

Журнал физической химии, 2019, T. 93, № 2, стр. 258-265
Спектрально-люминесцентные свойства и устойчивость дипиррометенатов цинка(II) различной структуры в протонодонорных средах в основном и электронно-возбужденных состояниях
А. А. Прокопенко a, *, Р. Т. Кузнецова a, Ю. В. Аксенова a, Е. Н. Тельминов a, М. Б. Березин b, Е. В. Антина b
a Томский государственный университет
Томск, Россия
b Российская академия наук, Институт химии растворов им. Г.А. Крестова
Иваново, Россия
* E-mail: alexpr898@gmail.com
Поступила в редакцию 16.04.2018
Аннотация
Изучены спектрально-люминесцентные свойства ряда дипиррометенатов цинка [Zn(dpm)2] с алкил-, фенил-, мезо-аза- и галоген-заместителями в лиганде в нейтральных и подкисленных соляной кислотой этанольных растворах. Исследована устойчивость комплексов в протонодонорных растворителях в основном и возбужденных состояниях, измерены квантовые выходы фотопревращений под действием лазерного излучения. В ряде случаев обнаружена и обсуждена связь фотохимической стабильности изученных дипиррометенатов с их свойствами в протонодонорных средах.
В последние годы существенно возрос интерес к изучению фотофизических и фотохимических свойств комплексов p- и d-элементов с дипиррометенами [1, 2] для их последующего использования в качестве современных оптических устройств: люминесцентных зондов [3, 4], лазерно-активных сред [5–8], оптических сенсоров [9, 10], фотосенсибилизаторов синглетного кислорода (1О2) [11] и т.п. Для целенаправленного выбора и последующего синтеза таких соединений для конкретных оптических устройств необходимо изучение связи структуры лигандов, типа комплексообразователя и растворителя со спектрально-люминесцентными и фотохимическими свойствами этих соединений в разных средах. В настоящее время наиболее изученными в этом направлении являются дифторборатные комплексы дипиррометенов (BODIPY) [1, 2], в то время как для комплексов дипиррометенов с цинком ([Zn(dpm)2]) исследования только начинаются [12–14], поэтому изучение фотоники комплексов [Zn(dpm)2] весьма актуально. Кроме этого, для практического использования в качестве оптических устройств необходимо выбирать наиболее устойчивые в разных электронных состояниях и кислотно-основных средах соединения.
Цель данной работы – изучение спектрально-люминесцентных, фотофизических и фотохимических свойств, а также устойчивости дипиррометенатов цинка(II) в протонодонорных средах, как в основном, так и в возбужденных электронных состояниях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объекты и методы. На рис. 1 представлены структурные формулы и обозначения изученных соединений. Комплексы цинка с дипиррометенами были синтезированы в Институте химии растворов им. Г.А. Крестова РАН (г. Иваново) по методикам, приведенным в [15–20], с соблюдением всех методов контроля структуры и чистоты соединений. Состав и структура соединений подтверждены данными ПМР, ИК-спектроскопии и элементного анализа [17–20]. В качестве растворителя использовали циклогексан (“ч.д.а.”) и 95% этанол.

Изучение устойчивости комплексов в основном и возбужденном состояниях в протонодонорных растворителях проводили в растворах этанола с последовательно изменяющимся содержанием концентрированной соляной кислоты, согласно методике, подробно изложенной в [21]. Полученные результаты сравнивали с данными для бис-дипиррометенатов цинка других структур [22, 23].
Спектрально-люминесцентные характеристики растворов измеряли при комнатной температуре и в замороженных растворах с помощью спектрометров СМ2203 (SOLAR, Беларусь) и Cary Eclipse (Varian). Квантовый выход флуоресценции определяли относительным методом по методикам, описанным в [7, 8, 14].
Характеристики фотостабильности изучали при возбуждении второй (λген = 532 нм) и третьей (λген = 355 нм) гармониками Nd:YAG-лазера. Квантовый выход фотопревращений определяли с погрешностью 5% по изменениям стационарных спектров поглощения, измеренных до и после облучения, согласно методике, подробно описанной в [7].
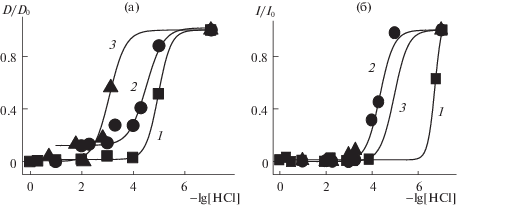
По точке перегиба кривых титрования определяли значения –lg[HCl]50, при которых концентрация комплексов [Zn(dpm)2] уменьшается за счет их распада на 50% с образованием протонированного лиганда и выходом комплексообразователя из комплекса, как показано в [21] для BODIPY-производных и [Zn2L2]-бис-геликатов [22]. На рис. 2 приведены изменения спектральных характеристик для одного из цинковых комплексов ([Zn((CH3)4-dpm)2]) в подкисленных этанольных растворах, аналогичные изменения при других значениях –lg[HCl] получены и для [Zn(дибромированных алкил-dpm)2]. Специфика картин спектральных превращений тетрафенил- и тетрафенил-аза-дипиррометенатов цинка(II) отражена на рис. 3 и 4. На основании этих изменений были построены кривые титрования для S0 и $S_{1}^{{{\text{ф л }}}}$ состояний (рис. 3–5).
Рис. 2.
Изменение спектров поглощения (а) и флуоресценции (λвозб = 460 нм, б) [Zn((CH3)4-dpm)2] в этаноле, 10–5 М при добавлении раствора 33% (10.8 М) HCl: 1 – нейтральный этанольный раствор, 2 и 3 – 0.0001 и 10%-ный раствор 33% HCl в воде.
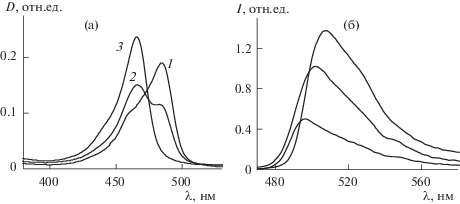
Рис. 3.
Изменение спектров поглощения (а) и флуоресценции (λвозб = 510 нм, б) [Zn((Ph)4-dpm)2] в этаноле, 10–5 М при добавлении от 0.000001 до 5% раствора 33% (10.8 М) HCl в воде. На вставках: а – экспериментальные кривые титрования, построенные по падению интенсивности поглощения на 525 нм (1) и по росту интенсивности поглощения на 550 нм (2); б – кривая титрования, характеризующая исчезновение флуоресценции комплекса.
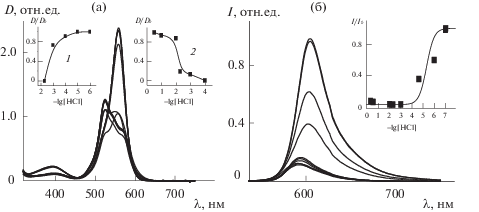
Рис. 4.
Изменение спектров поглощения [Zn((Ph)4–N-dpm)2] в этаноле, 10–5 М при добавлении от 0.000001 до 30% раствора 33% (10.8 М) HCl в воде. На вставках: экспериментальные кривые титрования, построенные по падению интенсивности поглощения на 650 нм (S0–S1) (1) и на 587 нм (S0–S2) (2); по росту интенсивности поглощения на 630 нм (3).

ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Спектрально-люминесцентные свойства
Спектрально-люминесцентные свойства некоторых изученных соединений приведены в [8, 14, 24], на рис. 2–4 и в таблице 1, где показано, что спектры поглощения комплексов лежат в области 485–590–650 нм. Введение в дипиррометеновый алкилзамещенный лиганд атомов галогенов в качестве заместителей вызывает несущественный длинноволновый сдвиг, уменьшает эффективность флуоресценции и приводит к увеличению фосфоресценции в замороженном этаноле [8] из-за увеличения выхода интеркомбинационной конверсии по механизму “тяжелого атома” при сравнении с комплексами цинка без галогенов в лиганде, что позволяет использовать такие комплексы для сенсорных и фотосенсибилизирующих синглетный кислород (1О2) сред. Эффективность интеркомбинационной конверсии в дипиррометенатах цинка(II) увеличивается по сравнению с BF2-аналогами, как показано в [8, 24].
Таблица 1.
Спектрально-люминесцентные, фотофизические свойства и характеристики стабильности комплексов [Zn(dpm)2] в протонодонорных средах в основном и электронно-возбужденных состояниях
| Соединение | $\lambda _{{{\text{п о г л }}}}^{{{\text{м а к с }}}}$, нм (нейтр. компл) | $\lambda _{{{\text{п о г л }}}}^{{{\text{м а к с }}}}$, нм (протонир по Nпирр лиганда) | $\lambda _{{{\text{ф л }}}}^{{{\text{м а к с }}}}{\text{/}}\lambda _{{{\text{ф о с ф }}}}^{{{\text{м а к с }}}}$, нм (нейтр. компл) | –lg[HCl]50 (S0) (протон акц. центр) | –lg[HCl]50 (S1)Ф–К | –lg[HCl]50 ($S_{1}^{{{\text{ф л }}}}$) | γфл (нейтр. компл) | φΦ × 105 (λвозб, нм) |
|---|---|---|---|---|---|---|---|---|
| [Zn((CH3)4-dpm)2] | 485 | 465 | 495/740 + + 830 | 5.0 (Nпирр) | 4.7 | 7 | 0.008 | 7.5 (355) |
| [Zn(Br2(CH3)4-dpm)2] | 504 | 489 | 526/744 | 4.4 (Nпирр) | 3.1 | 4.4 | 0.004 | 640 (355) |
| [Zn(Br2(C5H11)2 (CH3)2-dpm)2] | 511 | 463 | 517/745 | 3.2 (Nпирр) | 2.1 | 5.0 | 0.005 | 275 (355) |
| [Zn((Ph)4-dpm)2] | 525 + 570 | 555 | 602/830 | 2.8 | 0.05 | 1.1 (532) | ||
| 2.25 (Нdpm·НCl) | 0.97 | 5 | ||||||
| [Zn((Ph)4–N-dpm)2] | 587 + 650 | 630 | Нет фл в растворе/810 | 4.2 | 0.5 | Нет фл | Нет фл | 5 (532) |
| 3 | ||||||||
| 1.5 (Нdpm·НCl |
Замена в лиганде алкильных заместителей на фенильные существенно увеличивает длинноволновый сдвиг спектров (рис. 3, 4). Следует отметить, что для комплексов с фенильными циклами в лигандах [Zn((Ph)4-dpm)2], как с мезо-аза-замещением, так и с метиновой группой =СН– в мезо-положении в отличие от алкилпроизводных и галогенированных dpm-лигандов с цинком, а также BF2-дипиррометенатов с подобными тетрафенилсодержащими лигандами, в нейтральном растворе в видимой области наблюдается широкая полоса поглощения с двумя максимумами: более интенсивным коротковолновым (525 и 587 нм: S0–S2-переход) и менее интенсивным длинноволновым (S0–S1-переход: 570 и 650 нм), для [Zn((Ph)4-dpm)2] и [Zn((Ph)4–N-dpm)2] комплексов, соответственно (рис. 3, 4, таблица 1 и [14]). При этом максимумы S0–S1-переходов в дипиррометенатах бора и цинка с тетрафенильными заместителями практически совпадают: 568–570 нм для тетрафенил-BODIPY [14, 21] и [Zn((Ph)4-dpm)2] (рис. 3, таблица 1) и 648–650 нм для тетрафенил-аза-BODIPY [14, 21] и [Zn((Ph)4–N-dpm)2] (рис. 4, таблица 1).
Эта особенность может быть связана, с выходом из плоскости dpm “внешних” фенильных циклов (коротковолновый максимум) и взаимодействующих друг с другом по π-системам “внутренних” фенилов, расширяющих π-систему (длинноволновый максимум) (рис. 3, 4, таблица 1).
Следует обратить внимание на то, что в случае [Zn((Ph)4–N-dpm)2] щель между S1- и S2-состоянием (Δλ = 65 нм) увеличивается по сравнению с [Zn((Ph)4-dpm)2] (Δλ = 43 нм), что, как и предыдущее предположение, а также природа S0–S2-перехода, будут изучены дополнительно с участием квантово-химических расчетов.
Замена растворителя мало влияет на спектроскопические, но существенно изменяет фотофизические характеристики соединений: переход от циклогексана к этанолу увеличивает долю безызлучательных процессов в дезактивации энергии возбуждения за счет “перестроек” внутри ближней специфической сольватной оболочки комплексов в этаноле, которые тормозятся при замораживании растворов, увеличивая интенсивность люминесценции [8, 14]. Все изученные комплексные соединения, кроме [Zn((Ph)4-N-dpm)2], флуоресцируют, но имеют довольно низкие квантовые выходы флуоресценции: 0.4 в циклогексане и на 1–2 порядка меньше в этаноле (таблица 1). Для практического использования изучаемых комплексов необходимо изучение устойчивости в разных средах, как в основном состоянии, так и в электронно-возбужденных.
Устойчивость комплексов в протонодонорных средах
При подкислении растворов комплексы вступают в необратимые обменные реакции с соляной кислотой, и в результате изменяются электронные спектры поглощения и флуоресценции. Степень превращения комплексов в промежуточные и конечные продукты процессов протолитической диссоциации наглядно отражают изменения электронных спектров поглощения и флуоресценции. При добавлении и увеличении концентрации соляной кислоты в электронных спектрах поглощения этанольных растворов [Zn((CH3)4-dpm)2] и [Zn(Br2(CH3)4-dpm)2] интенсивная полоса комплекса преобразуется в полосу протонированного лиганда в составе соли с минеральной кислотой Hdpm·HCl с существенным коротковолновым сдвигом максимума и одним семейством изобестических точек (рис. 2, таблица 1), что подтверждает существование в растворе только двух устойчивых форм хромофоров. Такая картина спектральных превращений характерна для процессов протолитической диссоциации алкилзамещенных бис(дипиррометенатов) цинка(II) [22] в протонодонорных средах. Образование протонированной формы лиганда в качестве конечного продукта протолитического распада наблюдалось ранее и в реакциях алкил-BODIPY с кислотами [21].
Отнесение поглощения в сильнокислом растворе к поглощению протонированного по пиррольному азоту лиганда подтверждается совпадением спектров подкисленных растворов дипиррометенатов цинка и BF2 в соответствующих комплексах с одинаковыми по структуре лигандами: λmax = 465–467 нм для Zn-тетраметил- и тетраметил-BODIPY- лиганда и 485–489 нм для [Zn(Br2(CH3)4-dpm)2]-лиганда без комплексообразователей ([21], таблица 1). В то же время спектры поглощения соответствующих нейтральных дипиррометенатов различаются существенно (на 20 нм) ([14, 21, 22], таблица 1). В спектрах флуоресценции тоже наблюдаются изменения: при переходе лиганда в протонированную по пиррольному азоту форму интенсивность флуоресценции уменьшается из-за увеличения доли образующихся протонированных почти нефлуоресцирующих лигандов с незначительным коротковолновым сдвигом максимума в соответствии с изменениями поглощения (рис. 2). При этом введение атомов брома в α-положения пиррольных ядер повышает стабильность комплекса сильнее, чем для замещенных по β-позициям аналогов (таблица 1, рис. 5). Можно предположить, что вследствие высокой электроотрицательности атомов брома наблюдается понижение электронной плотности на координированных атомах азота и соответственно их основности, это затрудняет атаку координированных атомов азота протоном в начальной, лимитирующей процесс протолитической диссоциации хелатов. В результате описанный эффект доминирует над противоположным по знаку эффектом ослабления координационных связей Zn–N, вызванным тем же структурным фактором, т.е. оттоком электронной плотности на атомы брома.
На основании зависимости, полученной Ферстером [23]:
Стабильность [Zn(dpm)2]-комплексов продолжает увеличиваться при замене тетраметильных заместителей на более электроотрицательные тетрафенильные, что предположительно должно понижать электронную населенность связи Zn‒Nпирр по сравнению с алкилзамещенными лигандами и повышать стабильность комплексов (к отрыву комплексообразователя) в протонодонорных растворителях. Такое предположение требует подтверждения квантово-химическими расчетами.
В отличие от алкил- и галоген-замещенных dpm-лигандов, где при взаимодействии с протонодонорной сольватной оболочкой сразу наблюдается процесс протонирования пиррольного азота с последующим выходом Zn из комплекса, характеризуемый одной изобестической точкой (рис. 2), в комплексах цинка с Ph4-dpm-лигандами наблюдается две (для [Zn((Ph)4-dpm)2]) и даже три (для [Zn((Ph)4–N-dpm)2])-изобестических точки при изменении спектров поглощения по мере роста концентрации HCl (рис. 3, 4). Можно предположить, что взаимодействия [Zn(тетрафенил-dpm)2]-комплексов с протонами сольватной оболочки при меньших концентрациях протонов осуществляются с конкурирующим протоноакцепторным центром лиганда без выхода комплексообразователя из комплекса. Экспериментальные кривые титрования, построенные по падению поглощения комплекса на 525 и 570 нм (рис. 3) (при добавлении 10–6–10–4 М НС1), характеризуют уменьшение концентрации исходного [Zn((Ph)4-dpm)2] комплекса с исчезновением двух максимумов в видимой области и образованием промежуточного комплекса цинка(II) с лигандом, протонированным по альтернативному протоноакцепторному центру, который в дальнейшем может быть установлен с помощью расчетов. Этот процесс характеризуется значением (–lg[HCl]50(S0) = 2.8), рис. 3, сноска, кривая 1). При дальнейшем увеличении концентрации НСl до 10–3–10–2 М происходит протонирование дипиррометенового лиганда еще и по другому протоноакцепторному центру [Zn((Ph)4-dpm)2] (рост поглощения на 555–560 нм при –lg[HCl]50 = = 2.25 (рис. 3, кривая 2). Для определения структуры этого хромофора также необходимы квантово-химические исследования.
Экспериментальное изучение комплекса [Zn((Ph)4–N-dpm)2] в подкисленном этаноле характеризуется существованием как минимум трех процессов. Кривые титрования для этих процессов строятся: по уменьшению поглощения S0–S1-перехода на 650 нм (рис. 4, кривая 1): (‒lg[HCl]50(S0) = 4.2) при небольшом расширении полосы в длинноволновую сторону (до 670 нм). Следующая кривая 2 строится по уменьшению S0–S2-поглощения на 587 нм (рис. 4, кривая 2 – lg[HCl]50(S0) = 3). Наконец, кривая 3 строится по росту поглощения на 630 нм (‒lg[HCl]50(S0) = 1.5). Наличие на полученных спектральных картинах процессов протолитической диссоциации [Zn((Ph)4–N-dpm)2] трех семейств изобестических точек (рис. 4) свидетельствует о присутствии в растворах четырех форм дипиррометенового хромофора. Можно предположить, что по мере роста концентрации HCl в растворах реализуется система последовательных равновесий реакций протолитической диссоциации исходных гомолигандных хелатов до гетеролигандных [Zn(dpm)Cl], молекулярного лиганда Нdpm и, при значительном избытке кислоты, протонированной формы в виде соли Нdpm · НCl, что отражается в спектрах при соответствующих изменениях интенсивности и сдвигах максимумов полос поглощения. Нельзя исключать и возможность образования протонированной по мезо-азоту формы комплекса или лиганда. Расшифровка происходящих в этих системах процессов требует результатов отдельных исследований по получению в индивидуальном виде, идентификации и детальному изучению спектральных характеристик наиболее устойчивых из перечисленных выше форм соединений, а также квантово-химического моделирования обсуждаемых многостадийных процессов.
Насыщение роста поглощения на 555–560 нм ([Zn((Ph)4-dpm)2]) и 630 нм ([Zn((Ph)4–N-dpm)2]) и их коротковолновые сдвиги относительно S0–S1-поглощения комплексов (рис. 3, 4) указывают на завершение процесса выхода комплексообразователя из комплекса при высоких концентрациях НС1 (от 1 до 30%). Что касается отнесения промежуточных процессов к образованию определенных структур, так же, как и определения природы спектральных переходов S0–S1 и S0–S2 в комплексах цинка с тетрафенил-лигандами, на данном этапе, как указано выше, недостаточно данных для их однозначной интерпретации: необходимо применение квантово-химических ab-initio расчетов таких комплексов не только в основном, но и в возбужденных состояниях, а также значений молекулярного электростатического потенциала (МЭСП) в разных электронных состояниях, что выходит за рамки настоящей статьи.
Приведенные результаты показывают, что в комплексах тетрафенилдипиррометена с Zn наблюдается коротковолновый сдвиг S0–S1-полосы поглощения при переходе от нейтрального комплекса к протонированному лиганду (от 570 до 555 нм для [Zn((Ph)4-dpm)2] и от 650 до 630 нм для [Zn((Ph)4–N-dpm)2] ) с выходом комплексообразователя. Такой сдвиг, согласно [23], является причиной повышения стабильности дипиррометенатов в $S_{1}^{{{\text{Ф }} - {\text{К }}}}$-состоянии по сравнению с S0 (таблица 1), что может проявиться при изучении их фотостабильности в этанольных растворах.
Фотохимические свойства
Наряду со стабильностью в протонодонорных средах была изучена фотостабильность комплексов под действием лазерного облучения для успешного практического применения изученных комплексов в различных оптических устройствах. Фотостабильность очень важна, потому что именно фотопревращения ответственны за ресурсные характеристики этих соединений в оптических устройствах.
Как показали результаты, самым фотостабильным из цинковых комплексов при возбуждении в длинноволновой полосе (532 нм) является [Zn((Ph)4-dpm)2] (таблица 1), что согласуется с наиболее высокой стабильностью аналогичных тетрафенилзамещенных BODIPY комплексов [21] и обеспечивается структурой лиганда. Фотораспад в этом случае связан, по-видимому, не с отрывом комплексообразователя, а с менее вероятным образованием триплетно-возбужденных состояний дипиррометената цинка и менее эффективной генерацией 1О2 [8, 24], с которым цинковые комплексы могут далее реагировать. При замене заместителей с фенильных на метильные устойчивость комплексов и их фотостабильность падает, так как в данном случае возбуждение осуществляется в УФ (355 нм), поскольку линейное поглощение на 532 отсутствует (рис. 2, таблица 1), при этом может возрастать интеркомбинационная конверсия, как показано в [8, 14, 24]. Дибромзамещенные тетраметил- и диметил-дипентил-цинковые комплексы при УФ-возбуждении (355 нм) демонстрируют еще более низкую фотостабильность (квантовый выход фотопревращений выше на два порядка, таблица 1). Это еще раз подтверждает, что механизм фотопревращений дипиррометенатов сложный, обусловленный как стабильностью в протонодонорных средах, так и более эффективным образованием 1О2 галогенированными комплексами за счет увеличения выхода Т-состояний и последующего взаимодействия с ним исходных [Zn(Br2(CH3)4-dpm)2] и [Zn(Br2(С5Н11)2(СH3)2-dpm)2] комплексов. Самый фотостабильный из изученных здесь [Zn((Ph)4-dpm)2]-комплекс, а не наиболее устойчивый в (S1)Ф–К-состоянии в протонодонорных средах [Zn((Ph)4–N-dpm)2]-комплекс. Это означает, что фотопревращения комплексов [Zn-(dpm)2] осуществляются при суммарном участии специфической сольватной оболочки в возбужденном состоянии и эффективности образования в таких комплексах Т-состояний с последующей генерацией 1О2, для которой [Zn((Ph)4-N-dpm)2]-комплекс наиболее предпочтителен, как показано в [8, 24]. Для успешного практического использования [Zn-(dpm)2] необходимо найти способы повышения фотостабильности. Возможный путь – добавление в раствор дипиррометенатов диазабициклооктана (DABCO) [7, 21], известного тушителя триплетов и синглетного кислорода, кроме того, это соединение обладает щелочной реакцией, что предотвращает атаку протона.
ЗАКЛЮЧЕНИЕ
Выполненные исследования показали, что устойчивость комплексов [Zn(dpm)2] с разной структурой лиганда в протонодонорных растворителях, в целом, ниже, чем BODIPY-комплексов с аналогичными лигандами. Совпадение спектров поглощения протонированных лигандов алкил-дипиррометенатов бора(III) и цинка(II) с одинаковой структурой лигандов подтверждает предложенный механизм распада дипиррометенатов в протонодонорных средах, связанный с протонированием пиррольного азота с последующим выходом комплексообразователя из комплекса.
Комплексы цинка с алкилпроизводными лигандами имеют наименьшую устойчивость в протонодонорных средах по сравнению с другими замещенными: замена водорода в β-положении dpm и СН3-групп на более электроотрицательные атомы галогенов увеличивает стабильность таких комплексов в протонодонорных растворителях: ‒lg[HCl]50(S0) = 4.5 вместо 5. Замена метильных заместителей фенильными в [Zn((Ph)4-dpm)2] вызывает увеличение устойчивости: –lg[HCl]50 = = 2.25 вместо 5 в S0-состоянии.
Введение в мезо-положение лиганда атома азота в [Zn((Ph)4–N-dpm)2]-комплексе делает его самым устойчивым из комплексов цинка в протонодонорных средах при –lg[HCl]50(S0) = 1.5. В возбужденном франк-кондоновском состоянии устойчивость всех комплексов в протонодонорных средах повышается.
Полученные результаты подтверждают суммарный эффект устойчивости комплексов в возбужденных состояниях и их взаимодействия с фотосенсибилизированным 1О2. Для управления промежуточными процессами в тетрафенилзамещенных дипиррометенатах цинка в протонодонорных растворителях и целенаправленного выбора определенных структур для конкретных оптических устройств необходимо использовать комплексный подход, сочетающий совместное экспериментальное и квантово-химическое изучение возбужденных состояний таких комплексов.
Результаты исследования были получены в рамках выполнения государственного задания Минобрнауки России (проект № 4.6027.2017/8.9), при поддержке РФФИ (код проекта № 18-33-00284), а также Программы повышения конкурентоспособности ТГУ.
Список литературы
Loudet A., Burgess K. // Chem. Rev. 2007. V. 107. P. 4891.
Ziessel R., Ulrich G., Harriman A. // New J. Chem. 2007. V. 31. P. 496.
Кузнецова Р.Т., Аксенова Ю.В., Орловская О.О. и др. // ХВЭ. 2012. Т. 46. № 6. С. 464.
Bumagina N.A., Antina E.V., Sozonov D.I. // J. Luminesc. 2017. V. 183. P. 315.
Costela A., Garcia-Moreno I., Pintado-Sierra M. et al. // J. Phys. Chem. A. 2009. V. 113. P. 8118.
Kuznetsova R.T., Aksenova Yu.V., Tel’minov E.N. et al. // Opt. Spectr. 2012. V. 112. P. 746.
Kuznetsova R.T., Aksenova Yu.V., Solodova T.A. et al. // Quant. Electron. 2014. V. 44. P. 206.
Kuznetsova R.T., Aksenova Iu.V., Bashkirtsev D.E. et al. // J. Photochem. Photobiol. A: Chem. 2018. V. 354. P. 147.
Papkovsky D.B., Dmitriev R.I. // Chem. Soc. Rev. 2013. V. 42. P. 8700.
Ermolina E.G., Kuznetsova R.T., Aksenova Yu.V. et al. // Sens. Actuators. B. 2014. V. 197. P. 206.
Epelde-Elezcano N., Martinez-Martinez V., Pena-Cabrera E. et al. // RCS. Adv. 2016. V. 6. P. 41991.
Teets T.S., Partyka D.V., Updegraff J.B. III. et al. // Inorg. Chem. 2008. V. 47. P. 2338.
Baudron S.A. // Dalton Trans. 2013. V. 42. P. 7498.
Kuznetsova R.T., Aksenova Yu.V., Bashkirtsev D.E. et al. // High Energy Chem. 2015. V. 49. P. 16.
Дудина Н.А., Березин М.Б., Антина Е.В. и др. // Химия гетероциклических соединений. 2013. № 12. С. 1878.
Antina E.V., Berezin N.B., Dudina N.A. et al. // Russ. J. Gen. Chem. 2010. V. 80. № 6. P. 1214.
Berezin M.B., Semeikin A.S., Yutanova S.L. et al. // Ibid.2012. V. 82. P. 1287.
Berezin M.B., Semeikin A.S., Antina E.V. et al. // Ibid. 1999. V. 69. P. 1949.
Antina E.V., Guseva G.B., Dudina N.A. et al. // Russ. J. Inorg. Chem. 2010. V. 55. P. 1172.
Dudina N.A., Nikonova A.Yu., Antina E.V. et al. // Chem. Heterocycl. Comp. 2014. V. 49. P. 1740.
Aksenova Iu.V., Kuznetsova R.T., Tel’minov E.N. et al. // Rus. J. Phys. Chem. A. 2016. V. 90. № 2. P. 349.
Антина Л.А., Гусева Г.Б., Вьюгин А.И., Антина Е.В. // Журн. физ. химии. 2012. Т. 86. № 11. С. 1759.
Гусева Г.Б., Антина Е.В., Березин М.Б. // Коорд. химия. 2003. Т. 29. № 10. С. 745.
Aksenova Iu.V., Kuznetsova R.T., Pozdnyakov I.P. et al. // J. Photochem. Photobiol. A: Chem. 2017. V. 344. P. 206.
Паркер С. Фотолюминесценция растворов. М.: Мир, 1972. 510 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии