

Журнал физической химии, 2019, T. 93, № 11, стр. 1731-1736
Способы определения “мертвого” времени для газохроматографической системы
В. Е. Ширяева a, Т. П. Попова a, А. Ю. Канатьева a, А. А. Королев a, А. А. Курганов a, *
a Российская академия наук, Институт нефтехимического синтеза им. А.В. Топчиева
119991 Москва, Россия
* E-mail: kurganov@ips.ac.ru
Поступила в редакцию 08.01.2019
После доработки 08.01.2019
Принята к публикации 19.02.2019
Аннотация
Рассмотрены различные методы определения мертвого времени системы в газовой хроматографии. Показано, что мертвое время, находимое из аэродинамических зависимостей оказывается минимальным, но не учитывает вклад экстраколоночных эффектов. Мертвое время находимое экспериментально по времени удерживания “неудерживаемого” сорбата – метана, оказывается максимальным и зависит от интенсивности взаимодействия сорбат–стационарная фаза. Мертвое время, находимое из чисто эмпирической корреляционной зависимости логарифма чистого времени удерживания гомологов от числа атомов углерода занимает промежуточное положение между теоретическим и экспериментальным значениями. Показано, что термодинамически обоснованной является корреляция числа атомов углерода в гомологическом ряду не с чистым, а с относительным временем удерживания сорбатов и предложены соответствующие корреляционные соотношения, позволяющие находить не только мертвое время системы, но и такие параметры как фазовое соотношение и изменение свободной энергии метиленового звена при сорбции на стационарной фазе.
Как правило, описание результатов любого газохроматографического разделения требует вычисления таких параметров удерживания сорбата, как исправленный удерживаемый объем, чистое время удерживания, фактор удерживания или разделения и т.п. Определение любого из этих параметров неминуемо требует знания так называемого мертвого времени системы, которое, согласно номенклатуре IUPAC, есть “время необходимое подвижной фазе, чтобы пройти через колонку (время пребывания неудерживаемого соединения в хроматографической колонке)” [1]. В согласии с этим определением уже давно в газовой хроматографии был выбран набор соединений, считающихся “неудерживаемыми” на большинстве традиционных стационарных фаз [2]. Это прежде всего воздух и легкие инертные газы, но поскольку эти сорбаты требуют детектор по теплопроводности, широко используется также метан, детектируемый пламенно-ионизационным детектором (ПИД). Однако точность определения мертвого времени с этими сорбатами оказывается недостаточной, например, для априорного вычисления параметров удерживания или идентификации соединений в газовой хроматографии поскольку все эти газы в некоторой степени растворяются в стационарных фазах. В публикации [3] приведено сравнение времени выхода метана с мертвым временем ГХ системы, определенным из времен удерживания гомологического ряда углеводородов С1–С4 для двух типов стационарных фаз: сквалана и динонилфталата. Для обоих фаз время выхода метана превышало мертвое время на 7% и 0.5%, соответственно. Авторы [4] отмечают влияние неточности определения мертвого времени по метану на прямолинейность зависимости логарифма времени удерживания гомологов от числа атомов углерода. Соответственно, были разработаны другие методы для нахождения точной величины tM. Все эти методы основаны на той или иной форме линейного выражения [5]:
где tRi – время удерживания сорбата i, а и b – константы, а ψi – некоторая экстраполируемая функция для сорбата i. В качестве функции ψ наиболее широко использовалось число атомов углерода в серии углеводородных [6] или других [7] гомологов. Однако этот подход неприменим при использовании гомологического ряда инертных газов и для корреляции были привлечены такие физические параметры, как: теплота сорбции [8], поляризуемость [9, 10], потенциал Леннарда–Джонса [11], или потенциал Кирквуда–Мюллера [12].Поскольку построение прямолинейной зависимости, задаваемой уравнением (1), требует apriori знания величины tM, то этот процесс необходимо включает итеративную процедуру. Однако, проведение итеративных вычислений без использования вычислительной техники достаточно трудоемко и было предложено несколько вариантов алгебраического нахождения tM, используя времена удерживания трех последовательных [13] или любых трех [14] членов гомологического ряда. В ряде обзоров [3, 15] представлено детальное сравнение предложенных алгебраических и графических решений с итеративными методами. Авторы пришли к выводу, что итеративные методы более предпочтительны, хотя и требуют большего времени для вычислений. В наши дни, с развитием вычислительной техники, итеративные процедуры включены во все пакеты математических программ, их использование не требует больших затрат времени и сравнение с графическими и алгебраическими методами вычисления tM имеет лишь историческое значение.
Успешная корреляция времени удерживания сорбатов в гомологических рядах подвигла исследователей расширить спектр коррелируемых свойств и уравнение 1 было представлено в более общем виде [16]:
где Φ может быть логарифмом удельного, исправленного или относительного объема, или логарифмом времени удерживания сорбата, или просто индекс удерживания; n – число атомов углерода в молекуле гомолога. Константам a и b в выражении (2), в отличие от выражения (1), приписывается определенный физический смысл [16]: a – это константа, отражающая вклад в удерживание энергии взаимодействия функциональных групп с сорбентом, а b – это константа, характеризующая вклад энергии взаимодействия СН2-группы со стационарной фазой, правда авторы не указывают, как они пришли к такому заключению.В дальнейшем, самые разные хроматографические параметры были скоррелированы с самыми разными физическими параметрами или даже с их комбинациями, и в обзоре [16] приведены десятки подобных эмпирических корреляций. Общее выражение для исследованных корреляций может быть дано следующим выражением:
где Φ – некий хроматографический параметр (индекс Ковача, объем удерживания, время удерживания и т.д.); ψ – некоторое физическое свойство или комбинация физических свойств, с которыми коррелируется хроматографический параметр; a и b – константы. Качество подобных корреляций, по мнению Grajek [17], зависит от того насколько хорошо выполняется линейность, задаваемая уравнением (3). Общей чертой всех корреляций, описываемых выражением (3), является то, что они чисто эмпирические зависимости и справедливы лишь в той области значений параметров, для которой они были проверены. Другая отличительная черта корреляционных соотношений – это несогласованность размерностей правой и левой частей (3). Так, например, в выражении (1), правая часть безразмерна, а левая часть содержит логарифм размерной (время) величины. Равным образом в (2) правая часть имеет размерность энергии, а левая часть, если это индекс Ковача, – безразмерна. Некоторые авторы предлагают выходить из подобной ситуации за счет введения размерных единичных величин, позволяющих сбалансировать размерности в обоих частях соотношения [18]. Однако, вероятно, все же лучше постараться вывести корреляцию, основываясь на известной для данного хроматографического параметра зависимости. Целью данной работы и является вывод термодинамически обоснованного выражения, позволяющего находить мертвое время ГХ-системы и сравнение получаемых при этом результатов с данными традиционно используемых методов.ТЕОРИЯ МЕТОДА
Уравнение, устанавливающее взаимосвязь между фактором удерживания сорбата k и константой распределения сорбата между подвижной и неподвижной фазами (константа Генри К), было получено DeVault для несжимаемой подвижной фазы путем решения масс-балансового уравнения [19]:
где φ = VS/VM – фазовое отношение, т.е. отношение объема стационарной фазы в колонке VS к объему подвижной фазы VM.Позже было показано, что это же выражение справедливо и для сжимаемой подвижной фазы [20, 21] и, используя известное из термодинамики соотношение ΔG = –RTln K, нетрудно установить связь времени удерживания сорбата с изменением свободной энергии ΔG:
(5)
${\text{ln}}({{k}_{i}}{\text{/}}\varphi ) = {\text{ln}}\frac{{{{t}_{{{\text{Ri}}}}} - {{t}_{{\text{M}}}}}}{{{{t}_{{\text{M}}}}}} = - \frac{{\Delta {{G}_{i}}}}{{RT}}.$Если изменение свободной энергии может быть представлено для гомологического ряда сорбатов R-(CH2)n-H в виде линейного соотношения
(6)
$\Delta {{G}_{i}} = \Delta {{G}_{{\text{o}}}} + n\Delta {{G}_{{{\text{C}}{{{\text{H}}}_{{\text{2}}}}}}},$(7)
${{t}_{R}} = {{t}_{{\text{M}}}}(1 + \varphi {\kern 1pt} {\text{*exp}}( - \frac{{{\Delta }{{G}_{o}} + {\Delta }{{G}_{{{\text{C}}{{{\text{H}}}_{2}}}}}{\kern 1pt} {\text{*}}n}}{{RT}}$Уравнение (7) можно представить в линейной форме
(8)
${\text{ln}}\frac{{{{t}_{R}} - {{t}_{{\text{M}}}}}}{{{{t}_{{\text{M}}}}}} = \ln k = \ln \varphi - \frac{{{\Delta }{{G}_{o}} + {\Delta }{{G}_{o}}n}}{{RT}}$Как следует из выражения (8), число метиленовых звеньев в гомологической серии линейно коррелирует с логарифмом фактора удерживания сорбатов, а не с логарифмом чистого времени удерживания, как это предполагалось в корреляции (1). Кроме того, наклон линейной зависимости и отрезок, отсекаемый на оси ординат, – оба эти параметра зависят от температуры, при которой проводилось разделение.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все хроматографические измерения проведены на газовом хроматографе фирмы Shimadzu модель GC-210 с газом-носителем гелием в изотермическом режиме при 70°С. Капиллярная колонка Rtx-1 длиной 30 м и внутренним диаметром 0.25 мм покрытая слоем стационарной фазы SE-30 (полидиметилсилоксан), толщиной 0.25 мкм приобретена у фирмы RESTEK LLC (США). Температура испарителя 250°С, деление потока 1 : 50, детектор ПИД при температуре 250°С.
Стандартная смесь нормальных углеводородов С5-С12 была приготовлена растворением индивидуальных компонентов в хлористом метилене в концентрации от 0.2 до 1.5%.
Экспериментальные значения времени удерживания сорбатов корректировались в соответствии с законом Пуазейля–Дарси для сжимаемого флюида:
(9)
$\begin{gathered} {{t}_{R}} = {{t}_{{\text{M}}}}(k + 1) = \frac{{{{L}^{2}}\eta (k + 1)}}{{{{K}_{V}}}} \times \\ \times \;\frac{{4({{P}^{3}} - 1)}}{{3{{{({{P}^{3}} - 1)}}^{2}}{{p}_{o}}}} = \frac{{{{L}^{2}}\eta (k + 1)}}{{{{K}_{V}}}}\mathcal{H}{\text{,}} \\ \end{gathered} $(10)
$\mathcal{H} = \frac{1}{{j{\kern 1pt} {\kern 1pt} {\text{'*}}({{p}_{i}} - {{p}_{o}})}} = \frac{{4{\kern 1pt} {\text{*(}}{{P}^{3}} - 1{\text{)}}}}{{3{\kern 1pt} {\text{*}}{{{({{P}^{2}} - 1)}}^{2}}{{p}_{o}}}},$ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Теоретическое мертвое время
Время tM, которое газ-носитель затрачивает на прохождение по хроматографической колонке, может быть рассчитано по уравнению Пуазейля–Дарси, с поправкой j', введенной Halash [22] для учета сжимаемости газа:
(11)
$({{p}_{i}} - {{p}_{o}})j{\kern 1pt} ' = \frac{{L\eta }}{{{{K}_{V}}}}\bar {u} = \frac{{{{L}^{2}}\eta }}{{{{K}_{V}}}}\frac{1}{{{{t}_{{\text{M}}}}}}.$Чтобы вычислить мертвое время по формуле (11) $(t_{{\text{M}}}^{{{\text{th}}}})$ необходимо знать проницаемость колонки KV, которая в случае наполненных колонок зависит от плотности упаковки колонки, размера гранул и т.д. и обычно определяется экспериментально. Для полых капиллярных колонок величина KV может быть рассчитана по формуле KV = = $d_{{\text{c}}}^{2}{\text{/32}}$ [22], что позволяет определить теоретическое мертвое время tM капиллярных колонок чисто расчетным путем. Для капиллярной колонки, использованной в данной работе, мертвое время системы, рассчитанное по формуле (9), приведено в табл. 1, и, как видно из таблицы, оно наименьшее по сравнению с величинами tM, найденными другими методами. Следует, однако, отметить, что нам не удалось найти публикации, в которых мертвое время рассчитывалось бы подобным образом при определении термодинамических параметров сорбции. Вероятно, хроматографисты испытывают некоторое недоверие к таким величинам как диаметр колонки, толщина слоя неподвижной фазы и т.д., используемым при расчете теоретического мертвого времени, и предпочитают определять мертвое время системы tM экспериментальными методами. Основная область приложения теоретического мертвого времени $t_{{\text{M}}}^{{{\text{th}}}}$ – это теоретическое моделирование для предсказания и оптимизации времен удерживания в температурно-программируемой ГХ ([23] и ссылки там), поскольку для теоретического мертвого времени однозначно определены такие эффекты как температурная зависимость, влияние природы газа-носителя и т.д.
Таблица 1
. Мертвое время системы (tM, с)
| pi, атм | (11), $t_{{\text{M}}}^{{{\text{th}}}}$ | По метану*, $t_{{\text{M}}}^{{{\text{C}}{{{\text{H}}}_{{\text{4}}}}}}$ | (1)**, $t_{{\text{M}}}^{{\text{1}}}$ | (7)**, $t_{{\text{M}}}^{{\text{7}}}$ | $t_{{\text{M}}}^{{{\text{C}}{{{\text{H}}}_{{\text{4}}}}}}$–$t_{{\text{M}}}^{{{\text{11}}}}$ | $t_{{\text{M}}}^{{\text{7}}}$–$t_{{\text{M}}}^{{{\text{11}}}}$ |
|---|---|---|---|---|---|---|
| 1.3 | 340.1 | 357.0 | 353 | 353 | 16.8 | 12.9 |
| 1.4 | 256.0 | 268.2 | 265 | 265 | 12.2 | 9.0 |
| 1.5 | 205.6 | 215.1 | 213 | 213 | 9.5 | 7.4 |
| 1.6 | 172.1 | 179.8 | 177.8 | 177.8 | 7.7 | 5.7 |
| 1.7 | 148.2 | 154.5 | 152.9 | 152.9 | 6.4 | 4.7 |
| 1.8 | 130.3 | 135.7 | 134.2 | 134.2 | 5.4 | 3.9 |
| 1.9 | 116.3 | 121.0 | 119.7 | 119.7 | 4.6 | 3.3 |
| 2.0 | 105.2 | 109.2 | 108.1 | 108.1 | 4.0 | 2.9 |
| 2.1 | 96.1 | 99.6 | 98.6 | 98.6 | 3.5 | 2.5 |
| 2.2 | 88.5 | 91.6 | 90.7 | 90.7 | 3.1 | 2.2 |
| 2.3 | 82.1 | 84.8 | 84 | 84 | 2.7 | 1.9 |
| 2.4 | 76.6 | 79.0 | 78.2 | 78.2 | 2.4 | 1.6 |
| 2.5 | 71.8 | 74.0 | 73.2 | 73.2 | 2.2 | 1.4 |
| 2.6 | 67.6 | 69.5 | 68.9 | 68.9 | 2.0 | 1.3 |
| 2.8 | 60.6 | 62.2 | 61.6 | 61.6 | 1.6 | 1.0 |
| 3.0 | 55.0 | 56.2 | 55.7 | 55.7 | 1.3 | 0.7 |
Экспериментальное определение мертвого времени
Самый распространенный метод экспериментального определения мертвого времени системы – это измерение времени выхода “неудерживаемого” сорбата, в качестве которого при использовании пламенно-ионизационного детектора обычно применяют метан. Время выхода метана на исследуемой колонке $t_{{\text{M}}}^{{{\text{C}}{{{\text{H}}}_{{\text{4}}}}}}$ приведено в табл. 1, и оно превосходит теоретическое мертвое время $t_{{\text{M}}}^{{{\text{th}}}}$ на 2–5%. Интуитивно, можно было бы ожидать, что эта разница будет увеличиваться с ростом входного давления, но, как видно из табл. 1, она экспоненциально уменьшается с ~17 c до ~1.3 c, когда входное давление pi возрастает с 130 кПа до 300 кПа (рис. 1). В то же время относительное расхождение в величинах мертвого времени, найденных экспериментально и рассчитанных теоретически, уменьшается практически линейно с ростом входного давления на колонке (рис. 2). Это уменьшение можно связать с уменьшением времени вхождения пробы из испарителя в колонку, при возрастании входного давления и, соответственно, скорости потока газа-носителя. Другой фактор, потенциально способный вызвать изменение удерживания метана при возрастании давления газа-носителя, это возрастание конкурентной сорбции газа носителя при возрастании давления, если она в принципе возможна. Дополнительную информацию по этому вопросу можно получить из экстраполяционных методов определения мертвого времени, которые были отмечены выше.
Рис. 1.
Превышение мертвого времени, найденного экспериментально $t_{{\text{M}}}^{{{\text{C}}{{{\text{H}}}_{{\text{4}}}}}}$ (1) (или экстраполяцией по уравнению 7 $t_{{\text{M}}}^{{\text{7}}}$ (2)) над теоретически рассчитанной величиной $t_{{\text{M}}}^{{{\text{th}}}}$, р – относительное давление.
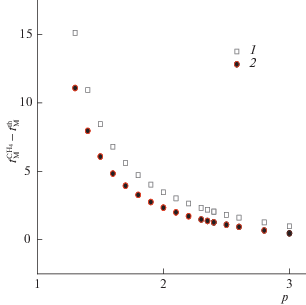
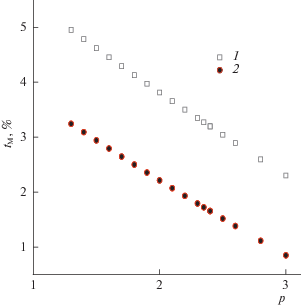
Рис. 2.
Относительное превышение мертвого времени (tM, %), найденного экспериментально $t_{{\text{M}}}^{{{\text{C}}{{{\text{H}}}_{{\text{4}}}}}}$ (1) (или экстраполяцией по уравнению (7) $t_{{\text{M}}}^{{\text{7}}}$ (2)) над теоретически рассчитанной величиной $t_{{\text{M}}}^{{{\text{th}}}}$.
Экстраполяционнные методы определения мертвого времени
Экстраполяционные зависимости, задаваемые уравнениями (1) или (7), предполагают, что нулевому члену гомологического ряда требуется все же больше времени, для того чтобы покинуть колонку, чем мертвое время системы. Нулевой член гомологического ряда углеводородов Н-(СН2)n-Н – это молекула водорода (n = 0), которая крайне слабо взаимодействует с полидиметилсилоксановой стационарной фазой. Соответственно, можно ожидать что мертвое время системы tM, находимое экстраполяционными методами, будет свободно от дополнительного вклада, связанного с возможной сорбцией реперного аналита. Действительно, мертвое время системы tM, получаемое при аппроксимации экспериментальных времен удерживания углеводородов С5–С10 уравнениями (1) и (7) ($t_{{\text{M}}}^{{\text{1}}}$ и $t_{{\text{M}}}^{{\text{7}}}$ соответственно, рис. 3), оказывается меньше, чем время удерживания метана $t_{{\text{M}}}^{{{\text{C}}{{{\text{H}}}_{{\text{4}}}}}}$ (табл. 1). Тем не менее оно превосходит время $t_{{\text{M}}}^{{{\text{th}}}}$ необходимое газу-носителю, чтобы пройти через колонку, и разница между этими величинами экспоненциально уменьшается при возрастании входного давления в колонке (рис. 2). В то же время, относительное расхождение между этими величинами линейно уменьшается с ростом давления (рис. 3). Этот же эффект, как отмечалось выше, наблюдался и для мертвого времени, определенного по метану и, вероятно, он связан с вкладом экстраколоночных эффектов в общее мертвое время, необходимое газу носителю, чтобы пройти по колонке. Несколько неожиданным оказалось то обстоятельство, что мертвое время системы, находимое по уравнениям (1) и (7), оказалось полностью идентичным (табл. 1). Различие между эмпирической моделью, задаваемой уравнением (1), и термодинамической моделью, задаваемой уравнением (7), наблюдалось в величинах констант, входящих в состав уравнений. Обе модели интерпретируют константы, находящиеся под знаком экспоненты, как величины пропорциональные изменению свободной энергии при сорбции начального члена ряда (а в уравнении (1); ΔGo/RT в (7)) или изменению свободной энергии, приходящейся на одну СН2-группу (параметр b в (1), $\Delta {{G}_{{{\text{C}}{{{\text{H}}}_{2}}}}}$/RT в уравнении (7)). Однако, как показала аппроксимация экспериментальных точек, измеренных при разных давлениях, уравнением (1) “константа” a константой не является, а меняется от –0.11 до –1.94 с ростом входного давления от 1.3 до 3 атм. Константа b = +0.762 ± 0.001 практически не зависит от давления, но ее положительное значение плохо согласуется с самопроизвольным характером сорбции, которая обычно происходит с выделением энергии, т.е. коэффициент b должен быть отрицательным.
Рис. 3.
Аппроксимация экспериментальных точек уравнением (1). Сорбаты: углеводороды С5–С10, газ–носитель – гелий, давление на входе в колонку 3 атм, температура колонки 70°С, nс – число атомов углерода.

Определяемые аппроксимацией экспериментальных точек параметры уравнения (7) лишены этих недостатков. Оба параметра в экспоненциальном множителе не зависят от давления. Величина первого из них $\Delta {{G}_{{{\text{C}}{{{\text{H}}}_{2}}}}}$/RT составляет –0.762 ± ± 0.002. Соответственно, изменение свободной энергии, приходящееся на одну СН2-группу составляет примерно –2.2 кДж/моль, что согласуется с данными, приводимыми в литературе [17].
Второй параметр ΔGо, соответствующий изменению свободной энергии при сорбции начального члена ряда оказался равным нулю. Это можно было предполагать заранее, учитывая, что первый член гомологического ряда алканов с n = 0 это водород, который не удерживается стационарной фазой. Предэкспоненциальный параметр уравнения (7) – это фазовое соотношение φ = = VS/VM. Эта константа также не зависит от давления в колонке, а ее величина (3.9 ± 0.2) × 10–3 соответствует толщине пленки неподвижной фазы 0.16 мкм, что заметно меньше величины, сообщенной производителем (0.25 мкм). Обычно толщину пленки неподвижной фазы определяют весовым методом и точность определения оставляет желать лучшего. Так что вопрос о том, какая из двух величин является более правильной, остается пока открытым.
В любом случае впервые представленное уравнение (7), основанное на строгом термодинамическом базисе, позволяет не только определять мертвое время системы, но и получить информацию о фазовом соотношении в колонке и энергетике взаимодействия сорбата со стационарной фазой. Традиционно использовавшееся уравнение (1) не отвечает строгим термодинамическим критериям и может использоваться только при постоянном давлении, поскольку параметр b в (1) зависит от давления. Еще менее надежно, особенно для легких аналитов, определять мертвое время по “неудерживаемому” компоненту. Отождествляя мертвое время ГХ системы с временем выхода “неудерживаемого” компонента экспериментатор может внести заметную ошибку в хроматографические величины, описывающие ГХ разделение, как это уже обсуждалось в обзоре [3] для (1).
Работа выполнена в соответствии с планом научных исследований ИНХС РАН.
Список литературы
Davankov V.A. // Pure and Applied Chemistry. 2001. V. 73. P. 969.
Hilmi A.K. // J. Chromatogr. 1965. V. 17. P. 407.
Wainwright M.S., Haken J.K. // Ibid. 1980. V. 184. P. 1.
Groenendijk H., van Kemenade A.W.C. // Chromatographia. 1968. V. 1. Is. 11–12. P. 472.
Parcher J.F., Selim M.I. // Anal. Chem. 1979. V. 51. P. 2154.
Guardino X., Albaiges J. // J. Chromatogr. 1976. V. 118. P. 13.
Ashes J.R., Mills S.C., Haken J.K. // Ibid. 1978. V. 166. P. 391.
Masukawa S., Kobayashi R.J. // J. Gas Chromatogr. (J. Chromatogr. Sci.) 1968. V. 6. P. 257.
Masukawa S., Kobayashi R. // Ibid. 1968. V. 6. P. 461.
Masukawa S., Alyea J.I., Kobayashi R. // Ibid. 1968. V. 6. P. 266.
Hori Y., Kobayashi R. // J. Chem. Phys. 1971. V. 54. P. 1226.
Nakahara T., Chappelear P.S., Kobayashi R. // Ind. Eng. Chem. Fundam. 1977. V. 16. P. 220.
Peterson M., Hirsch J. // J. Lipid Research. 1959. V. 1. P. 132.
Gold H.J. // Anal. Chem. 1962. V. 34. P. 174.
Smith R.J., Haken J.K., Wainwright M.S. // J. Chromatogr. 1985. V. 334. P. 95.
Набивач В.М., Дмитриков В.П. // Успехи химии. 1993. Т. 62. С. 27.
Grajek H. // J. Chromatogr. A. 2007. V. 1145. P. 1.
Очков В.Ф. “Физические и экономические величины в Mathcad и Maple” (Серия “Диалог с компьютером”). М.: Финансы и статистика, 2002 (http://twt.mpei.ac.ru/ochkov/Units/Forword_book.htm)
De Vault D. // J. Am. Chem. Soc. 1943. V. 65. P. 532
James A.T., Martin A.J.P. // Biochem. J. 1952. V. 50. P. 679.
Kurganov A. // J. Chromatogr. A. 2007. V. 1150. P. 100.
Ettre L.S., Hinshaw J.V. Basic Relationships of Gas Chromatography. Advanstar. Cleveland. USA. 1993.
Blumberg L.M. // J. Chomatogr. 2017. V. 1491. P. 159.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии