

Журнал физической химии, 2019, T. 93, № 12, стр. 1886-1893
Динамический эффект растворителя в реакциях фотоиндуцированного внутримолекулярного протонно-связанного переноса электрона
Т. В. Михайлова a, В. А. Михайлова a, *, А. И. Иванов a
a Волгоградский государственный университет
Волгоград, Россия
* E-mail: mikhailova.va@volsu.ru
Поступила в редакцию 14.02.2019
После доработки 14.03.2019
Принята к публикации 09.04.2019
Аннотация
Исследован динамический эффект растворителя (ДЭР) в реакциях сверхбыстрого протонно-связанного переноса электрона (ПСПЭ), протекающих в неравновесных условиях. В частности, выявлена зависимость эффекта от угла между направлениями координат реакции φ, соответствующих этапам фотовозбуждения системы и формирования продукта. Установлены следующие закономерности, присущие таким реакциям: 1) наибольшая величина ДЭР должна наблюдаться в области сильной экзергоничности реакции, а в области слабой экзергоничности эффект мал; 2) максимум зависимости константы скорости ПСПЭ от величины экзергоничности реакции смещается в область меньших значений с ростом матричного элемента перехода; 3) зависимость эффективного времени реакции от угла φ имеет выраженный минимум, положение которого зависит от энергетических параметров реагентов.
Фотоиндуцированный протонно-связанный перенос электронов (ПСПЭ) – важнейшая стадия широкого спектра процессов преобразования энергии в химических и биологических системах. Понимание основополагающих принципов этого процесса на уровне, позволяющем настраивать и контролировать его сверхбыструю динамику, имеет решающее значение для проектирования возобновляемых и устойчивых источников энергии, таких как устройства для искусственного фотосинтеза и фотоэлектрохимические элементы. В связи с фундаментальностью этих процессов интерес к их всестороннему исследованию как экспериментаторов, так и теоретиков с каждым годом непрерывно растет [1–15].
В работах [6–8] показано, что теория неадибатических переходов, развитая для описания кинетики переноса электрона (ПЭ), может успешно применяться для описания ПСПЭ. Это означает, что многие закономерности, присущие кинетике ПЭ, должны наблюдаться в реакциях ПСПЭ. Один из таких эффектов – зависимость кинетики ПЭ в донорно-акцепторных системах от динамических свойств растворителя – динамический эффект растворителя (ДЭР) [16, 17]. Данный эффект по-разному проявляется в термических и сверхбыстрых неравновесных процессах переноса заряда (ПЗ). В термических реакциях ПЗ он оказывается сильным в нормальной области Маркуса (область слабой экзергоничности реакции), а в инвертированной области Маркуса (область сильной экзергоничности реакции) – ослаблен [18, 19]. В случае сверхбыстрых реакций ПЗ, протекающих в неравновесном режиме, области экзергоничности реакции, где ДЭР сильный и слабый, меняются местами [20]. Следовательно, область проявления ДЭР в реакциях ПЭ может служить надежным маркером режима, в котором они протекают [21]. Другая отличительная особенность реакций ПЭ – влияние электронного матричного элемента ${{V}_{{el}}}$ на величину ДЭР: в неравновесных реакциях с ростом ${{V}_{{el}}}$ происходит значительное ослабление эффекта, а в случае термических реакций – его монотонное возрастание [20].
Естественно ожидать, что динамические свойства полярного растворителя оказывают значительное влияние на кинетику иных реакций ПЗ, например, сверхбыстрого внутримолекулярного ПСПЭ [1–15]. Эта сложная реакция включает несколько стадий и может протекать различными путями. Сверхбыстрый фотоиндуцированный ПСПЭ экспериментально обнаружен в огромном количестве химических соединений [9–12]. Один из примеров – молекулярная система 3-гидроксифлавона (3HF), а также некоторые ее производные [9–12]. В различных молекулярных структурах ПСПЭ протекает в разных последовательностях: в некоторых молекулярных структурах переход в возбужденное состояние под действием оптического импульса накачки сопровождается значительным внутримолекулярным ПЭ, а затем на следующей стадии происходит перенос протона. В других молекулярных соединениях перенос протона предшествует ПЭ [9–12].
Детальные исследования систем, в которых возможен внутримолекулярный ПСПЭ, позволили получить информацию об их физических и энергетических особенностях, а также количественные данные о динамике населенностей возбужденных состояний в средах различной полярности [9–12]. В частности, в работах [9–12] было представлено подробное описание ряда молекулярных соединений, в которых может протекать ПСПЭ, и их свойств. Было зафиксировано несколько состояний, заселяемых в ходе реакции, каждое из которых соответствовало конкретной протонно-электронной конфигурации. Особенность ПСПЭ – его двухстадийность. Описание таких реакций требует введения двух координат реакции, связанных с каждым из этапов реакции. В общем случае координаты реакции не являются независимыми, что отражается в их неортогональности. Угол между направлениями координат реакции в пространстве ядерных степеней свободы служит мерой их корреляции и может влиять на эффективную константу скорости многостадийного процесса [22–26]. Это приводит к влиянию предыдущей стадии на кинетику последующей, т.е. нарушается принцип независимости элементарных реакций Оствальда [27]. Ранее нами была разработана теория ПСПЭ [15], учитывающая эту особенность, в рамках которой удалось корректно воспроизвести кинетику сверхбыстрого внутримолекулярного ПСПЭ и показать, что данный процесс в различных растворителях полностью или частично протекает в неравновесном режиме. Реакции ПСПЭ, как правило, являются сверхбыстрыми, поэтому отмеченные эффекты должны ярко проявляться. Систематические исследования влияния вязкости растворителя на ПСПЭ до сих пор не проводились. Однако имеется информация, показывающая, что переход от ацетонитрила к дихлорметану, имеющих существенно различную вязкость, приводит к заметному изменению кинетики процесса [10, 15].
Основная цель данной статьи – исследование ДЭР в кинетике реакций сверхбыстрого ПСПЭ, протекающих в неравновесных условиях. В частности, выявить зависимость ДЭР от угла между направлениями координат реакции, соответствующих этапам фотовозбуждения системы и стадии формирования продукта ПСПЭ. На примере конкретного молекулярного соединения (4'‑N,N-диэтиламин-3-гидроксифлавона [10]), энергетические параметры которого можно определить из стационарных спектров поглощения и люминесценции, выявить область параметров, где наиболее сильно проявляется данный эффект.
ДВУХСТАДИЙНАЯ МОДЕЛЬ ПСПЭ
В ряде работ Ш. Хаммес–Шиффер и ее коллег [6–8] кинетика ПСПЭ была описана в рамках неадиабатических переходов между несколькими диабатическими состояниями, каждое из которых соответствовало конкретной электронно-протонной конфигурации. Наличие таких состояний подтверждается экспериментальными данными: в стационарных спектрах протонно-электронных систем [10] наблюдалась двойная эмиссия, связанная с переходами из начального возбужденного состояния ($PE{\text{*}}$) и состояния продукта ($PCET{\text{*}}$). Именно это позволяет интерпретировать процесс ПСПЭ в рамках трехуровневой модели в соответствии со схемой [15]:
(1)
$PE\;\xrightarrow{{h\nu }}\;PE{\text{*}}\;\xrightarrow{{{{k}_{{PCET}}}}}\;PCET{\kern 1pt} {\text{*}}{\text{.}}$Рис. 1.
Изолинии поверхностей свободной энергии ${{U}_{j}}$ ($j = 1,2,3$) на координатной плоскости ${{Q}_{1}},{{Q}_{2}}$. Пунктирные линии изображают пересечения термов возбужденного состояния (2) и состояния продукта (3) и их колебательные подуровни. Серой стрелкой схематично показано движение волнового пакета (заштрихованный овал) по терму ${{U}_{2}}$ к его минимуму.

Следует отметить, что введенный угол φ проявляется только в сверхбыстрых процессах, когда вторая стадия протекает параллельно с релаксацией ядерных степеней свободы, порожденной на предыдущем этапе сложной реакции. В результате появляется зависимость кинетики элементарной реакции от характеристик предшествующей стадии, т.е. проявляется нарушение принципа независимости элементарных реакций Оствальда [28, 29], который выполняется для относительно медленных реакций, протекающих в термическом режиме.
Следуя разработанной модели [15], профили поверхностей свободной энергии ${{U}_{j}}$ ($j = 1,2,3$) на координатной плоскости ${{Q}_{1}}$, ${{Q}_{2}}$ для рассматриваемых диабатических состояний реакции выбираются в виде:
(2)
$ + \;\frac{{Q_{2}^{2}}}{2} + \Delta {{G}_{{12}}} + \sum\limits_{\alpha = 1}^N {{{n}_{\alpha }}\hbar {{\Omega }_{\alpha }}} ,$(3)
$E_{{rm}}^{{(23)}} = E_{{rm}}^{{(12)}} + E_{{rm}}^{{(13)}} - 2\sqrt {E_{{rm}}^{{(12)}}E_{{rm}}^{{(13)}}} \cos \theta ,$Релаксация растворителя на каждой стадии характеризуется автокорелляционной функцией $X(t) = \exp \left( { - {t \mathord{\left/ {\vphantom {t {{{\tau }_{L}}}}} \right. \kern-0em} {{{\tau }_{L}}}}} \right)$ с одним эффективным временем релаксации ${{\tau }_{L}}$ [30, 31]. Предлагаемая модель также учитывает реорганизацию внутримолекулярных высокочастотных колебательных мод как на стадии возбуждения молекулярной системы, так и на стадии образования продукта. Предполагается, что в возбужденном состоянии заселен лишь основной колебательный подуровень. Это предположение выполняется, если частота импульса накачки близка к красной границе. Возбужденные колебательные подуровни продукта с квантовыми числами ${{m}_{\alpha }}$ заселяются в процессе релаксации неравновесных переходов 2 → 3, протекающей параллельно с релаксацией растворителя, и быстро релаксируют с константой скорости $1{\text{/}}\tau _{v}^{{({{m}_{\alpha }})}} = {{m}_{\alpha }}{\text{/}}{{\tau }_{v}}$, где $\tau _{v}^{{}}$ – время колебательной релаксации. Здесь используется механизм одноквантовой необратимой колебательной релаксации ${{m}_{\alpha }} \to {{m}_{\alpha }} - 1$ [32, 33]. Ранее было показано [32], что константа скорости переноса заряда слабо зависит от спектральной плотности (набора частот высокочастотных колебаний и их вклада в энергию реорганизации) при условии, что полная энергия реорганизации этих мод ${{E}_{{rv}}} = \sum\nolimits_{\alpha = 1}^M {{{E}_{{rv\alpha }}}} $ постоянна, а их количество N ≥ 5. В модели используется пять высокочастотных внутримолекулярных колебательных мод [32, 33]. В качестве универсальной спектральной плотности будет использован спектр донорно-акцепторного комплекса, состоящего из фенилциклопропана (PhCP) в качестве донора и тетрацианоэтилена (TCNE) в качестве акцептора [33].
Временная эволюция рассматриваемой системы описывается набором дифференциальных уравнений для функций распределения вероятностей возбужденного $\rho _{2}^{{(\vec {n})}}$ и конечного состояний системы $\rho _{3}^{{(\vec {m})}}$ [15, 34–36]:
(4)
$\begin{gathered} + \;\sum\limits_\alpha {\frac{{\rho _{2}^{{(\vec {n}')}}}}{{\tau _{\nu }^{{({{n}_{\alpha }} + 1)}}}}} - \sum\limits_\alpha {\frac{{\rho _{2}^{{(\vec {n})}}}}{{\tau _{\nu }^{{({{n}_{\alpha }})}}}}} , \\ \frac{{\partial \rho _{3}^{{(\vec {m})}}}}{{\partial t}} = {{{\hat {L}}}_{3}}\rho _{3}^{{(\vec {m})}} + \sum\limits_{\vec {n}} {{{k}_{{\vec {n}\vec {m}}}}(\rho _{2}^{{(\vec {n})}} - \rho _{3}^{{(\vec {m})}})} + \\ \end{gathered} $(5)
${{\hat {L}}_{j}} = \frac{1}{{{{\tau }_{L}}}}\sum\limits_{k = 1}^2 {\left( {1 + (Q_{k}^{{}} - Q_{k}^{{(j)}})\frac{\partial }{{\partial Q_{k}^{{}}}} + {{k}_{B}}T\frac{{{{\partial }^{2}}}}{{\partial Q_{k}^{2}}}} \right)} .$Параметры Зусмана ${{k}_{{\vec {n}\vec {m}}}}$ = $2\pi V_{{23}}^{2}{{F}_{{\vec {n}\vec {m}}}}\delta \left( {{{U}_{{^{{\mathop 2\limits_{} }}}}}} \right.$ – ‒ ${{U}_{3}} + \sum\nolimits_{\alpha = 1}^M {{{n}_{\alpha }}\hbar {{\Omega }_{\alpha }}} $ – $\left. {\sum\nolimits_{\alpha = 1}^M {{{m}_{{\mathop \alpha \limits_{} }}}\hbar {{\Omega }_{\alpha }}} } \right){\text{/}}\hbar $ описывают переходы между колебательными подуровнями возбужденного состояния $U_{2}^{{(\vec {n})}}\,$ и продукта $U_{3}^{{(\vec {m})}}\,$ [34–36], где $\hbar $ – постоянная Планка, $\delta \left( Q \right)$ – дельта-функция Дирака, ${{V}_{{23}}}$ – матричный элемент перехода,
Система уравнений (4) решалась численно с начальными условиями
(6)
$\begin{gathered} \rho _{2}^{{(\vec {0})}}({{Q}_{1}},{{Q}_{2}},t = 0) = A\exp [ - {{{{U}_{1}}} \mathord{\left/ {\vphantom {{{{U}_{1}}} {{{k}_{B}}T}}} \right. \kern-0em} {{{k}_{B}}T}}] \times \\ \times \;\exp \{ - {{(\hbar {{\omega }_{e}} - U_{2}^{{(\vec {0})}} + {{U}_{1}})}^{2}}\tau _{e}^{2}{\text{/}}2{{\hbar }^{2}}\} , \\ \rho _{2}^{{(\vec {n} \ne \vec {0})}}({{Q}_{1}},{{Q}_{2}},t = 0) = 0,\quad \rho _{3}^{{(m)}}({{Q}_{1}},{{Q}_{2}},t = 0) = 0. \\ \end{gathered} $Количественное описание кинетики ПСПЭ в рамках многоканальной стохастической модели требует знания основных энергетических параметров модели: параметра экзергоничности и энергии реорганизации среды для каждой стадии ($ - \Delta {{G}_{{12}}}$, $ - \Delta {{G}_{{13}}}$, $E_{{rm}}^{{(12)}}$, $E_{{rm}}^{{(13)}}$), энергии реорганизации высокочастотных внутримолекулярных колебательных мод ${{E}_{{rv}}}$. Значения данных параметров могут быть получены с помощью фитинга экспериментальных стационарных спектров поглощения и флуоресценции молекулярных соединений, например, 4'-N,N-диэтиламин-3-гидроксифлавона [10]. Энергию реорганизации среды $E_{{rm}}^{{(12)}}$ и величину параметра экзергоничности $ - \Delta {{G}_{{12}}}$ на стадии фотовозбуждения можно определить из стационарных спектров поглощения и флуоресценции, связанных с переходом 1 → 2, а спектр флуоресценции, соответствующий переходу 1 → 3, определяет значения энергетических параметров $E_{{rm}}^{{(13)}}$ и $ - \Delta {{G}_{{13}}}$ [15]. При расчетах были использованы два набора энергетических параметров, соответствующих соединению 4'-N,N-диэтиламин-3-гидроксифлавон в ацетонитриле (ACN, ${{\tau }_{L}}$ = = 0.26 пс) и дихлорметане (DCM, ${{\tau }_{L}}$= 0.56 пс), которые имеют различные полярности и динамические свойства [9–12]. Важно отметить, что в компьютерном моделировании значения некоторых параметров принимаются фиксированными: общая энергия реорганизации высокочастотных колебаний ${{E}_{{rv}}}$ = 0.1 эВ и время их релаксации ${{\tau }_{v}}$ = = 100 фс.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Численное решение системы (3)–(4) методом броуновского моделирования [34–39] позволило рассчитать кинетику населенности возбужденного состояния протонно-электронной системы
и вычислить эффективное время ${{\tau }_{{eff}}}$ или эффективную скорость $k_{{PCET}}^{{}}$ ПСПЭ ${{\tau }_{{eff}}}\, = \,k_{{PCET}}^{{ - 1}}\, = \,\int {{{P}_{{PE*}}}(t)dt} $. Результаты численного моделирования кинетики ПСПЭ, полученные с использованием кода [37], представлены на рис. 2–5. Зависимость эффективной скорости ПСПЭ ${{k}_{{PCET}}}$ от параметра экзергоничности $ - \Delta {{G}_{{23}}}$ для угла θ = 90° показана на рис. 2. Зависимость имеет колоколообразный вид с двумя ярко выраженными областями реакции: в “нормальной” области Маркуса ($ - \Delta {{G}_{{23}}}$ < 0.2 эВ) величина эффективной скорости ПСПЭ увеличивается с ростом параметра экзергоничности, в то время как в инвертированной области ($ - \Delta {{G}_{{23}}}$ > > 0.2 эВ) наблюдается ее монотонное уменьшение. Это – закон энергетической щели Маркуса, первоначально предсказанный для термических реакций. Для рассматриваемых реакций предсказывается влияние матричного элемента перехода ${{V}_{{23}}}$ на положение максимума кривой: максимум смещается в область меньших значений экзергоничности реакции с ростом ${{V}_{{23}}}$. Эта особенность присуща только реакциям, протекающим в неравновесном режиме, параллельно с релаксацией растворителя. Отметим, что в термических реакциях положение максимума определяется только величиной энергии реорганизации ядерных степеней свободы. Также в “нормальной” области Маркуса величина эффективной скорости ПСПЭ выше в менее полярном растворителе (DCM), а при преодолении максимума зависимости ${{k}_{{PCET}}}$ ($ - \Delta {{G}_{{23}}}$) наблюдается противоположный тренд. Кроме того, с ростом ${{V}_{{23}}}$ максимум колоколообразной зависимости ${{k}_{{PCET}}}$ ($ - \Delta {{G}_{{23}}}$) смещается в сторону больших значений $ - \Delta {{G}_{{23}}}$, а его величина уменьшается. Отметим также, что по мере уменьшения угла θ максимум функции ${{k}_{{PCET}}}$ ($ - \Delta {{G}_{{23}}}$) смещается в область меньших значений экзергоничности, а его величина возрастает [15].Рис. 2.
Зависимости эффективной скорости реакции ПСПЭ ${{k}_{{PCET}}}$ от параметра экзергоничности $ - \Delta {{G}_{{23}}}$ для конфигурации θ = 90° в ACN (сплошные линии, φ ≈ 37°, $E_{{rm}}^{{(12)}}$ = 0.25 эВ, $E_{{rm}}^{{(23)}}$ = 0.4 эВ, $E_{{rm}}^{{(13)}}$ = 0.14 эВ) и DCM (пунктирные линии, φ ≈ 41°, $E_{{rm}}^{{(12)}}$ = 0.19 эВ, $E_{{rm}}^{{(23)}}$ = 0.34 эВ, $E_{{rm}}^{{(13)}}$ = 0.14 эВ).
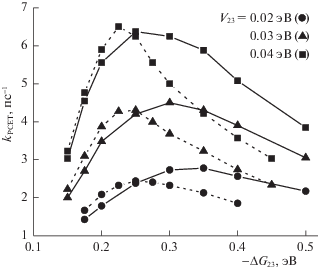
Рис. 3.
Зависимости эффективного времени реакции ПСПЭ ${{\tau }_{{eff}}}$ от матричного элемента перехода ${{V}_{{23}}}$ в ACN (сплошные линии, φ ≈ 37°, $E_{{rm}}^{{(12)}}$ = 0.25 эВ, $E_{{rm}}^{{(23)}}$ = 0.2 эВ, $E_{{rm}}^{{(13)}}$ = 0.14 эВ) и DCM (пунктирные линии, φ ≈ 41°,$E_{{rm}}^{{(12)}}$ = 0.19 эВ, $E_{{rm}}^{{(23)}}$ = 0.17 эВ, $E_{{rm}}^{{(13)}}$ = = 0.14 эВ) при θ = 90°.

Рис. 4.
Зависимости эффективного времени реакции ПСПЭ ${{\tau }_{{eff}}}$ от времени продольной релаксации ${{\tau }_{L}}$. Расчетные параметры: $ - \Delta {{G}_{{23}}}$ = 0.4 эВ, $E_{{rm}}^{{(12)}}$ = 0.25 эВ, $E_{{rm}}^{{(13)}}$ = 0.14 эВ, ${{V}_{{23}}}$ = 0.015 эВ (пунктирные линии) и 0.03 эВ (сплошные линии). Величина $E_{{rm}}^{{(23)}}$ вычислялась по формуле (3): $E_{{rm}}^{{(23)}}$ = 0.2 эВ (θ = 60°, φ ≈ 46°), 0.45 эВ (θ = 100°, φ ≈ 33°), 0.58 эВ (θ = 120°, φ ≈ 26°).
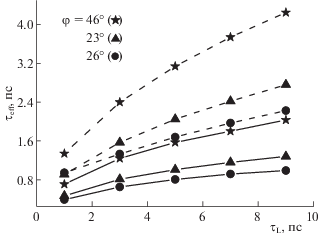
Рис. 5.
Зависимости эффективного времени реакции ПСПЭ ${{\tau }_{{eff}}}$ от угла φ в ACN (сплошные линии, $E_{{rm}}^{{(12)}}$ = = 0.25 эВ, $E_{{rm}}^{{(13)}}$ = 0.14 эВ) и DCM (пунктирные линии, $E_{{rm}}^{{(12)}}$ = 0.19 эВ, $E_{{rm}}^{{(13)}}$ = 0.14 эВ).
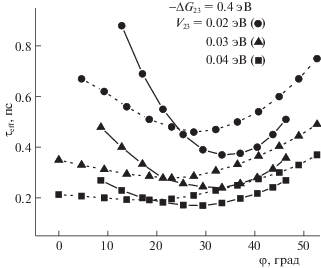
Вторая закономерность состоит в том, что наиболее яркое проявление ДЭР предсказывается в области сильной экзергоничности реакции, а в области слабой экзергоничности эффект ослабевает. Подчеркнем, что в термических реакциях переноса электрона тенденция обратная, ДЭР наблюдается только в области слабой экзергоничности [18]. Переходя к третьей закономерности, отметим, что в реакциях, протекающих в термическом режиме, проявление ДЭР возможно только при достаточно больших значениях электронного матричного элемента. С увеличением электронного матричного элемента константа скорости перестает зависеть от его величины и начинает зависеть от ${{\tau }_{L}}$, т.е. формируется раствороконтролируемый режим реакции [17]. Данные рис. 3 демонстрируют противоположный тренд: при малых значениях ${{V}_{{23}}}$ (≈0.01 эВ) с ростом ${{\tau }_{L}}$ наблюдается рост эффективного времени ПСПЭ почти в 2 раза (сравните сплошные и пунктирные линии с одинаковыми символами). С ростом ${{V}_{{23}}}$ зависимость ${{\tau }_{{eff}}}$(${{\tau }_{L}}$) значительно ослабевает. При увеличении экзергоничности реакции значение ДЭР увеличивается (сравните линии с разными символами). Для объяснения этой закономерности отметим, что рассматриваемые нами реакции ПСПЭ протекают в основном в неравновесном режиме. В этом случае, как было показано ранее [20], скорость процесса зависит от эффективности нетермических переходов, определяемой вероятностью перехода ${{W}_{{CT}}}$ при прохождении волнового пакета через область пересечения поверхностей свободной энергии [28]:
(7)
${{W}_{{CT}}} = \frac{{2\pi V_{{CT}}^{2}{{\tau }_{L}}}}{{\hbar (1 + g)\left| {\Delta {{G}_{{CT}}} - {{E}_{{rm}}}} \right|}},$(8)
$g = \frac{{2\pi V_{{CT}}^{2}{{\tau }_{L}}}}{\hbar }\left[ {\frac{1}{{\left| {\Delta {{G}_{{CT}}} + {{E}_{{rm}}}} \right|}} + \frac{1}{{\left| {\Delta {{G}_{{CT}}} - {{E}_{{rm}}}} \right|{{f}_{V}}}}} \right],$(9)
${{f}_{V}} = {{\left[ {1 + \frac{{8{{E}_{{rm}}}{{k}_{B}}T{{\tau }_{L}}}}{{{{{(\Delta {{G}_{{CT}}} - {{E}_{{rm}}})}}^{2}}{{\tau }_{V}}}}} \right]}^{{1/2}}},$Данные рис. 4 демонстрируют описанную выше тенденцию: с ростом матричного элемента перехода ${{V}_{{23}}}$ наблюдается значительное уменьшение ДЭР (сравните сплошные и пунктирные линии с одинаковыми символами). Заметим также, что с увеличением угла φ (или уменьшением θ) наблюдается спад величины ДЭР (сравните линии с различными символами). Поскольку скорость термического ПСПЭ не может зависеть от угла φ между направлениями координат реакции, соответствующих этапам фотовозбуждения системы и формирования продукта, то представленная на рис. 5 зависимость ${{\tau }_{{eff}}}(\varphi )$ является мерой неравновесности ПСПЭ. Следует также отметить немонотонную зависимость эффективного времени реакции ${{\tau }_{{eff}}}$ от угла φ. Численное моделирование показало, что значение угла φ, при котором ${{\tau }_{{eff}}}$ достигает минимального значения, зависит как от полярности растворителя, так и от параметра экзергоничности $ - \Delta {{G}_{{23}}}$. Подобное поведение объясняется зависимостью положения минимума от энергии реорганизации $E_{{rm}}^{{(23)}}$, которая монотонно увеличивается с ростом θ (или с уменьшением φ) [15].
Таким образом, проведенное моделирование кинетики сверхбыстрого ПСПЭ в рамках стохастической многоканальной модели показало, что для типичных значений энергетических параметров реакция протекает преимущественно в неравновесном режиме. Характерной особенностью неравновесных реакций является то, что наибольшая величина ДЭР должна наблюдаться в области сильной экзергоничности реакции, а в области слабой экзергоничности эффект мал. Второй особенностью неравновесных реакций ПСПЭ является зависимость положения максимума константы скорости ПСПЭ как функции экзергоничности реакции от матричного элемента перехода. Третьей особенностью неравновесных реакций является зависимость константы скорости от угла φ между направлениями координат реакции, соответствующих этапам фотовозбуждения системы и формирования продукта. Эти закономерности следует учитывать при проектировании оптических органических молекулярных устройств.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований (код проекта № 19-03-00175).
Список литературы
Cukier R.I., Nocera D. // Annu. Rev. Phys. Chem. 1998. V. 49. P. 337. https://doi.org/10.1146/annurev.physchem.49.1.337
Adams D.M., Brus L., Chidsey C.E.D. et al. // J. Phys. Chem. B. 2003. V. 107. P. 6668. https://doi.org/10.1021/jp0268462
Martín C., Ziółek M., Douhal A. // J. Photochem. Photobiol. C. 2016. V. 26. P. 1. https://doi.org/10.1016/j.jphotochemrev.2015.12.001
Weller A. // Z. Elektrochem. Ber. Bunsen-Ges. Phys. Chem. 1952. V. 56. P. 662. https://doi.org/10.1002/bbpc.19520560714
Kumpulainen T., Lang B., Rosspeintner A. et al. // Chem. Rev. 2017. V. 117. P. 10826. https://doi.org/10.1021/acs.chemrev.6b00491
Hammes-Schiffer S. // Acc. Chem. Res. 2001. V. 34. P. 273. https://doi.org/10.1021/ar9901117
Hammes-Schiffer S., Stuchebrukhov A.A. // Chem. Rev. 2010. V. 110. P. 6939. https://doi.org/10.1021/cr1001436
Hammes-Schiffer S., Goyal P. // ACS Energy Lett. 2017. V. 2. P. 512. https://doi.org/10.1021/acsenergylett.6b00723
Hsieh C.C., Cheng Y.M., Hsu C.J. et al. // J. Phys. Chem. A. 2008. V. 112. P. 8323. https://doi.org/10.1021/jp804216u
Hsieh C.C., Jiang C.M., Chou P.T. // Acc. Chem. Res. 2010. V. 43. P. 1364. https://doi.org/10.1021/ar1000499
Chou P.T., Yu W.S., Cheng Y.M. et al. // J. Phys. Chem. A. 2004. V. 108. P. 6487. https://doi.org/10.1021/jp048415k
Chou P.T., Pu S.C., Cheng Y.M. et al. // J. Phys. Chem. A. 2005. V. 109. P. 3777. https://doi.org/10.1021/jp044205w
Иванов А.И., Михайлова В.А. // Успехи химии. 2010. Т. 79. № 12. С. 1139. Ivanov A.I., Mikhailova V.A. // Russ. Chem. Rev. 2010. V. 79. P. 1047. https://doi.org/10.1070/RC2010v079n12ABEH004167
Feskov S.V., Mikhailova V.A., Ivanov A.I. // J. Photochem. Photobiol. C. 2016. V. 29. P. 48. https://doi.org/10.1016/j.jphotochemrev.2016.11.001
Mikhailova T.V., Mikhailova V.A., Ivanov A.I. // J. Phys. Chem. C. 2018. V. 122. P. 25247. https://doi.org/10.1021/acs.jpcc.8b09097
Zusman L.D. // Chem. Phys. 1980. V. 49. P. 295. https://doi.org/10.1016/0301-0104(80)85267-0
Зусман Л.Д. // Успехи химии. 1992. Т. 61. С. 29. Zusman L.D. // Russ. Chem. Rev. 1992. V. 61. P. 15. https://doi.org/10.1070/RC1992v061n01ABEH000963.
Yudanov V.V., Mikhailova V.A., Ivanov A.I. // J. Phys. Chem. A. 2012. V. 116. P. 4010. https://doi.org/10.1021/jp301837t
Юданов В.В., Михайлова В.А., Иванов А.И. // Хим. физика. 2013. Т. 32. № 5. С. 3. Yudanov V.V., Mikhailova V.A., Ivanov A.I. // Russ. J. Phys. Chem. В. 2013. V. l17. P. 187. https://doi.org/10.1134/S1990793113050114.https://doi.org/10.7868/S0207401X13050129
Mikhailova T.V., Mikhailova V.A., Ivanov A.I. // J. Phys. Chem. B. 2016. V. 120. P. 11987. https://doi.org/10.1021/acs.jpcb.6b09363
Mikhailova T.V., Mikhailova V.A., Ivanov A.I. // J. Phys. Chem. B. 2017. V. 121. P. 4569. https://doi.org/10.1021/acs.jpcb.7b02537
Najbar J., Tachiya M. // J. Phys. Chem. 1994. V. 98. P. 199. https://doi.org/10.1021/j100052a033
Cho M., Silbey R.J. // J. Chem. Phys. 1995. V. 103. P. 595. https://doi.org/10.1063/1.470094
Ando K., Sumi H. // J. Phys. Chem. B. 1998. V. 102. P. 10991. https://doi.org/10.1021/jp982659l
Fedunov R.G., Ivanov A.I. // J. Chem. Phys. 2005. V. 122. P. 064501. https://doi.org/10.1063/1.1844371
Khokhlova S.S., Mikhailova V.A., Ivanov A.I. // J. Chem. Phys. 2006. V. 124. P. 114507. https://doi.org/10.1063/1.2178810
Temkin O.N. Homogeneous Catalysis with Metal Complexes: Kinetic Aspects and Mechanisms. J. Wiley: Chichester, 2012.
Mikhailova V.A., Ivanov A.I. // J. Phys. Chem. C. 2017. V. 121. P. 20629. https://doi.org/10.1021/acs.jpcc.7b06106
Mikhailova V.A., Malykhin R.E., Ivanov A.I. // Photochem. Photobiol. Sci. 2018. V. 17. P. 607. https://doi.org/10.1039/C7PP00464H
Horng M.L., Gardecki J.A., Papazyan A.A. et al. // J. Phys. Chem. 1995. V. 99. P. 17311. https://doi.org/10.1021/j100048a004
Maroncelli M., Kumar V.P., Papazyan A.A. // J. Phys. Chem. 1993. V. 97. P. 13. https://doi.org/10.1021/j100103a004
Ionkin V.N., Ivanov A.I. // Chem. Phys. 2009. V. 360. P. 137. https://doi.org/10.1016/j.chemphys.2009.04.024
Kelly A.M. // J. Phys. Chem. A. 1999 V. 103. P. 6891. https://doi.org/10.1021/jp991530o
Fedunov R.G., Feskov S.V., Ivanov A.I. et al. // J. Chem. Phys. 2004. V. 121. P. 3643. https://doi.org/10.1063/1.1772362
Gladkikh V., Burshtein A.I., Feskov S.V. et al. // J. Chem. Phys. 2005. V. 123. P. 244510. https://doi.org/10.1063/1.2140279
Ionkin V.N., Ivanov A.I. // J. Phys. Chem. A. 2009. V. 113. P. 103. https://doi.org/10.1021/jp806589m
Nazarov A.E., Fedunov R.G., Ivanov A.I. // Computer Physics Communications. 2017. V. 210. P. 172. https://doi.org/10.1016/j.cpc.2016.09.015
Nazarov A.E., Eloev G.G., Ivanov A.I. // J. Photochem. Photobiol. A: Chem. 2018. V. 358. P. 207. https://doi.org/10.1016/j.jphotochem.2018.03.017
Feskov S.V., Ivanov A.I. // J. Chem. Phys. 2018. V. 148. P. 104107. https://doi.org/10.1063/1.5016438
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии