точкой при формулировке новых идей относительно механизма формирования оксида графита, роли тех или иных реагентов, природы (выделяемых при определенных условиях) промежуточных продуктов и, конечно же, строения слоев оксида графита и природы тех функциональных групп, которые и определяют свойства этого вещества.
Его сложно назвать соединением, поскольку его состав заметно зависит от условий синтеза: размера исходных графитовых частиц, применяемых для их окисления композиций реагентов и их концентрации, соотношения количеств графита и окислителя, а также длительности процесса окисления и методики последующей промывки образцов. Традиционно выделяют три основных метода синтеза оксида графита, имеющих много вариаций и модификаций. Исторически первым был метод Броди [1], в котором роль окислителя играл хлорат калия в среде дымящей азотной кислоты (KClO3 + конц. HNO3). В методике Штауденмайера [2] был использован хлорат калия в смеси концентрированных азотной и серной кислот (KClO3 + конц. HNO3 + конц. H2SO4). Метод Хаммерса [3] основан на ином окислителе, перманганате калия, в среде концентрированной серной кислоты в присутствии нитрата натрия (KMnO4 + конц. H2SO4 + NaNO3). При этом, как справедливо отмечено в [4], последний метод правильнее было бы называть методом Шарпи–Хаммерса, поскольку та же самая композиция перманганата калия и серной кислоты была полувеком ранее применена Шарпи [5]. В одной из эффективных модификаций метода Шарпи–Хаммерса в качестве окислительной композиции была недавно предложена следующая: KMnO4 + смесь 9 : 1 конц. H2SO4 : H3PO4 (которую мы условно будем называть методом Тура) [6].
Согласно приведенным в [7] данным, соотношение C/O в различных образцах оксида графита попадает в диапазон от 3.5 (метод Броди) до 2.9 (метод Шарпи–Хаммерса), включая промежуточную величину 3.3, характерную для метода Штауденмайера. Таким образом, наиболее окисленные образцы графита могут быть получены при использовании перманганата калия и серной кислоты в рамках более быстрого (при использовании небольших исходных частиц графита) и экологичного (без выделения оксидов азота и хора) процесса.
Для выделения оксида графита из любой из указанных сред требуется его длительное промывание в воде, поэтому получить безводный (сухой) оксид графита практически невозможно: образцы, высушенные при комнатной температуре над оксидом фосфора P2O5, содержат не менее 5–10% воды. Согласно рекомендации [7], рационально использовать образцы с содержанием воды 10–17%, что отвечает нормальному давлению паров воды в лабораторных условиях. Наличие воды влияет как на оценки относительного количества углерода и кислородсодержащих функциональных групп (при выяснении состава вещества), так и на свойства образца, что не только усложняет определение типов и относительного количества различных функциональных групп в оксиде графита, но и вносит неопределенность в результаты последующей модификации, механизм которой, очевидно, зависит не только от природы, количества и взаимного расположения (и влияния) функциональных групп, но и от их доступности (в том числе, при наличии межфрагментных связей и сольватных оболочек).
При определении состава и вероятного строения слоев оксида графита обычно используют данные рентгеновской фотоэлектронной спектроскопии (РФС), спектроскопии твердотельного ядерного магнитного резонанса (прежде всего, 13С ТТ ЯМР) и спектроскопии поглощения в инфракрасной области (ИК-спектры). Каждый из методов имеет свои достоинства и недостатки в плане выявления структурных особенностей и функциональных групп в образцах оксида графита. И ни один не дает исчерпывающей информации о строении оксида графита, которое до сих пор не определено в степени, которая была бы достаточной для корректного выбора процедур его целенаправленной модификации.
РФС дает общее представление об элементном составе образца с детализацией относительно различных степеней окисления атомов каждого изучаемого типа, т.е. фактически вариантов ближайшего окружения атомов. Анализ поверхностных (в среднем порядка 5 нм толщиной) слоев образца в случае оксида графита дает вполне надежную информацию в силу слоистого строения материала и предположительно близкой по типу химической модификации внешнего и внутренних графитовых слоев при окислении. Обзорный C 1s-спектр практически любых образцов оксида графита имеет характерную структуру, включающую два основных пика при 286.5 эВ (доминирующий) и 284.5 эВ (примерно вдвое меньшей интегральной интенсивности) и плечо при 288.2 эВ. Основной пик относят к эпоксидам и третичным спиртам; пик меньшей интенсивности – к не окисленному углероду, а плечо – к карбоксильным группам, хотя не исключено, что оно обусловлено наложением сигналов кетонов и карбоксилов [4].
В некоторых работах для деконволюции обзорного спектра используют большее число базисных функций, особенно в области высокочастотного плеча, соотнося соответствующие пики с наличием, например, различных карбонил-содержащих функциональных групп, т.е. карбоксилов, кетонов и собственно карбонилов. Поскольку в целом эта процедура не имеет жестких критериев выбора необходимого и достаточного числа базисных функций, сложно оценивать корректность различных вариантов интерпретации. Ясно лишь, что в образцах оксида графита всегда присутствуют фрагменты не окисленного графита, доминируют атомы углерода, формирующие одинарные связи с атомами кислорода (С–О–С или С–ОН), и в небольшом количестве присутствуют атомы, формирующие связи С=О, причем природу этих групп определить сложно в силу того, что их ближайшее окружение в случае фрагмента окисленного графитового листа заведомо отлично от такового в индивидуальных небольших органических молекулах, сигналы в C 1s-спектрах которых попадают в интервал от 287.7 до 289.2 эВ [8, 9].
Более детальную информацию о наличии различных структурных мотивов в оксиде графита дает спектроскопия ТТ ЯМР. Согласно результатам, полученным в [10, 11], основные сигналы при значениях химического сдвига 132 (130), 71.2 (70) и 60.2 (60) м.д. соотнесены с не окисленными атомами углерода и атомами, вовлеченными в С–ОН- и С–О–С-(1,2-эфирные или эпоксидные) фрагменты соответственно. О том, что это именно эпоксиды, а не 1,3-эфиры, по мнению авторов, свидетельствует снижение интенсивности сигналов после обработки иодидом калия, который не реагирует с 1,3-эфирами, но реагирует с эпоксидами. Сигнал карбоксильных атомов углерода (ожидаемый вблизи 190 м.д.) не был зафиксирован, что возможно при отсутствии или очень малом количестве подобных функциональных групп. При этом общий состав был определен в [10] как C8(O)1.7(OH)1.7. В более поздних работах [12, 13] в спектрах более высокого разрешения помимо указанных сигналов были выделены три пика, более чем на порядок меньшей интенсивности, соотнесенные с кетонами (191 м.д.), карбоксилами (167 м.д.) и лактолами или геминальными диолами (101 м.д.) [12], а двумерный корреляционный спектр ТТ ЯМР позволил выделить наиболее вероятные близко расположенные группы, которыми оказались последовательности COH···COC···COC и COH···COH···COC при приблизительно втрое меньшей доле фрагментов COC···COC и COH···COH, непосредственно связанных с не окисленным углеродом [13]. При этом предположительное присутствие в структуре терминальных фенольных и карбоксильных групп и кетонов по-прежнему не имеет непосредственного подтверждения, как и относительное количество дефектов и сохранившихся после окисления ароматических фрагментов.
Третий основной источник информации о наличии определенных функциональных групп в структуре оксида графита – ИК-спектроскопия. Спектры различных образцов оксида графита в целом похожи. В них можно выделить три основные области: высокочастотную (3600–2200 см–1), средний диапазон (1730–1615 см–1) и характеристичную (1500–700 см–1). В области высоких частот в основном должны присутствовать сигналы валентных колебаний OH-связей, причем основной вклад вносят колебания молекул воды, поскольку при использовании дейтерированной воды кардинально снижается интенсивность сигнала в этой области, смещаясь в область 2700–1900 см–1 [14]. Пик около 1725 см–1 относят к валентным колебаниям карбонильных групп, причем определить тип этих групп (кетоны, альдегиды или карбоксилы) невозможно. Сигнал при 1620 см–1 часто соотносят с деформационными колебаниями воды, прочно связанной со структурой оксида графита. Наконец, характеристичная область спектра является результатом наложения большого числа колебаний различных функциональных групп, среди которых доминирующими полагают сигналы гидроксильных и эпоксидных групп. Но, как показано в нашей предыдущей работе [15], интенсивность и частота сигналов в спектре поглощения в случае этих двух типов групп сильно зависит от их взаимного расположения, наличия водородных связей между ними и наличия и количества гидратирующей воды, что не позволяет на основании интенсивности определенных сигналов в характеристичной области делать выводы об относительном количестве эпоксидных и гидроксильных групп в структуре оксида графита.
Таким образом, несмотря на большое количество экспериментальной информации, точно определить типы, характер взаимного расположения и относительное количество различных функциональных групп в структуре оксида графита по-прежнему практически невозможно. Можно лишь при определенных допущениях говорить о наиболее вероятных вариантах. Именно так и поступало большинство авторов, предлагавших на протяжении почти 90 лет различные модели строения оксида графита.
В первой модели, предложенной Тиле [16], предполагалось наличие исключительно гидроксильных групп, расположенных попарно над и под плоскостью графитового листа, что отвечало брутто-составу C6(OH)3. Несколькими годами позже Хофманн с соавторами [17] предложили кардинально иной вариант: на основании среднего соотношения C/O, близкого к трем, и не очень большой энергии связи С–О был сделан вывод о наличии исключительно эпоксидных групп в структуре оксида графита, а присутствие водорода объяснено прочно связанной водой. Позднее [18] они допустили, что при гидролизе части эпоксидных групп могут возникать гидроксильные группы, таким образом, тоже входящие в состав оксида графита. Еще десятилетием позже Руесс [19], указывая на то, что в случае оксида графита не реализуется типичная для эпоксидов реакция раскрытия циклов, предложил заменить все эпоксиды на 1,3-эфирные группы при сохранении небольшого количества гидроксильных групп (третичные спирты). Но в силу того, что исключительно третичные спирты не могли объяснить высокую кислотность образцов оксида графита, Хофманн, Боэм и Клаусс [20] расширили модель, допустив наличие в системе енольных фрагментов, что возможно лишь при условии разрыва части С=С-связей углеродной плоскости, и, как следствие, приводит к вероятному наличию и кетонов (возникающих в результате кето-енольной перегруппировки). Еще одним десятилетием позже Шольц и Боэм [21] фрагментарно объединили некоторые предыдущие идеи, допустив наличие в оксиде графита относительно регулярно чередующихся третичных спиртов, кетонов, а также восстановленных и не окисленных участков углеродной плоскости. Спустя еще два десятилетия Накажима с соавт. [22, 23] предложили новую модель, которая фактически оказалась незначительной модификацией модели Тиле: в нерегулярной в целом структуре предполагалось наличие преимущественно гидроксильных групп, в том числе частично депротонированных. Наконец, после публикации относительно надежных экспериментальных данных ЯМР- и ИК-спектроскопии оксида графита Лерф и Клиновски [24] предложили модель, которая предполагает наличие нерегулярно расположенных эпоксидных и гидроксильных групп, перемежающихся с не окисленными участками поверхности графитового листа, по периферии которого расположены карбоксильные группы.
Как видим, эта модель не привнесла почти ничего нового в постепенно формировавшуюся концепцию строения оксида графита. В некотором смысле она оказалась лишь рационализацией того, что ей предшествовало: были исключены менее вероятные или присутствующие в значительно меньших количествах функциональные фрагменты и сохранены те, наличие которых с большой вероятностью подтверждено практически всеми экспериментальными методами. При этом, например, вопрос о том, почему эпоксиды, сигналы которых наблюдаются и в ЯМР-, и в ИК-спектрах, не участвуют в типичных реакциях раскрытия цикла, остается без ответа. Не больше ясности и в вопросе, насколько вероятны структурные мотивы, включающие енолы или кетоны во внутренней части графитового листа, т.е. насколько вероятны локальные разрывы отдельных С=С-связей или формирование дефектов слоя, отвечающих потере одного или большего числа атомов углерода. Вместе с выяснением того, какие функциональные группы и в каком относительном количестве присутствуют на внешней границе углеродного листа, эти проблемы составляют тот блок вопросов, без решения которых корректное определение структурных мотивов оксида графита и надежное прогнозирование условий его целенаправленной химической модификации невозможны.
Решить такую задачу на основании исключительно экспериментальных данных невозможно в силу указанных выше (при обсуждении применяемых физических методов исследования) причин, а именно, невозможности однозначно соотнести сигналы в спектрах с определенными функциональными группами, поскольку в зависимости от локального окружения и наличия сольватирующих молекул воды эти сигналы могут смещаться в пределах перекрывающихся спектральных диапазонов. Более того, любое отнесение сигналов основано на сопоставлении с имеющимися базами данных для известных соединений, но все эти соединения являются относительно небольшими и ни в одном из них нет достаточно протяженных либо окисленных, либо не окисленных областей графитового листа, которые неизбежно и по-разному влияют на распределение электронной плотности и соответственно на упругость связей в интересующих функциональных группах. О степени и характере такого влияния можно судить лишь на основании теоретических неэмпирических расчетов модельных систем, размер которых дает возможность учесть основные ожидаемые эффекты реальной структуры. Но в случае расчетов есть свои проблемы. Теоретически выяснить механизм и предпочтительные пути окисления графита при обработке его той или иной композицией (на основе хлората или перманганата калия в присутствии концентрированной серной или азотной кислоты) – задача, практически не решаемая. Она требует построения очень больших моделей, включающих значительные области не менее чем двух графитовых листов, большое количество молекул кислоты, которые обеспечивают расслаивание графита, и собственно молекулы реагента и воды. Более того, это должно быть динамическое моделирование, причем с имитацией определенных физических (температурных) условий. Последнее легко осуществимо в варианте классической молекулярной динамики канонического ансамбля молекул, которая, однако, не позволяет моделировать химические превращения. Для описания же процессов перераспределения электронной плотности и разрыва/образования химических связей требуется вариант неэмпирической молекулярной динамики, в которой силы, действующие на ядра, определены решением квантово-химических уравнений, а потому применимой лишь к ограниченным по размерам системам. В результате и при теоретическом подходе исследователи вынуждены допускать (в качестве начального приближения) наличие тех или иных функциональных групп на поверхности углеродного слоя, не уточняя пути и условия их формирования. О природе этих групп судят (как и при создании моделей оксида графита) на основании доступных экспериментальных данных и концепций органической химии. Указанная выше неопределенность обоих источников информации приводит к необходимости учета различных комбинаций разных функциональных групп. И судить о том, какие комбинации более вероятны, оказывается возможным лишь при сопоставлении теоретических результатов с аналогичными данными, извлеченными из эксперимента, и выявлении сходных мотивов. Именно такой подход и был реализован в нашей работе и позволил прояснить некоторые из перечисленных выше вопросов строения оксида графита.
ЭКСПЕРИМЕНТ И МЕТОДЫ ТЕОРЕТИЧЕСКОГО МОДЕЛИРОВАНИЯ
Как было указано в предыдущем разделе, при построении надежной модели оксида графита необходимо учитывать не просто наличие тех или иных функциональных групп, но рассматривать возможные их комбинации, а при изучении отдельных групп допускать наличие различных вариантов ближайшего окружения, причем граничные эффекты не должны вносить ощутимое возмущение в моделируемые свойства. Это возможно при достаточно большом размере модельной системы. Поэтому в качестве базовой структурной единицы нами был выбран фрагмент графитового листа, включающий 54 атома углерода и представляющий собой центральный шестиугольник, окруженный двумя полными кольцевыми рядами структурных шестиугольников. При изучении свойств функциональных групп на внутренней поверхности углеродного слоя внешнее кольцо из двенадцати шестиугольников играет роль окружения, причем отсутствующие атомы углерода заменены терминальными атомами водорода. При изучении границ углеродных слоев часть таких атомов водорода заменена интересующими функциональными группами. На внутренней поверхности бездефектного углеродного листа могут быть расположены эпоксидные и гидроксильные группы. На периферии слоя могут быть локализованы как гидроксильные группы, так и кетоны или карбоксилы. Для выяснения же того, какие именно группы и в каких комбинациях присутствуют в реальных образцах оксида графита, рассчитанные спектры поглощения модельных систем были сопоставлены с зарегистрированным экспериментальным спектром образца оксида графита.
Для исключения вкладов терминальных атомов водорода в расчетный спектр и имитации общей жесткости углеродного каркаса на стадии решения колебательной задачи соответствующие атомы были заменены эффективными частицами с массами 100 а.е. С целью учета погрешности численных оценок частот в гармоническом приближении был использован масштабирующий множитель 0.98, который представляется обоснованным именно для характеристичной области частот. При визуализации сигналы рассчитанного спектра были уширены лоренцевыми функциями с шириной на полувысоте 50 см–1 для имитации реального спектра сложной системы при нормальной температуре.
Квантовохимические расчеты были выполнены в приближении метода функционала плотности с использованием обменно-корреляционного функционала B3LYP и двухэкспонентного базиса гауссовых атомных функций (6-31G), дополненного поляризационными функциями на всех атомах функциональных групп (6-31G(d,p)). Такой подход обеспечивает корректное описание молекулярных систем, построенных из атомов элементов первого и второго периодов, в том числе при наличии межфрагментных водородных связей. Расчеты были выполнены с помощью программных пакетов Firefly [25] и GAMESS US [26] с использованием вычислительных ресурсов суперкомпьютерного комплекса Ломоносов Московского государственного университета [27] и визуализированы с помощью программного пакета Chemcraft [28].
Оксид графита был получен в соответствии с методикой, предложенной в [6]. Чистый графит в количестве 2 г был внесен в смесь 9 : 1 концентрированных серной (240 мл H2SO4, 95%) и фосфорной (26 мл H3PO4, 85%) кислот. Затем, небольшими порциями к полученной смеси добавляли перманганат калия, общим количеством 12 г. Процесс длился 24 ч. Продукт промывали водой при центрифугировании на скорости 10 000 оборотов в минуту и сушили при пониженном давлении. Выделенный оксид графита представлял собой зеленоватый желто-коричневый слоистый материал.
Согласно анализу методом РФС, образец содержит (ат. %) 67.9 С, 31.3 O, 0.4 N, 0.3 S, 0.1 Mn и характеризуется соотношением C/O = 2.17, что отвечает достаточно большой глубине окисления. ИК-спектр поглощения данного образца был зарегистрирован с помощью спектрометра с фурье-преобразованием Nicolet IR200 (Thermo Scientific) в режиме внутреннего отражения (KBr) при угле падения 45° и общем числе сканирований 20 с разрешением 4 см–1. Спектр представлен на рис. 1.

В зарегистрированном спектре оксида графита присутствуют все три типичные области поглощения: высокочастотная (3600–2400 см–1), средняя (с четкими пиками при 1733 и 1615 см–1) и характеристичная, в которой наиболее четкие пики при 1367, 1225, 1082 и 984 см–1. Природа пиков в средней и характеристичной областях и представляла основной интерес.
НЕЭМПИРИЧЕСКОЕ МОДЕЛИРОВАНИЕ ФРАГМЕНТОВ ОКСИДА ГРАФИТА
Квантово-химические расчеты были выполнены для модельных фрагментов оксида графита, построенных на основании экспериментальных данных о составе образцов (соотношении C/O в них) в предположении наличия гидроксильных, эпоксидных, кето- и карбоксильных групп, а также молекул воды.
В простейшей модели C54H18O3 (рис. 2), где базовый графитовый фрагмент C54 был модифицирован введением трех эпоксидных групп в центральной части, геометрические параметры этих групп –C–O–C– оказались близки к таковым для типичных небольших эпоксидов: при расстоянии между ядрами углерода, чуть меньшем длины одинарной связи (1.53–1.54 Å), два одинаковых в каждом треугольнике расстояния C–O составили в среднем 1.463 Å. В спектре этой модельной системы все линии имеют сравнительно небольшую интенсивность, и наиболее заметны сигналы при 1158, 1200, 1340 и 1661 см–1. Заметим сразу, что в области 980 см–1, где в экспериментальном спектре виден достаточно интенсивный пик, часто соотносимый именно с эпоксидными группами, предсказываемая вероятность поглощения модельной системы минимальна. Сигнал при 1158 см–1 соответствует дышащим колебаниям (в основном не окисленного) углеродного каркаса, при котором искажаются его шестиугольные кольца, и связанным с ними аналогичным искажениям эпоксидных треугольников. Собственно вызванные искажением каркаса деформационные колебания эпоксидов и обусловливают заметную интенсивность сигнала благодаря смещениям ядер кислорода, приводящим к изменению дипольного момента структуры. Сходна природа и у сигналов при 1200 и 1340 см–1. Первый отвечает таким искажениям углеродного каркаса, которые инициируют качания присоединенных к нему эпоксидных групп. Второй соответствует искажению каркаса, коррелирующему с асимметричной деформацией эпоксидных треугольников. Но в силу преимущественно тангенциального по отношению к поверхности смещения ядер кислорода в обоих случаях результирующая интенсивность сигналов заметно меньше, чем у пика 1158 см–1. Наконец, сигнал при 1661 см–1 отвечает исключительно валентным колебаниям изолированных С–С-связей. Заметим, что из всех перечисленных сигналов, только пик при 1340 см–1 попадает в окрестность экспериментального сигнала заметной интенсивности, и все пики, включая этот, почти на порядок слабее, чем экспериментальные в средней и характеристичной областях спектра. Это значит, что отдельные эпоксидные группы, модифицирующие графитовую плоскость, не могут приводить к появлению характерных линий в спектре поглощения оксида графита.
Рис. 2.
Структура модельной системы C54H18O3 и ее рассчитанный спектр поглощения в диапазоне 2000–500 см–1.

Еще одним, в некотором смысле предельным вариантом модельной структуры, является графитовый фрагмент, модифицированный исключительно гидроксильными группами, причем так, что группы попарно расположены над и под его плоскостью, и с каждой стороны от плоскости удалены друг от друга настолько, что не образуют водородные связи между собой, за единственным исключением (С54H18(OH)8, рис. 3). Если две ОН группы присоединены к соседним ядрам углерода, будучи расположены по разные стороны от углеродной плоскости, они имеют одинаковые геометрические параметры: расстояния С–O составляют 1.46 Å, а расстояния О–Н – 0.97 Å. В тех ситуациях, когда ОН-группа либо удалена на большее расстояние от соседних, либо, наоборот, вовлечена в водородную связь с ближайшей к ней группой, ее конфигурация более искажена: расстояние С–О увеличено до 1.49–1.50 Å, что неизбежно сказывается на частоте валентных колебаний.
Рис. 3.
Структура модельной системы C54H18(OH)8 и ее рассчитанный спектр поглощения в диапазоне 2000–500 см–1.

Представляется вполне ожидаемым, что при наличии гидроксильных групп в той области модельного спектра, где проявляются их колебания, интенсивности сигналов существенно выше: по сравнению с системой C54H18O3 они возрастают практически на полпорядка. Появляется интенсивный пик при 987–942 см–1, наполовину меньшей интегральной интенсивности при 1176 см–1, наиболее интенсивный сигнал при 1372 см–1 и, наконец, слабый пик (сравнимый по интенсивности со спектром системы, содержащей эпоксиды) при 1596 см–1. Заметим, что последний пик и по природе такой же, как сигнал в средней области системы C54H18O3, т.е. относится к колебаниям отдельных связей C=C, причем, как и в указанной системе, его интенсивность заметно ниже той, что характерна для экспериментального спектра, что служит подтверждением гипотезы о том, что полоса в средней частотной области спектра может быть обусловлена, в том числе, и валентными колебаниями С=С-связей, но их вклад не является определяющим. Сигналы же в характеристичной области имеют следующую природу. Наиболее интенсивный сигнал при 1372 см–1 отвечает дышащим колебаниям углеродного каркаса и сильно связанным с ними деформационным колебаниям гидроксильных групп, которые и обусловливают большое изменение дипольного момента и, как следствие, высокую вероятность соответствующего колебательного возбуждения. Наконец, близкий к нему по интегральной интенсивности сигнал в области 987–942 см–1 тоже обусловлен деформационными колебаниями гидроксильных групп, но в данном случае сильно связанных с локальными искажениями пирамидальных фрагментов поверхности С4.
Как видим, относимый к эпоксидам экспериментальный пик поглощения в окрестности 980 см–1 может иметь совершенно иную природу, а потому его наличие никак не может рассматриваться как свидетельство присутствия в структуре оксида графита значительного количества эпоксидных групп. В экспериментальную полосу с пиком 1367 см–1 тоже существенный вклад могут вносить возбуждения колебаний именно гидроксильных групп, когда их согласованной деформации способствует подходящее искажение расположенных между ними участков углеродной поверхности.
При включении в модельную систему функциональных групп обоих типов, т.е. гидроксильных и эпоксидных, результат (спектр поглощения) существенно зависит от того, в какой степени эти группы взаимодействуют. Система С54H18O3(OH)6 (рис. 4) была построена так, что все эпоксидные и гидроксильные группы были удалены друг от друга, причем три гидроксильные группы были расположены на границе углеродного фрагмента, а три – в его внутренней части; два эпоксидных треугольника были расположены на общих ребрах периферийных углеродных шестиугольников, а один – на общем ребре шестиугольников внешнего и внутреннего колец. Примечательно, что первые две эпоксидные группы сильно асимметричны: одно расстояние C–O равно 1.45 Å, а второе составляет 1.48–1.51 Å, что сближает их по виду с предполагавшимися в модели [22, 23] депротонированными гидроксильными группами. Третья же –С–О–С-группа более похожа не на эпоксид, а на 1,2-эфир по причине увеличенного расстояния С–С, составляющего 1.56 Å при расстояниях С–О, по-прежнему равных 1.46 Å. В рассчитанном спектре поглощения такой системы есть четыре наиболее интенсивных пика в характеристичной области (1370, 1320, 1037 и 886 см–1) и два пика, существенно меньшей интенсивности, около 1595 и 1171 см–1. Последние, как и в предыдущих модельных системах, отвечают валентным колебаниям отдельных C–C-связей и связанных шестиугольных колец. Пики 1370 и 1320 см–1 обусловлены возбуждением деформационных колебаний соответственно периферийных и внутренних гидроксильных групп, в обоих случаях сильно связанных с асимметричными искажениями пирамид С3 или C4. Возбуждение аналогичного колебания периферийных гидроксильных групп, но связанного с симметричным искажением углеродных пирамид С3 или С4, обусловливает наличие сигналов при 1037 и 886 см–1, которые попадают в область заметного поглощения в случае экспериментального спектра, но смещены в длинноволновую область относительно основных пиков на 50 и 100 см–1. В последнем случае выявляется и небольшой вклад искажений эпоксидных (1,2-эфирных) треугольников, который никак не может определять высокую интегральную интенсивность спектрального сигнала.
Рис. 4.
Структура модельной системы C54H18O3(OH)6 и ее рассчитанный спектр поглощения в диапазоне 2000–500 см–1.

Система С54H8O2(OH)14(H2O) (рис. 5) была построена принципиально иначе. В ней существенно увеличено число гидроксильных групп, причем значительная их часть вовлечена в водородные связи. Из восьми гидроксльных групп, расположенных на периферии углеродного листа, две пары дополнены расположенными рядом кето-группами, а четыре образуют водородно-связанную цепочку. Еще шесть гидроксильных групп локализованы на внутренней части углеродного фрагмента, по три с каждой стороны от его средней плоскости, причем в одном случае они образуют Н-связанный треугольник, а во втором – Н-связанную цепочку, в которую дополнительно включена молекула воды. Рассчитанный спектр этой модельной системы кардинально отличается от обсуждавшихся выше. Интенсивности всех сигналов выше почти на порядок. Наиболее высокими оказываются пики при 1403 и 1340 см–1; вдвое меньше, но тоже существенно больше, чем ранее, интенсивность сигнала 1611 см–1, и еще меньшей, но по-прежнему заметной высоты пик при 1136 см–1. Существенно возросший по величине сигнал при 1611 см–1 обусловлен возбуждением не только валентных колебаний С=С-связей, но и валентными колебаниями кетонов С=О и в меньшей степени деформационными колебаниями гидроксильных групп, непосредственно H-связанных с кетонами.
Рис. 5.
Структура модельной системы C54H8O2(OH)14(H2O) и ее рассчитанный спектр поглощения в диапазоне 2000–500 см–1.
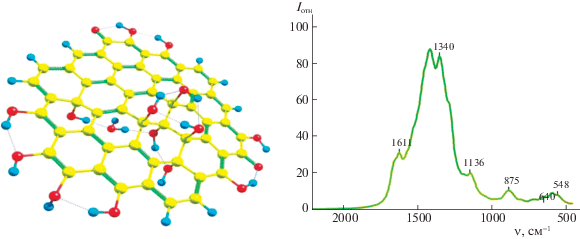
Именно колебания кето- и гидроксильных групп и обусловливают кратно возрастающую интенсивность спектрального отклика благодаря заметным изменениям дипольного момента структуры. Что же касается наиболее интенсивных пиков рассчитанного спектра при 1403 и 1340 см–1, то они оба обусловлены деформационными колебаниями гидроксильных групп, образующих водородные связи с соседями. Так, пик 1403 см–1 отвечает согласованным колебаниями трех расположенных рядом гидроксильных групп, образующих водородно-связанный треугольник во внутренней части структуры. Пик же 1340 см–1 соответствует колебаниям периферийных гидроксильных групп, вовлеченных в водородные связи либо друг с другом, либо с кето-группой. Во всех случаях цепочки водородных связей отвечают критерию возникновения сопряжения [29], когда закономерно чередуются ковалентные (O–H) и водородные (H…O) связи. Такое сопряжение облегчает согласованные смещения протонов водородных связей, повышая вероятность соответствующего возбуждения благодаря кратно возрастающему локальному изменению дипольного момента структуры. Учитывая сравнимую интенсивность линий экспериментального спектра в средней и характеристичной областях и то, что число кето-групп, расположенных преимущественно по границам углеродных листов, должно быть заметно меньше числа гидроксильных групп, приходящихся на единицу их поверхности, кажется обоснованным следующий вывод: в структуре оксида графита относительно маловероятно наличие фрагментов, включающих гидроксильные группы, вовлеченные в сопряженные цепочки водородных связей. В противном случае интенсивность сигналов в характеристичной области должна была быть существенно выше наблюдаемой в эксперименте.
Наконец, для формирования более корректного представления о локализации и взаимном расположении различных кислород-содержащих функциональных групп в образцах оксида графита нами была рассмотрена модельная система С54H10O(OH)5(COOH)5 (рис. 6), которая помимо гидроксильных групп, расположенных как во внутренней части структуры, так и на границе углеродного листа (причем формирующих лишь локальные водородные связи), включала периферийные карбоксильные группы, расположенные либо рядом друг с другом, либо рядом с кето- или гидроксильной группой. В рассчитанном спектре такой системы выделяются следующие пики: 1733 и 1672 см–1 в средней области и 1539, 1448, 1380, 1244 и 1141 см–1 в характеристичной области.
Рис. 6.
Структура модельной системы C54H10O(OH)5(COOH)5 и ее рассчитанный спектр поглощения в диапазоне 2000–500 см–1.
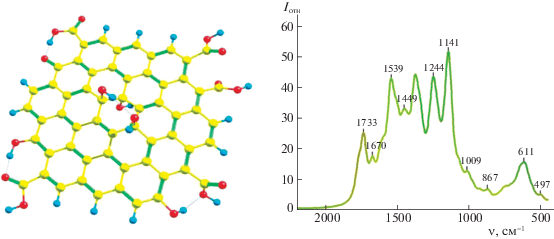
Как и следовало ожидать, в сигналы в средней области спектра заметный вклад вносят колебания карбоксильных групп. При более высокой частоте наблюдается комбинированное колебание карбоксильных групп, в котором растяжение (не вовлеченного в водородные связи) C=O фрагмента взаимодействует с деформацией C–O–H-угла, независимо от того, вовлечен ли OH фрагмент как донор или акцептор в водородные связи с соседними группами или нет. При более низкой частоте (1672 см–1) проявляются опять-таки сильно связанные валентные колебания C=C-связей каркаса и деформационные колебания периферийных гидроксильных групп и карбоксильной группы, в которой C=O-фрагмент как акцептор протона образует связь с соседней гидроксильной группой.
Сигнал в окрестности 1540 см–1 обусловлен деформационными колебаниями Н-связанных периферийных C–O–H-фрагментов либо гидроксильных, либо карбоксильных групп, сильно скоррелированных с искажениями углеродного каркаса. Аналогичную природу имеет менее интенсивный сигнал при 1450 см–1, но он в большей степени обусловлен деформациями гидроксильных групп на внутренней поверхности углеродного листа. Деформации C–O–H-фрагментов карбоксильных групп вносят заметный вклад и в сигнал при 1380 см–1, но в этом случае эти движения сильно связаны не только с делокализованными искажениями углеродного каркаса, но и с локальными растяжениями соответствующих связей C‒C(O)(OH). Наконец, частоты в окрестности 1140 см–1 отвечают дышащим колебаниям углеродных колец C6, когда расширение последовательно соединенных в одну полосу колец сопровождается сжатием аналогичной полосы соседних колец, и такое чередование охватывает практически весь углеродный каркас, инициируя искажение гидроксильных фрагментов на его периферии.
Таким образом, в отсутствие эпоксидных групп и Н-связанных гидроксильных групп в рассчитанном спектре модельной системы отсутствуют пики в области 980–1080 см–1. Включение же в систему карбоксильных групп приводит к появлению более четких сигналов в области 1240–1380 см–1, где проявляется сильная взаимосвязь деформации их C–O–H-фрагментов и более или менее локальных искажений углеродного каркаса, и при 1730 см–1, где растяжение не вовлеченного в водородные связи C=O-фрагмента таких групп сопряжено с изменением соответствующего C–O–H-угла.
ВЫВОДЫ
Квантово-химическое моделирование систем СnHp(O)k(OH)l(COOH)m, имитирующих структуру локальных фрагментов окисленного графитового листа, включающего эпоксидные и гидроксильные группы во внутренней его части и гидроксильные, карбоксильные и кето-группы на границе, позволило прояснить природу большей части сигналов в экспериментальном спектре поглощения оксида графита в его средней и характеристичной областях.
В характеристичной области спектра пик в окрестности 980 см–1 не может быть обусловлен возбуждением колебаний отдельных эпоксидных групп. Заметный вклад в данный сигнал вносят деформационные колебания гидроксильных групп, расположенных на внутренней части углеродного листа, которые сильно сопряжены с искажениями соответствующих C4–пирамид. Вблизи 1100 см–1 может проявляться возбуждение дышащих колебаний не окисленного углеродного каркаса и инициируемое им искажение периферийных гидроксильных групп. В сигнал с пиком 1370 см–1 заметный вклад должны вносить деформационные колебания C–O–H-фрагментов карбоксильных групп, взаимодействующие с валентными колебаниями соответствующих связей C–C(O)(OH) и искажениями углеродного каркаса, как локальными (отдельные C=C-связи), так и охватывающими относительно большие участки не окисленной углеродной поверхности.
В средней области спектра пик 1615 см–1 обусловлен валентными колебаниями связей углерод–углерод, но большая его интенсивность обусловлена сопряженными валентными колебаниями C=O-фрагментов и деформацией Н-связанных с ними гидроксильных групп. В отсутствие карбонильных фрагментов в системе сигнал в данной области спектра имеет весьма малую интенсивность. Интегральное же поглощение при более высоких частотах (пик при 1733 см–1) обеспечивают колебания карбоксильных групп, которые могут быть представлены суперпозицией растяжения C=O-связи и изменения угла C–O–H.
Список литературы
Brodie M.B.C. // Ann. Chim. Phys. 1855. V. 45. P. 351.
Staudenmaier L. // Ber. Dtsch. Chem. Ges. 1898. V. 31. № 2. P. 1481.
Hummers W.S., Offeman R.E. // J. Am. Chem. Soc. 1958. V. 8. № 6. P. 1339.
Graphene Oxide: Fundamentals and Applications. Dimiev A.M., Eigler S., Eds., 2017. United Kingdom: John Wiley & Sons.
Charpy G. // C. R. Hebd. Sệances Acad. Sci. 1909. V. 148. № 5. P. 920.
Marcano D.C., Kosynkin D.V., Berlin J.M. et al. // ACS Nano 2010. V. 4. P. 4806.
Boehm H.P., Scholz W. // Liebigs Ann. Chem. 1966. V. 691. № 1. P. 1.
Briggs D., in Handbook of X-ray Photoelectron Spectroscopy. V. 3. Wagner C.D., Riggs W.M., Davis L.E., Moulder J.F., Muilenberg G.E. Perkin-Elmer Corp.: Eden Prairie, 1979.
Hofflund G.B. In Handbook of Surface and Interface Analysis. New York; Marcel Dekker. 1998.
Mermoux M., Chabre Y., Rousseau A. // Carbon. 1991. V. 29. № 3. P. 469.
Lerf A., He H., Forster M., Klinowski J. // J. Phys. Chem. B. 1998. V. 102. № 23. P. 4477.
Gao W., Alemany L.B., Ci L., Ajayan P.M. // Nature Chem. 2009. V. 1. № 5. P. 403.
Casabianca L.B., Shaibat M.A., Cai W.W. et al. // J. Am. Chem. Soc. 2010. V 132. № 16. P. 5672.
Dimiev A.M., Alemany L.B., Tour J.M. // ACS Nano 2013. V. 7. № 1. P. 576.
Shilyaeva E.A., Novakovskaya Yu.V. // Struct. Chem. 2019. V. 30. № 2. P. 583.
Thiele H. // Z. Anorg. Allg. Chem. 1930. V. 190. № 1. P. 145.
Hofmann U., Frenzel A., Csalan E. // Liebigs Ann. Chem. 1934. V. 510. № 1. P. 1.
Hofmann U., Konig E. // Z. Anorg. Allg. Chem. 1937. B. 234. № 4. S. 311.
Ruess G. // Monatsh. Chem. 1947. V. 76. № 3. P. 381.
Clauss A., Plass R., Boehm H.P., Hofmann U. // Z. Anorg. Allg. Chem. 1957. V. 291. № 5–6. P. 205.
Scholz W., Boehm H.P. // Z. Anorg. Allg. Chem. 1969. B. 369. № 3–6. S. 327.
Nakajima T., Mabuchi A., Hagiwara R. // Carbon 1988. V. 26. № 3. P. 357.
Nakajima T., Matsuo Y. // Carbon 1994. V. 32. № 3. P. 469.
Lerf A., He H., Forster M., Klinowski J. // J. Phys. Chem. B. 1998. V. 102. № 23. P. 4477.
Granovsky A.A., Firefly Version 8, www http://classic.chem.msu.su/gran/firefly/index.html.
Schmidt M.W., Baldridge K.K., Boatz J.A. et al. // J. Comput. Chem. 1993. V. 14. P. 1347.
Sadovnichy V., Tikhonravov A., Voevodin Vl., Opanasenko V. “Lomonosov”: Supercomputing at Moscow State University. in Contemporary High Performance Computing: From Petascale toward Exascale, Boca Raton, USA, CRC Press: 2013. P. 283.
Chemcraft − Graphical Software for Visualization of Quantum Chemistry Computations. https://www. chemcraftprog.com
Novakovskaya Yu.V. // Struct. Chem. 2012. V. 23. P. 1253.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии