

Журнал физической химии, 2019, T. 93, № 2, стр. 313-320
Фотодиссоциация протонированных форм амино- и диметиламиноазобензола по данным резонансной рамановской спектроскопии
Ю. А. Михеев a, *, Ю. А. Ершов b
a Российская академия наук, Институт биохимической физики им. Н.М. Эмануэля
Москва, Россия
b Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
* E-mail: mik@sky.chph.ras.ru
Поступила в редакцию 15.03.2018
Аннотация
Приведен анализ литературных данных по резонансной рамановской спектроскопии амино- и диметиламиноазобензола на основе ранее сформулированных представлений о ридимерном строении аминоазокрасителей. Показана несостоятельность традиционной мономерно-хиноидной теории таутомерии и цветности при отнесении полос в рамановских спектрах протонированных форм амино- и диметиламиноазобензола. Резонансные рамановские спектры адекватно охарактеризованы в рамках ридимерного строения протонированных форм. Установлен ранее неизвестный факт, что при фотовозбуждении протонированных ридимеров происходит их переход в сильные кислоты.
Проведенный в работах [1–5] анализ UV–Vis-спектров аминоазокрасителей, в частности, аминоазобензола (AAB), диметиламиноазобензола (DAB) и различных их C- и аминопроизводных, обнаружил наличие ранее неизвестного фундаментального свойства: аминоазокрасители в своем основном состоянии являются субнаночастицами – ридберговскими димерами (ридимерами). Ридимеры (например, AAB2 и DAB2) стабилизируются межмономерной ковалентной связью нового типа. Эта связь образуется в результате спаривания электронов, промотированных с sp2-орбиталей атомов N азогрупп на ридберговские 3s-орбитали азогрупп. При этом характерная для данных азокрасителей цветность связана не с их мономерными хиноидными таутомерами, как полагали более 100 лет, а с катионами фениламинильного типа – хромогенными группами ридимеров.
Ридимеры, попадая в среду с достаточно высокой кислотностью, распадаются на протонированные мономерные фрагменты с хиноидным строением, которые, вопреки сложившемуся мнению, не имеют окраски, так как не поглощают Vis-света. При этом углубление окраски растворов азокрасителей, наблюдаемое при низких кислотностях, обусловлено протонированием ридимерных аминогрупп без распада ридимеров. Для таких ридимеров характерны специфические хромогены – смежные катионы фениламинильного типа. В отдельности каждый из этих катионов сам по себе имеет полосу поглощения в Vis-области спектра. Вместе с тем, ридимерная фиксация катионов в смежном положении (при отсутствии между ними π-сопряжения по Хюккелю в основном состоянии) превращает их в спаренные резонаторы с вырожденными π*-уровнями. Между ними в фотовозбужденном состоянии возникает энергетическое взаимодействие по типу квантово-волновых резонаторов (механизм Симпсона [6]). При этом происходит расщепление вырожденных π*-уровней и появление Vis-полос, батохромно смещенных относительно Vis-полос одиночных катионов [1–5]. Такой механизм характерен также для смежных катионов фениламинильного типа, образующихся в дипротонированном азобензоле (phN+H–HN+ph) [7] и фотовозбужденном азобензоле [8].
Результаты работ [1–5] открывают возможность корректировать представления, сложившиеся в предшествующем длительном периоде накопления экспериментальных данных. Такая коррекция проведена в настоящей работе. Здесь рассмотрены результаты исследования аминоазобензола (AAB) и диметиламиноазобензола (DAB) и их протонированных форм, полученные в работе [9] методом резонансной рамановской спектроскопии (RRS). Следует отметить, что этот вид спектроскопии был впервые зафиксирован как научное открытие в “Государственном реестре открытий СССР” и получил название “Явление резонансного комбинационного рассеяния света” [10].
В методе RRS [9, 10] используется фактор значительного усиления интенсивности комбинационного рассеяния (в ~100 раз) при приближении частоты падающего света к максимуму полосы поглощения спектра вещества. Метод RRS интересен не только тем, что позволяет определять RRS-полосы ридимеров AAB2 и DAB2, но и тем, что излучение, генерирующее RRS, может одновременно индуцировать фотохимические реакции ридимеров и демонстрировать образование новых частиц по их собственным RRS-полосам. Следует отметить, что возможность фотоиндуцированных реакций в условиях метода RRS для AAB и DAB в [9] не рассматривалась.
Резонансные рамановские (RR) спектры AAB2 и DAB2 [9]
Авторы [9] преследовали цель показать эффективность метода RRS для демонстрации обратимых превращений протонированных форм азокрасителей AAB и DAB. Обсуждение RRS-результатов, полученных для протонированных форм, они вели на основе общепринятой схемы таутомерного равновесия между аммониевыми и азониевыми мономерами Am+ ↔ Az+:

Регистрацию RR-спектров AAB2, DAB2 и их протонированных форм в [9] осуществляли, используя растворы красителей в ацетонитриле (CH3CN) при концентрации ~10–4 моль/л (в расчете на индивидуальные молекулы). Для реализации равновесия Am+ ↔ Az+ в растворы вводили минимальные количества кислоты HСl, достаточные для получения монопротонированных (в расчете на молекулы) катионов, контролируя процесс по UV–Vis-спектрам. Полученные для растворов красителей в ацетонитриле UV–Vis-спектры ([9], рис. 1) представлены в настоящем сообщении на рис. 1 с их реальным отнесением к ридимерам [1–5]. Так, кривые 1, 1 ' характеризуют непротонированные ридимеры красителей AAB2 и DAB2, кривые 2, 2 ' – дипротонированные по аминогруппам ридимеры (AAB+H)2 и (DAB+H)2.
Рис. 1.
Спектры UV–1Vis-поглощения аминоазобензола (1) и диметиламиноазобензола (1'), а также их протонированных форм (2 и 2 ' соответственно). Данные [9].
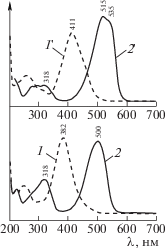
В [9] желтый цвет растворов исходных красителей традиционно считали свойством молекул (схема 1). Молекулам AAB приписывали полосу поглощения с максимумом 382 нм (рис. 1, кривая 1), а молекулам DAB – полосу 411 нм (рис. 1, кривая 1 '). Возникающие при протонировании красителей UV-полосы при 318 нм традиционно отнесли к мономерным формам с аммониевыми катионами Am+, а Vis-полосы при 500 нм (AAB+H)2 и 515, 535 нм (DAB+H)2 – к мономерным Az+-формам с хиноидной структурой (рис. 1, кривые 2, 2 '). При всей неадекватности трактовок работы [9], вид UV–Vis-спектров на рис. 1 свидетельствует о практически полном смещении равновесия в сторону протонированных форм. Это доказывается отсутствием в спектрах 2, 2 ' поглощения непротонированных ридимеров со спектрами 1 (AAB2) и 1 ' (DAB2).
Возбуждение RR-спектров в [9] осуществляли с помощью пучков непрерывного излучения лазера (на Ar+) с длинами волн 351, 458 и 514 нм. Мощность пучков находилась в пределах 25–50 мВт (~1016–1017 фотон/с). Образцы, на которые фокусировали излучение, охлаждали во избежание локального нагрева. Для спектрального анализа измеряли интенсивность света, рассеянного под углом 90°, в качестве референтной служила ацетонитрильная RRS-полоса с волновым числом ν = 920 см–1. RR-спектры, опубликованные в [9, рис. 3 ], приведены на рис. 2. Обсуждение этих спектров и квантово-химические расчеты авторы [9] вели в привязке к мнимым мономерным структурам схемы 1, сфокусировав свое внимание главным образом на свойствах AAB и их катионов (AAB+HAm ↔ AAB+HAz). При этом использовали традиционные представления об n,π*-состояниях азокрасителей, неадекватность которых показана в [1, 4].
Рис. 2.
Резонансные рамановские спектры непротонированных аминоазобензола (1, 2) и диметиламиноазобензола (кривые 1', 2'), а также их протонированных форм (соответственно 3, 4 и 3', 4'). Данные [9].

Изложенное выше и то, что результаты метода RRS объективно отражают свойства ридимеров азокрасителей, требует нового подхода к рассмотрению RRS-данных [9] на базе результатов [1–5].
RRS-полосы непротонированных ридимеров
RR-спектры непротонированных красителей (рис. 2, кривые 1, 2 (AAB2) и 1', 2' (DAB2)), получены при возбуждении лазерным светом с длинами волн λe = 514 (кривые 1, 1') и 458 нм (кривые 2, 2'). Все они имеют отчетливое сходство, а низкая интенсивность спектров 1, 1' обусловлена большей удаленностью λe = 514 нм от максимумов Vis-полос поглощения AAB2 (382 нм) и DAB2 (411 нм) по сравнению с λe = 458 нм (кривые 2, 2').
В спектре AAB2 (рис. 2, кривая 2) наиболее сильные полосы отнесены к связям: 1143 см–1– ν(C–NAZO); 1421 см–1– ν(N=N); 1467 см–1– ν(N=N), δ(С–H), здесь ν – валентные колебания (stretch), а δ – плоскостные деформационные колебания (bend). (Те же полосы ν(C–NAZO) 1143 [11] и 1142 см–1 [12] вместе с сильной полосой 1440 см–1 [11, 12] присутствуют также в RRS-системе азобензола AB). Отнесение слабых полос (рис. 2, кривая 2): 1189 см – 1– δ(С–H); 1312 см–1– δ(С–H), ν(С–NH2); 1596, 1603 см–1 – bend/stretch C–C в Рh-кольцах: ν(С–С) + δ(С–С–H). (В RRS-системе AB тоже присутствуют слабые δ(С–H) 1183, 1315 см–1 и “ν(С–С) + δ(С–С–H)” 1594, 1593 см–1 [11, 12].)
На рис. 2 видно, что спектр 2' (DAB2) качественно повторяет спектр 2 (AAB2) и, вероятно, поэтому авторы [9] не уделили спектру 2' особого внимания, хотя на нем отметили без объяснения очень слабую полосу 1623 см–1, которая по частоте совпадает с RRS-полосами 1624 см–1 в спектре 3 и 1623 см–1 в спектре 3', отнесенными к сигналам хиноидных структур протонированных мономеров.
e-Таутомеры непротонированных AAB2 и DAB2
Формулы строения непротонированных e-таутомеров [1–5] приведены на схеме 2.

На схеме 2 символом R обозначены атомы H или CH3-группы; точками под атомами N азогруппы обозначены электроны на sp2-орбиталях, а точками над N азогруппы – электроны на pz-орбиталях. Между азогруппами мономеров показаны ридберговские ковалентные связи из двух электронов, промотированных с sp2-орбиталей азогрупп на R3s-орбитали. В каждом мономере sp2-орбиталь, потерявшая электрон, наделяет свой атом N положительным зарядом. На pz-орбиталь этого атома N стягивается электрон с pz-орбитали соседнего атома N (имеющего неподеленную электронную пару на sp2-орбитали). И получается, что потерявший sp2-электрон атом N, восстанавливает электронейтральность, заряжая положительным зарядом pz-орбиталь соседнего атома N; получив положительный заряд, pz-орбиталь (в каждом мономере) поляризует электронную pz-систему в сопряженных с ней фенильном (Ph) или фениленовом (Pħ) кольцах, образуя катионы фениламинильного типа.
В схеме 2а на аминных азотах (R2N) обозначены pz-орбитали, подающие электроны в соседние фениленовые кольца. Фениленовые кольца, получив
электрон, становятся донорами электронов для катионов PhN+ в противолежащих мономерах (согласно схеме 2, PhN+≡Ph(+)…(+)N(••)…). В результате этого в ридимерах AAB2, DAB2 возникает по два комплекса с переносом заряда (КПЗ), усиливающих связь между мономерами.
Характерные для КПЗ кулоновские и обменные взаимодействия между фениленовыми анионами
и фениламинильными катионами PhN+ в итоге создают более устойчивые e-таутомеры (схема 2б) с катионами  . В них между противолежащими катионами обоих мономеров действуют одноэлектронные
связи по типу связей, существующих в катионе ${\text{H}}_{2}^{ + }$ [3–5, 13].
. В них между противолежащими катионами обоих мономеров действуют одноэлектронные
связи по типу связей, существующих в катионе ${\text{H}}_{2}^{ + }$ [3–5, 13].
Индуцированные по схеме 2б положительные заряды делокализуются в орто- и пара-положениях колец Ph(+) и Pħ(+), как в бензильных и фениламинильных катионах [7, 8]. Катионы подобного типа имеют не только связывающие и разрыхляющие молекулярные π-орбитали (МО), но и вакантные несвязывающие МО (НСМО) с нулевой энергией. В таких катионах переход электронов с высших занятых π-орбиталей (ВЗМО) происходит на НСМО под действием VIS-света (π → π*-переход). В результате этого в спектрах красителей AAB2 и DAB2 появляются VIS-полосы при 400–410 нм [1–5, 7, 8] (рис. 1 спектры 1, 1 ').
Согласно [3–5], VIS-поглощение при 400 нм у ридимеров DAB2, растворенных в гептане, обусловлено наложением двух близко расположенных VIS-полос с λmax = 396 и 384 нм. Они принадлежат неодинаковым по строению катионам фениламинильного типа и претерпевают батохромное смещение при замене гептана этанолом, сливаясь в одну интенсивную уширенную VIS-полосу при λmax = 408 нм. Сходная полоса с λmax = 411 нм наблюдается в спектре раствора DAB2 в ацетонитриле (рис. 1, кривая 1 '). Естественно ожидать, что такая же ситуация имеет место с VIS-полосой в спектре раствора AAB2 (рис. 1, кривая 1).
e-Таутомеризация и RRS-полосы AB и ридимеров AAB2, DAB2
e-Таутомеризация ридимеров (схема 2) связана с обменом электронов между орбиталями: (:sp2) + R3s ↔ (·sp2) + ${\text{R}}_{{3{\text{s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$), как в ситуации с AB [8]. Согласно [8], для AB характерен процесс e-таутомеризации, протекающий под влиянием ридберговской 3s-орбитали (R3s), которая исполняет роль акцептора электрона, находящегося на sp2-орбитали того или другого атома азота азогруппы. Благодаря этому обеспечивается чередование актов акцептирования и возврата sp2-электрона с участием то одного, то другого атома N азогруппы (e-волновой механизм) (схема 3).

Действие этого механизма вызывает непрерывную осцилляцию электрона между электронейтральной формой молекул AB (схема 3A) и двумя поляризованными формами (схема 3B), отличающимися положением заряда в молекуле, причем частота этих актов на многие порядки превосходит частоту электромагнитных UV–Vis-колебаний [1, 8]. Следует отметить, что вследствие своей ультравысокой частоты e-волновая поляризация подключается к актам UV–Vis-абсорбции одновременно, с частотой электромагнитных колебаний, тем самым усиливая интенсивность RRS-полос AB, AAB2, DAB2.
В схеме 3 e-таутомер (A) относится к преобладающей по концентрации неполяризованной форме AB с UV-полосой поглощения при 320 нм. Форма B невозбужденного AB имеет низкую концентрацию и обеспечивает AB слабую π → π*-полосу поглощения при 440 нм (S0 → S1), эту полосу долгое время приписывали n → π*-переходу.
Указанные выше RRS-полосы AB возбуждали непрерывным лазерным излучением в Vis-полосе при 440 нм [11]. В [12] использовали пикосекундные импульсы UV-возбуждения в полосе 273 нм и зондовые импульсы в Vis-полосе поглощения с λe = 410 нм транзитного S1-состояния. (В [8] показано, что S1-состояние AB это π,π*-состояние B* (схема 3)). В [12] установлено, что возбуждение AB в полосе 273 нм (переход S0 → S2) сопровождается мгновенным переходом из франк-кондоновского (FC) состояния S2 в FC-состояние S1 (B*), испускающее RRS-полосу ν(N=N) = 1428 см–1 и релаксирующее в состояние S0, которое имеет RRS-полосу ν(N=N) = 1440 см–1.
Таким образом, появление в спектре AB этих интенсивных RRS-полос, как и полос ν(C–NAZO) = = 1143 [11] и 1142 см–1 [12], отражает осцилляцию электронов между sp2- и R3s-орбиталями азогрупп [8].
В молекуле AB, в отличие от ридимеров AAB2 и DAB2, процесс переполяризации фенильных колец создает одинаковые фениламинильные катионы (cхема 3B): ${{(}^{{( + )}}}{\text{ph}}{\kern 1pt} \underline \cdots {{}^{{( + )}}}{\text{N}}{\kern 1pt} \underline \cdots {\text{N}}{\kern 1pt} - {\kern 1pt} {\text{ph}}){\text{R}}_{{{\text{3s}}}}^{{( - )}}$ ↔ $({\text{ph}}{\kern 1pt} - {\kern 1pt} {\text{N}}{\kern 1pt} \underline \cdots {\text{N( + )}}{\kern 1pt} \underline \cdots {\text{p}}{{{\text{h}}}^{{( + )}}}){\text{R}}_{{{\text{3s}}}}^{{( - )}}$.
В ридимерах процесс переполяризации охватывает оба мономера и межмономерные ковалентные связи из R3s-электронов. Эти R3s-электроны, вовлекаясь в e-обмен (:sp2) + R3s ↔ (·sp2) + ${\text{R}}_{{3{\text{s}}}}^{{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{ \bullet } }}$), способны создавать энергетически неустойчивые e-таутомеры с хиноидными мономерами (схема 2в). Такие таутомеры имеют крайне низкую стационарную концентрацию и обладают в спектре 2 ' (рис. 2) очень слабой RRS-полосой ν = 1623 см–1.
Схема 2, позволяет связать дублетные RRS-полосы в спектрах 2, 2' на рис. 2, отнесенные в [9] к ν(N=N): 1421, 1467 см–1 (AAB2) и 1411, 1462 см–1 (DAB2), с чередованием e-таутомеров (2a) ↔ (2б). Азогруппы в (2a) должны испытывать некоторое сжатие из-за кулоновского притяжения колец с отрицательным и положительным зарядами. Наоборот, в (2б) азогруппы должны испытывать некоторое растяжение из-за отталкивания связанных с ними положительно заряженных колец. Не исключено, что e-таутомеризация “a ↔ б” (схема 2), изменяя электромеханическое состояние азогруппы, приводит к появлению двух RRS-полос ν(N=N): 1421, 1467 см–1 (AAB2) вместо полосы с ν(N=N) 1440 см–1, характерной для AB (и, соответственно 1411, 1462 см–1 в случае DAB2).
Строение, RRS-полосы и UV–Vis-спектры протонированных ридимеров
Представляет интерес заново обсудить отнесение полос RR-спектров, полученных в [9] с λe = 514 нм для протонированных красителей (AAB+H)2 и (DAB+H)2 (рис. 2, кривые 3, 3'). Как уже отмечалось, авторы [9] в своем обсуждении и расчетах RRS-полос ограничились примером с (AAB+H)2 (рис. 2, кривая 3), базируясь на неадекватном механизме Am+ ↔ Az+ (схема 1). Наиболее существенным RRS-полосам на кривой 3 авторы дают следующее отнесение, см–1: 896–δ(N–N); 1171, 1177 – δ(C–H); 1276 – ν(C–NAZO); 1328 – δ(C–H) и ν(N–N); 1492 – δ(C–H); 1594 – ν(N–N), причем одинарные связи N–N и C–NAZO приписаны хиноидной структуре Az+. Кроме того, полоса 1594 см–1 отнесена еще к внутренним валентным и деформационным движениям атомов в бензеноидном кольце хиноидного таутомера Az+, а полоса 1624 см–1 – к таким же движениям в хиноидном кольце таутомера Az+ (схема 1). Была также отмечена особенно высокая интенсивность полос 1594 и 1624 см–1 на рис. 2 (спектр 3') у мнимого хиноидного таутомера Az+.
Выше отмечалось, что UV–Vis-спектры данных протонированных красителей (рис. 1, кривые 2, 2 ') свидетельствуют о практически полном смещении равновесия протонирования в сторону ридимеров (AAB+H)2, (DAB+H)2. Согласно [1–5], e-таутомеры этих ридимеров характеризуются формулами строения, представленными на схеме 4.

На схеме 4 показано, что e-таутомерный обмен в ридимерах (4a) ↔ (4б) идет путем ультрабыстрой переполяризации мономеров с вовлечением в этот обмен и R3s-электронов. Именно такие e‑таутомеры генерируют UV–Vis-спектры 2, 2 ', представленные на рис. 1 [1–3, 5]. В частности, отмеченные на схеме 4 светлыми треугольниками смежные пары фенильных и фениленовых колец (не имеющие зарядов) ответственны за UV-полосы при 318 нм дипротонированных ридимеров (AAB+H)2, (DAB+H)2 (рис. 1, кривые 2, 2 '). Эти спаренные кольца взаимодействуют друг с другом в возбужденном состоянии по механизму Симпсона [1–3, 5].
Отмеченные черными треугольниками смежные пары фенильных и фениленовых колец ответственны за уширенную Vis-полосу ридимера (AAB+H)2 при 500 нм (рис. 1, спектр 2) и две налагающиеся друг на друга Vis-полосы ридимера (DAB+H)2 с максимумами при 515 и 535 нм (рис. 1, спектр 2 '). Указанные Vis-полосы тоже появляются в результате взаимодействия положительно заряженных парных фенильных и парных фениленовых фрагментов по механизму Симпсона [1–5].
Таким образом, UV-полосы при 320 нм и Vis-полосы с λmax ≥ 500 нм принадлежат не мономерным катионам Am+ и Az+, а протонированным по аминогруппам ридимерам.
Идентификация RRS-полос, генерируемых светом с λ = 514 нм
Согласно утверждению авторов [9, табл. 4 ], все полосы в спектре 3 (рис. 2) принадлежат хиноидным формам (к ним же, по умолчанию, отнесены и полосы спектра 3'). Это утверждение нельзя признать оправданным в отношении всех полос спектра 3 в связи с реальным отсутствием хиноидных структур в основном состоянии протонированных ридимеров [1–5]. В спектре 3 могут присутствовать как RRS-полосы протонированных ридимеров (схема 4), так и RRS-полосы хиноидных структур, образующихся в результате фотохимической реакции.
В связи с этим для идентификации RRS-полос следует привлечь данные инфракрасной спектроскопии (IRS). Дело в том, что области частот колебаний химических связей (группировок) в методе RRS должны соответствовать областям частот колебаний тех же связей (группировок) в IRS-области. Появление RRS-полос (в форме смещения их частот ν от частоты возбуждающего электромагнитного излучения νe) определяется способностью связей (группировок) испытывать поляризацию, тогда как IR-полосы отражают наличие дипольных моментов у связей (группировок). Вместе с тем, несмотря на разную электрофизическую природу, единая механика колебаний атомов предопределяет наличие общих частотных областей для RRS и IRS-полос. Иначе говоря, учет данных IRS может облегчить идентификацию RRS-полос.
В связи с изложенным следует обратить внимание на очень интенсивную RRS-полосу ν = 1623 см–1 (рис. 2, кривая 3') и менее интенсивную ν = 1624 см–1 (рис. 2, кривая 3), которые четко характеризуются как сигналы хиноидных форм [9]. Согласно данным IRS, указанные частоты присутствуют в том же интервале 1639–1600 см–1, где находится сильная IR-полоса, характеристичная для анилиновых производных с C=N-связями [14]. В [15] указано, что валентным колебаниям двойной связи C=N, сопряженной с фенильным кольцом в бензилиденанилине C6H5–N=CH–С6H5 соответствует полоса 1627 см–1, практически совпадающая по частоте с RRS-полосами хиноидных форм [9].
На рис. 2 в спектрах 3, 3' рядом с полосами с $\text{v}$ =1623 и 1624 см–1 находятся полосы 1595 и 1594 см–1, которые отнесены к внутренним валентным и деформационным колебаниям атомов в бензеноидном кольце хиноидного таутомера Az+ [9]. Существенно, что интенсивность полос 1595 и 1594 см–1 в спектрах 3, 3' (рис. 2) значительно выше, чем у таких же полос в спектрах непротонированных ридимеров (рис. 2, кривые 2, 2').
Таким образом, сопоставление данных IRS и RRS подтверждает факт образования хиноидных структур в условиях облучения ридимеров лазерным светом с λe = 514 нм.
Все остальные полосы в спектре 3 (рис. 2) тоже отнесены к RR-полосам мономерной формы Az+ (хиноидный таутомер) [9], что весьма сомнительно. Например, полоса δ(C–H) = 1492 см–1 (рис. 2, спектр 3), отнесенная к Az+ (схема 1) [9], характерна для колебаний связей в бензольных кольцах AB: ν(С–С) + δ(CCH) [11]. В спектре 3' (рис. 2) эта ситуация имеет место для полосы 1494 см–1. Иначе говоря, полосы при 1492 и 1494 см–1 могут принадлежать бензольным кольцам ридимеров (отмечены светлыми треугольниками в схеме 4).
Сомнительно и отнесение полосы 896 см–1 (спектр 3, рис. 2) к δ(N–N) хиноидной структуры Az+ (схема 1). Дело в том, что интенсивность референтной RRS-полосы растворителя – ацетонитрила (ν = 920 см–1) в спектре 3' значительно ниже, чем в спектре 3. Это связано с тем, что высоты всех полос на графике спектра 3' были по необходимости снижены. Если бы в спектре 3' референтная полоса (ν = 920 см–1) имела ту же интенсивность, что и в спектре 3, то полоса 900 см–1 превысила бы полосу 896 см–1, а полоса реальной хиноидной структуры (ν = 1623 см–1) не поместилась бы на рисунке. При этом полоса при 896 см–1 примерно равна по высоте хиноидной полосе 1624 см–1 (спектр 3, рис. 2), тогда как полоса 900 см–1 несоизмеримо ниже полосы 1623 см–1 (спектр 3', рис. 2).
Иначе говоря, между интенсивностями полос при 896 (900) и 1624 см–1 (1623 см–1) нет прямой пропорциональности, необходимой в случае их принадлежности одной и той же хиноидной структуре. Таким образом, полосы 896 и 900 см–1 требуют другого отнесения.
Пользуясь данными метода IRS, можно принять, что за RRS-полосы 896 см–1 (900 см–1) ответственны маятниковые колебания групп H3N+–(R2HN+–), являющихся важнейшими элементами протонированных ридимеров (схема 4). Частота маятниковых колебаний H3N+–группы (установленная для аминокислот) составляет ~800 см–1 [14, с. 372]. В случае протонированных ридимеров эти положительно заряженные группы испытывают кулоновское отталкивание со стороны положительно заряженных фениленовых колец (схема 4). Обусловленное этим отталкиванием механическое напряжение может стать причиной повышения RRS-частоты до 900 см–1.
Из изложенного выше следует, что RRS-полосы в спектрах 3, 3' (рис. 2) принадлежат протонированым по аминогруппам ридимерам и хиноидным продуктам их фотохимического депротонирования.
Фотохимические реакции протонированных ридимеров
Реакцию, индуцированную непрерывным излучением лазера с длиной волны 514 нм можно выразить упрощенно в виде схемы, включающей в себя акты депротонирования и обратимого восстановления исходных ридимеров (схема 5)

Согласно схеме 5, поглощение света с λe = = 514 нм вызывает частичное депротонирование ридимеров с образованием в них одного мономерного хиноидного фрагмента. Она же объясняет причину появления RRS-полос при 1492 (в спектре 3) и 1494 см–1 (в спектре 3'), принадлежащих бензольным кольцам частично депротонированных ридимеров (отмечены светлыми треугольниками в схеме 5), но отсутствующих в спектрах 2 и 2 '.
Характерно, что фотохимическая реакция депротонирования (схема 5) протекает в среде с достаточно высокой кислотностью и параллельно с генерацией RRS-полос исходных протонированных ридимеров. Это свидетельствует о том, что вызванное фотовозбуждением ридимеров перераспределение электронной плотности и локализация избыточного положительного заряда на аминогруппах ридимеров (по типу a → б, схема 5) превращает ридимеры в очень сильные кислоты.
Усиление кислотной функции протонированных ридимеров при их фотовозбуждении – общее свойство протонированных ароматических аминов. Например, протонированный β-нафтиламин (${\text{NpNH}}_{3}^{ + }$) в основном состоянии имеет pK = 4.07 (равновесие: ${\text{NpNH}}_{3}^{ + }$ + H2O ↔ NpNH2 + H3O+). В возбужденном состоянии он имеет pK* на 5–6 единиц меньше, т.е. становится гораздо более сильной кислотой [16].
Сопоставляя спектры 3 и 3' (рис. 2), можно видеть, что интенсивность RRS-полосы хиноидной формы (DAB+H)2 (спектр 3', ν = 1623 см–1) значительно выше, чем у хиноидной формы (AAB+H)2 (спектр 3, ν = 1624 см–1). Из этого следует, что возбужденное состояние $({\text{DA}}{{{\text{B}}}^{ + }}{\text{H}})_{2}^{*}$ – значительно более сильная кислота, чем $({\text{AA}}{{{\text{B}}}^{ + }}{\text{H}})_{2}^{*}$.
Интересное изменение в RRS-системе происходит при возбуждении (AAB+H)2 непрерывным лазерным излучением с λe = 351 нм. Так, в спектре 4 практически нет полос, отнесенных в [9] к хиноидным структурам Az+ (схема 1), и превалируют полосы, отнесенные в [9] к нехиноидным формам (Am+) (схема 1): 1148 – ν(C–NAzo), δ(C–H); 1185, 1314 – δ(C–H); 1428, 1449, 1471 – ν(N=N), а также очень слабая полоса 1595. Характерно, что все эти полосы попадают в частотные RRS-области сигналов непротонированных ридимеров AAB2 (спектры 1, 2 на рис. 2).
Полученный с λe = 351 нм для (DAB+H)2 спектр 4' (рис. 2) еще более беден по числу полос, имеющих более низкую интенсивность относительно полос (AAB+H)2 (спектр 4, рис. 2). Вместе с тем, наблюдаемое качественное сходство спектров 4 и 4' друг с другом и со спектрами 2 и 2' (рис. 2) свидетельствует о том, что излучение с λe = 351 нм, несущее более высокую энергию, смещает равновесие депротонирования ридимеров сильнее, чем излучение с λe = 514 нм. Излучение с λe = 351 нм поглощается смежными незаряженными фенильными и фениленовыми кольцами (отмечены светлыми треугольниками на схемах 4 и 5). Энергия возбуждения этих колец, скорее всего, частично переносится на смежные заряженные кольца, вызывая быстрое депротонирование одного из ридимерных мономеров по направлению (5a) → (5б) в схеме 5.
Следующий акт поглощения излучения с λe = 351 нм уже ридимером (5б) приводит к депротонированию второго ридимерного мономера с образованием дихиноидного e-таутомера, изображенного на схеме 2в. Затем дихиноидный e-таутомер (энергетически крайне неустойчивый) быстро релаксирует в равновесное состояние ридимерных структур (2a) ↔ (2б), чьи RRS-полосы преобладают в спектрах 4, 4' (рис. 2). Относительно низкие интенсивности RRS-полос 4, 4' (рис. 2), особенно в спектре 4', можно связать с наличием побочных фотохимических реакций разрушения ридимеров, протекающих под действием достаточно жесткого UV-излучения с λe = 351 нм.
Таким образом, анализ опубликованных в [9] данных по резонансной рамановской спектроскопии, осуществленный в настоящей работе в рамках концепции ридимерного строения аминоазокрасителей [1–5], показывает применимость этой концепции не только для UV–Vis-спектроскопических исследований аминоазокрасителей, но и для RRS-исследований. В частности, учет строения протонированных ридимеров позволил уточнить отнесение полученных в [9] RRS-полос. Кроме того, он позволил установить наличие фотохимической реакции в условиях RRS с образованием хиноидных структур, что свидетельствует об усилении кислотных свойств протонированных ридимеров при их фотохимическом возбуждении.
Список литературы
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2017. Т. 91. № 4. С. 672.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2017. Т. 91. № 10. С. 1683.
Михеев Ю.А., Ершов Ю.А. // Там же. 2018. Т. 92. № 2. С. 267.
Михеев Ю.А., Ершов Ю.А. // 2018. Т. 92. № 8. С. 1251.
Михеев Ю.А., Ершов Ю.А. // 2018. Т. 92. № 10. С. 1552.
Robin M.B., Simpson W.T. // J. Chem. Phys. 1962. V. 36. № 3. P. 580.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Журн. физ. химии. 2015. Т. 89. № 2. С. 243.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // Там же. 2015. Т. 89. № 11. С. 1773.
Matazo D.R.C., Ando R.A., Borin A.C., Santos P.S. // J. Phys. Chem. A 2008. V. 112. № 19. P. 4437.
www.ross-nauka.narod.ru/06/06-151.html. Авторы: Шорыгин П.П., Иванова Т.М.: Гос. реестр открытий СССР. Научное открытие “Явление резонансного комбинационного рассеяния света”. Номер и дата приоритета № 151 от 18 июня 1952 г.
Stuart Ch.M., Frontiera R.R., Mathies R.A. // J. Phys. Chem. A. 2007. V. 111. № 48. P. 12072.
Fujino T., Tahara T. // J. Phys. Chem. A. 2000. V. 104. № 18. P. 4203.
Грей Г. Электроны и химическая связь. М.: Мир, 1967. С. 52.
Беллами Л.Б. Инфракрасные спектры сложных молекул. М.: Изд-во иностр. лит., 1963. С. 368.
Наканиси К. Инфракрасные спектры и строение органических соединений. М.: Мир, 1965. С. 207.
Паркер С. Фотолюминесценция растворов. М.: Мир, 1972. С. 314.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии