

Журнал физической химии, 2019, T. 93, № 10, стр. 1556-1563
Влияние кобальта на каталитические свойства платины в окислении СО: Эксперимент и квантово-химическое моделирование
Д. А. Пичугина a, *, Н. А. Никитина a, Н. Е. Кузьменко a, Д. И. Потемкин b, c
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
b Российская академия наук, Сибирское отделение, Институт катализа имени Г.К. Борескова
Новосибирск, Россия
c Новосибирский государственный университет
Новосибирск, Россия
* E-mail: daria@phys.chem.msu.ru
Поступила в редакцию 15.03.2019
После доработки 15.03.2019
Принята к публикации 09.04.2019
Аннотация
Каталитические свойства Pt и Pt–Co в окислении СО изучены экспериментально и квантово-химически. Моделированием методом функционала плотности показано, что окисление СО на Pt13 реализуется через стадию диссоциативной адсорбции кислорода. Рассчитана энергия активации диссоциации О2 на Pt13, равная 56 кДж/моль. Установлено, что при введении в состав кластера атома кобальта энергии активации стадий окисления СО и диссоциации О2 уменьшаются. Данными проведенных каталитических испытания на нанопорошках Pt и Pt0.5Co0.5 подтверждена высокая активность системы Pt–Co в окислении СО.
Квантово-химическое моделирование – современный метод исследования и способ прогнозирования свойств переходных металлов в каталитических реакциях, имеющих важное практическое значение [1, 2]. Например, методом функционала плотности был предсказан новый состав биметаллического катализатора на основе Co–Mo в синтезе аммиака [3]. Основное преимущество квантово-химического моделирования каталитических процессов – возможность изучать механизмы реакций с участием наночастиц переходных металлов, являющихся компонентами перспективных гетерогенных катализаторов, что позволяет избежать применения дорогостоящего экспериментального оборудования.
Важная задача – разработка низкотемпературных катализаторов окисления СО. Традиционно в этом процессе применяют платину, включая различные ее формы: наночастицы, атомные кластеры и изолированные атомы, стабилизированные на поверхности оксидного носителя [4–7]. Для окисления СО на платине известны различные механизмы: Ленгмюра–Хиншельвуда, Марса–Ван Кревелена, Или–Ридила [8]. Механизм Ленгмюра–Хиншельвуда включает стадии адсорбции СО и О2 (молекулярной или диссоциативной) на активном центре катализатора:
Образование СО2 осуществляется через карбонатный или пероксидный интермедиаты:
(3)
${\text{CO}}{\kern 1pt} {\text{*}} + {\text{O}}_{2}^{*}\;\xrightarrow{{{\text{Pt}}}}\;{\text{OOCO}}{\kern 1pt} {\text{*}}\;\xrightarrow{{{\text{Pt}}}}\;{\text{C}}{{{\text{O}}}_{2}} + {\text{O}}{\kern 1pt} {\text{*}},$(4)
${\text{CO}}{\kern 1pt} {\text{*}} + 2{\text{O}}{\kern 1pt} {\text{*}}\;\xrightarrow{{{\text{Pt}}}}\;{\text{OC(O)O}}{\kern 1pt} {\text{*}}\;\xrightarrow{{{\text{Pt}}}}\;{\text{C}}{{{\text{O}}}_{2}} + {\text{O}}{\kern 1pt} {\text{*}}.$Механизм окисления СО на платине определяется многими факторами: размером и формой частицы, типом носителя и др. [9]. Один из многообещающих способов влияния на каталитическую активность платины – введение в состав катализатора другого металла: железа, никеля, кобальта, меди и др. [10–14]. Установлено, что наночастицы Pt–M с упорядоченной структурой улучшают каталитические свойства [15]. В биметаллическом катализаторе происходит формирование особых центров, имеющих определенный состав атомов металлов (геометрический эффект) и заряд, возникающий вследствие перераспределения электронной плотности между двумя металлами (электронный эффект) [14, 16].
В данной работе представлены результаты квантово-химического исследования механизма окисления СО на Pt13 и Pt12Co, а также результаты расчета структуры кластеров, изучения взаимодействия с О2 и СО, моделирования окисления. Полученная теоретическая информация сопоставлена с данными экспериментальных исследований каталитических свойств нанопорошков Pt и Pt0.5Co0.5.
ЭКСПЕРИМЕНТАЛЬНАЯ И ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
Оптимизацию структуры и расчет энергии кластеров Pt13 и Pt12Co и их комплексов с О2 и СО проводили методом функционала плотности с функционалом РВЕ (Perdew–Burke–Ernzerhof) [17], использовали базисный набор и псевдопотенциалы SBKJC [18]. Вклад энергий нулевых колебаний определяли на основе частот колебаний, рассчитанных в гармоническом приближении. С помощью алгоритма оптимизации Берни [19] проводили локализацию структуры переходных состояний (ПС) и рассчитывали энергии активации (Еа) в стадиях окисления СО. Идентификацию ПС осуществляли методом IRC, включающим исследование зависимости энергии реакционной системы от координаты реакции [20]. Вычисления выполняли с использованием программы ПРИРОДА [21] на суперкомпьютере МГУ “Ломоносов” [22].
Окисление СО исследовали на нанопорошках Pt и Pt0.5Co0.5 (неупорядоченный твердый раствор), которые получали разложением в восстановительной атмосфере комплексных соединений [Pt(NH3)4](NO3)2 · 2H2O и [Pt(NH3)4][Co (C2O4)2(H2O)2] · 2H2O соответственно [23]. Выбор в качестве образцов нанопорошков обусловлен легкостью контролирования структуры и отсутствием влияния носителя.
Полученные монофазные нанопорошки Pt и Pt0.5Co0.5 детально охарактеризованы в [23, 24]. Дополнительно методом хемосорбции СО измеряли площадь поверхности образцов, которая составила 10 ± 2 м2/г.
Каталитические свойства в реакции окисления СО исследовали в проточном U-образном кварцевом реакторе (внутренний диаметр 3 мм) при атмосферном давлении и загрузке катализатора в реактор 25 мг (фракция ≤0.1 мм) [25]. Температуру измеряли хромель-алюмелевой термопарой, помещенной в центр слоя катализатора.
Каталитические эксперименты проводили в смеси состава, об. %: 1.0 CO, 1.0 O2 и Hе-баланс; при скорости подачи реакционной смеси 80 000 см3 г–1 ч–1. Состав и концентрации компонентов газовой фазы до и после реактора определяли при помощи хроматографа “ХРОМОС ГХ-1000”. Перед каждым экспериментом нанопорошки восстанавливали в течение 1 ч в Н2 при 200°С. Затем катализаторы выдерживали 15 мин в реакционной смеси при 25°С, после чего каталитические свойства определяли в нескольких циклах подъема/снижения температуры. Каталитические характеристики Pt были стабильны в течение не менее 20 ч в нескольких циклах повышения/снижения температуры. На Pt0.5Co0.5 наблюдалась медленная обратимая (путем повторного восстановления) дезактивация, вероятно, связанная с сегрегацией оксида кобальта на поверхности. Подробное исследование этого феномена выходит за рамки данной работы. Для сопоставления использовали характеристики Pt0.5Co0.5, полученные в первом цикле подъема/снижения температуры.
Протекание реакции окисления СО характеризовали конверсией СО (XCO), которую вычисляли по следующему выражению:
где [CO]вход и [CO]выход – концентрация CO на входе и выходе соответственно.ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Квантово-химическое исследование строения и каталитических свойств кластеров Pt13 и Pt12Co в окислении СО
На первом этапе проведен расчет структуры кластера Pt13. Несмотря на то, что данный кластер получен экспериментально [26], результаты расчета его строения квантово-химическими методами противоречивы [27–32]. Было рассмотрено шесть изомеров Pt13 в триплетном электронном состоянии. Оптимизированные структуры кластеров и рассчитанные энергии (относительно энергии наиболее устойчивого изомера 1 приведены на рис. 1. Изомер 1 представляет собой искаженную пирамиду, основание которой образовано восемью атомами платины. Подобное строение для Pt13 предсказано ранее [31, 32]. Следует отметить, что изомеры 2–4 незначительно отличаются по энергии (не более, чем на 18 кДж/моль) от изомера 1, что свидетельствует о возможной изомеризации кластера. Известно, что подобные структурно-нежесткие свойства частиц способствуют как активации реагентов, так и их дальнейшему превращению при каталитических процессах [33, 34].
Рис. 1.
Оптимизированные структуры кластеров Pt13 и Pt12Co. В скобках указаны относительные энергии изомеров в кДж/моль. Атом кобальта показан светло-серым цветом.
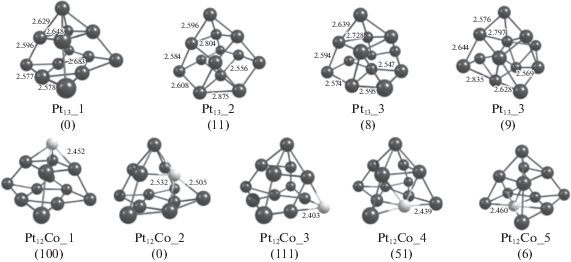
Был проведен расчет структуры и энергии биметаллических кластеров Pt12Со, различающихся расположением атома Co (рис. 1). Наиболее стабилен по энергии кластер Pt12Со_2, в котором кобальт занимает позицию на ребре. Можно отметить сохранение исходного строения монометаллического кластера, однако мультиплетность Pt12Со увеличивается до восьми. Этот факт отмечен в работе [35], где показано, что введение кобальта в состав кластера платины приводит к увеличению мультиплетности и изменению магнитных свойств частицы.
Далее было проведено моделирование окисления СО на Pt13 и Pt12Co_2 (далее обозначенный как Pt12Co). При взаимодействии Pt13 с О2 образуются комплексы супероксидного Pt13O2 и пероксидного Pt12OOPt типов (рис. 2). Согласно рассчитанным изменениям энергии при взаимодействии кластеров с кислородом (табл. 1), наиболее выгодно образование кластера Pt12OOPt, в котором мостиковая координация О2 осуществляется при участии вершинного и реберного атома платины. Было найдено переходное состояние разрыва связи О–О в Pt12OOPt. Рассчитанное значение энергии активации диссоциации О2 на Pt13 составило 56 кДж/моль.

Таблица 1.
Изменение энергии (ΔЕ, кДж/моль) стадий окисления СО на Pt13 и Pt12Co
| Стадия | Pt13 | Pt12Co |
|---|---|---|
| M + O2 → MO2 | –72 | – |
| M + O2 → M12OOM | –126 | –156 |
| М12OOМ → OМ13O | –20 | –34 |
| (56) | (29) | |
| М13 + CO → М13СO | –154 | –199 |
| М13O2 + СО → М13O2СО | –172 | – |
| М12OOМ + СО → М12OOМCO | –152 | –188 |
| М13СO + О2 → М12(СО)OOМ | –179 | –149 |
При взаимодействии кластера Pt12Co с О2 образуется комплекс CoPt11OOPt пероксидного типа, в котором кислород координирован по атомам платины (рис. 2). Диссоциация кислорода в CoPt11OOPt проходит через невысокий активационный барьер (29 кДж/моль), меньший, чем в Pt12OOPt.
Молекула СО взаимодействует с Pt13 и образуется стабильный комплекс Pt13СO. Среди рассмотренных 13 различных способов координации СО на Pt12Co наиболее выгодно образование комплекса CoPt12CO, в котором молекула СО связана с атомом платины на ребре.
При активации О2 и СО на Pt13 обнаружен эффект соадсорбции: присоединение СО к окисленным кластерам Pt13O2, Pt12OОPt более выгодно по энергии, чем к Pt13, а присоединение О2 к Pt13СO и более выгодно, чем к Pt13 (табл. 1). Эффект соадсорбции О2 и СО на Pt12Co не зафиксирован. Таким образом, можно предположить, что на первом этапе СО координируется на Pt13 с низким координационным числом, а затем происходит связывание О2 на центре “ребро–вершина” в пероксидной форме. Дальнейшее окисление CO рассмотрено из двух интермедиатов Pt13O2СО и Pt12(CО)OOPt.
Энергетическая диаграмма многостадийного окисления СО на Pt13 представлена на рис. 3. Первая стадия образования пероксидного интермедиата IM1 имеет наиболее высокую энергию активации. В этой связи для окисления СО на Pt13 с большей вероятностью будет реализовываться механизм через IM1' или механизм, включающий диссоциацию О2:
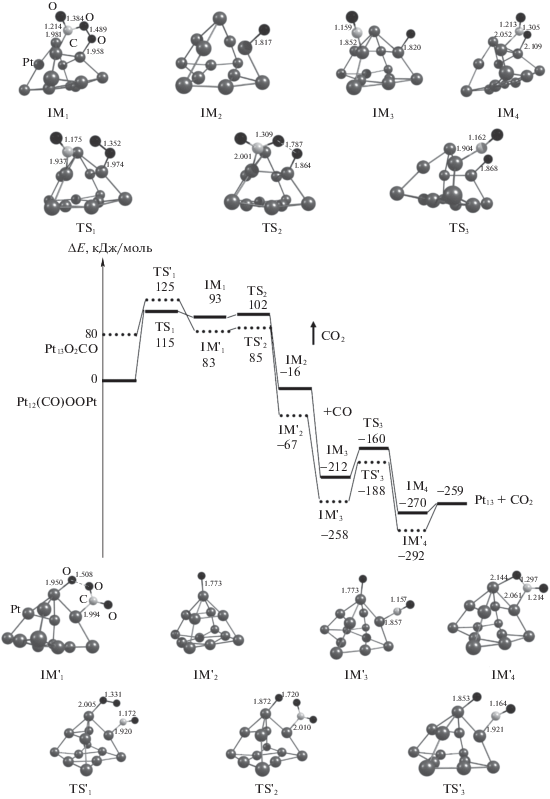
Энергетическая диаграмма окисления СО на Pt12Co из двух интермедиатов CoPt11(СO)OOPt и CoPt11OOPtCO представлена на рис. 4. В отличие от Pt13 процесс проходит не через карбонатный интермедиат, а через диссоциацию О2. Энергия активации первой стадии окисления СО значительно снижается по сравнению с Pt13. Путь окисления СО из CoPt11(СO)OOPt более кинетически и термодинамически благоприятен, чем из CoPt11OOPtCO. Можно отметить, что все стадии окисления СО на Pt12Co имеют невысокие энергии активации и могут протекать при невысокой температуре. Сравнивая рассчитанные величины для стадий окисления СО на Pt12Co и Pt13, можно сделать вывод об активности Pt12Co. Примечательно, что атом кобальта не входит в состав активного центра кластера: окисление СО на биметаллическом кластере проходит только на атомах платины. По-видимому, роль кобальта заключается в изменении электронных свойств кластера, в результате чего реализуется альтернативный более благоприятный механизм окисления.

Каталитические свойства моно- и биметаллических нанопорошков, содержащих платину и кобальт, в окислении СО
Проведено сравнение каталитических свойств нанопорошка Pt и неупорядоченного твердого раствора Pt0.5Co0.5. На рис. 5 представлены температурные зависимости конверсии СО на образцах Pt и Pt0.5Co0.5. Видно, что с увеличением температуры конверсия СО возрастает, достигает 100% и остается постоянной вследствие отсутствия побочных реакций. Важно отметить, что на Pt окисление начинается при 150°С, полная конверсия достигается при 194°С, а на Pt0.5Co0.5 окисление СО проходит при более низкой температуре (25°С), полная конверсия достигается при 93°С. Таким образом, Pt0.5Co0.5 более активен в окислении СО, чем Pt. Низкую активность монометаллической Pt в окислении СО при низких температурах обычно связывают с плотным покрытием поверхности Pt адсорбированными молекулами СО, которые препятствуют адсорбции и активации кислорода [23–25]. Предполагается, что для биметаллических Pt–M (M = Cu, Fe, Co, Ni и др.) появляется дополнительный низкотемпературный маршрут окисления СО, который реализуется при значительном покрытии поверхности металлов адсорбированными молекулами СО [10–14], отмечается важность “геометрического” эффекта [23, 24]. Подобный низкотемпературный маршрут окисления СО на Pt12Co, рассчитанный нами, подтверждает эту гипотезу. Проведенный в настоящей работе квантово-химический расчет показывает, что биметаллические системы Pt–Co активнее монометаллической Pt в окислении СО даже в условиях слабых покрытий и свободной поверхности.
Рис. 5.
Температурные зависимости конверсии СО при полном окислении СО на нанопорошках Pt (1) и Pt0.5Co0.5 (2).

Таким образом, при квантово-химическом моделировании окисления СО на кластере Pt13 и Pt12Co, включающем расчет структуры кластеров, расчет изменения энергии при их взаимодействии с участниками реакции О2 и СО, расчет энергии активации и изменения энергии в основных стадиях окисления СО, показано, что биметаллический кластер Pt12Co обладает лучшими каталитическими свойствами, чем Pt13. Увеличение каталитической активности достигается не геометрическим, а электронным эффектом, который заключается в изменении электронных свойств кластера платины при введении в его состав кобальта. Экспериментальные исследования окисления СО на наночастицах Pt и биметаллическом неупорядоченном твердом растворе Pt0.5Co0.5 подтвердили более высокие каталитические характеристики последнего и наличие низкотемпературной активности.
Работа выполнена с использованием оборудования Центра коллективного пользования высокопроизводительных вычислительных ресурсов МГУ имени М.В. Ломоносова. Экспериментальные исследования выполнены при поддержке государственного задания Института катализа СО РАН (проект АААА-А17-117041710088-0).
Список литературы
Hammami R., Dhouib A., Fernandez S. et al. // Catal. Today. 2008. V. 139. P. 227.
Weijing D., Weihong Z., Xiaodong Z. et al. // Energy Procedia. 2018. V. 152. P. 997.
Jacobsen C.J.H., Dahl S., Clausen B.S. et al. // J. Am. Chem. Soc. 2001. V. 123. P. 8404.
Yu W., Porosoff M.D., Chen J.G. // Chem. Rev. 2012. V. 112. P. 5780.
van Spronsen M.A., Frenkenb J.W.M., Groot I.M.N. // Chem. Soc. Rev. 2017. V. 46. P. 4347.
Cisternas J., Holmes P., Kevrekidis I.G. et al. // J. Chem. Phys. 2003. V. 118. P. 3312.
Śmiechowicz I., Kocemba I., Rogowski J. et al. // React. Kinet. Mech. Cat. 2018. V. 124. № 2. P. 633.
Baxter R.J., Hu P.J. // Chem. Phys. 2002. V. 116. P. 4379.
Merki D., Hu X. // Energy Environ. Sci. 2011. V. 4. P. 3878.
Komatsu T., Tamura A. // J. Catal. 2008. V. 258. P. 306.
Fu Q., Li W. X., Yao Y. X. et al. // Science. 2010. V. 328. P. 1141.
Zhang C., Lv W., Yang Q. et al. // Appl. Surf. Sci. 2012. V. 258. P. 7795.
Mu R., Fu Q., Xu H. et al. // J. Am. Chem. Soc. 2011. V. 133. P. 1978.
Sato K., Ito A., Tomonaga H. et al. // Chempluschem. 2019. B пeчaти. https://doi.org/10.1002/cplu.201800542
Xu D., Liu Z., Yang H. et al. // Angew. Chem. Int. Edit. 2009. V. 48. P. 4217.
Singh J., Nelson R.C., Vicente B.C. et al. // Phys. Chem. Chem. Phys. 2010. V. 12. P. 5668.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865.
Stevens W.J., Krauss M., Basch H. et al. // Can. J. Chem. 1992. V. 70. № 2. P. 612.
Schlegel H.B. // J. Comput. Chem. 1982. V. 3. № 2. P. 214.
Gonzalez C., Schlegel H.B. // J. Chem. Phys. 1989. V. 90. № 4. P. 2154.
Лайков Д.Н., Устынюк Ю.А. // Изв. РАН. Сер. хим. 2005. № 3. С. 804.
Sadovnichy V., Tikhonravov A., Voevodin Vl., Opanasenko V. // Contemporary High Performance Computing: From Petascale toward Exascale, CRC Press, Chapman & Hall/CRC Computational Science, Boca Raton, USA, 2013. P. 283.
Potemkin D.I., Filatov E. Yu., Zadesenets A.V. et al. // Catal. Commun. 2017. V. 100. P. 232.
Потемкин Д.И., Сапарбаев Э.С., Задесенец А.В. и др. // Катализ в промышленности. 2017. Т. 17. № 5. С. 383. [Potemkin D.I., Saparbaev E.S., Zadesenets A.V. et al. // Catal. Ind. 2018. V. 10. P. 62.]
Potemkin D.I., Konishcheva M.V., Zadesenets A.V. et al. // Kinet. Catal. 2018. V. 59. P. 514.
Imaoka T., Kitazawa H., Chun W.-J. et al. // J. Am. Chem. Soc. 2013. V. 135. P. 13089.
Watari N., Ohnishi S. // Phys. Rev. B. 1998. V. 58. P. 1665.
Sun J., Xie X., Cao B. et al. // Comput. Theor. Chem. 2017. V. 1107. P. 127.
Kumar V., Kawazoe Y. // Phys. Rev. B. 2008. V. 77. P. 205418.
Sun Y., Zhang M., Fournier R. // Phys. Rev. B. 2008. V. 77. P. 075435.
Zhang M., R. Fournier // Phys. Rev. A. 2009. V. 79. P. 043203.
Rodríguez-Kessler P.L., Rodríguez-Domínguez A.R. // J. Chem. Phys. 2015. V. 143. P. 184312.
Compton O.C., Osterloh F.E. // J. Am. Chem. Soc. 2007. V. 129. № 25. P. 7793.
Wallace R. // Comptes Rendus. Chimie 2011. V. 14. № 12. P. 1117.
Aguilera-Granja F., Longo R.C., Gallego L.J. et al. // J. Chem. Phys. 2010. V. 132. P. 184507.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии