

Журнал физической химии, 2019, T. 93, № 4, стр. 558-564
Ступенчатый механизм реакции рений(V)порфирина с пиридином и химическое строение донорно-акцепторного комплекса
Н. Г. Бичан a, Е. Н. Овченкова a, Т. Н. Ломова a, *
a Институт химии растворов им. Г.А. Крестова Российской академии наук
Иваново, Россия
* E-mail: tnl@isc-ras.ru
Поступила в редакцию 19.07.2018
Аннотация
Методами химической термодинамики и УФ-, видимой, ИК-, 1H ЯМР- и масс-спектрометрии изучена сложная реакция (5,15-бис(4'-метоксифенил)-3,7,13,17-тетраметил-2,8,12,18-тетраэтилпорфинато)(оксо)(хлоро)рения(V) (O=Re(Cl)P) с пиридином (Py) и химическое строение продукта. Установлена природа, стехиометрия и определены количественные параметры двухсторонних ступенчатых реакций в ходе сложной реакции. На первой стадии имеет место обратимое замещение иона Cl‒ молекулой пиридина с константой K1, равной (4.7 ± 1.1) × 102 л/моль с образованием катионного комплексного соединения [O=Re(Py)P]+Cl–, на второй – обратимое присоединение двух молекул пиридина ([O=Re(Py)3P]+Cl–) с константой K2 = (0.10 ± 0.03) л2/моль2. Изученная реакция является модельной по отношению к процессам в самособирающихся системах на основе металлопорфиринов и пиридильных производных наноформ углерода для формирования активных слоев с фотоиндуцированным разделением зарядов в гибридных солнечных элементах.
Металлоорганические соединения со связями M–O и M–N привлекают широкое внимание как активные промежуточные соединения в биологически важных реакциях [1, 2] и успешно используются в качестве эффективных катализаторов во многих промышленных процессах [3, 4]. Высокое окислительное состояние металла в катализаторе позволяет проводить реакцию в условиях “открытых колб”, без необходимости строгого исключения воздуха и влаги [5]. Богатая координационная химия рения используется в дизайне радиофармацевтических препаратов [6–8], поскольку его изотопы 186Re и 188Re испускают излучение. Рений также широко используется в качестве нерадиоактивной модели технеция, которая находит применение в ядерной медицине [9].
В литературе описывается синтез, свойства и применение соединений рения с карбенами [10], гетероциклическими молекулами [5, 11, 12], каликсаренами [6], порфиринами [10, 13–15]. Разнообразные возможности модификации соединений рения делают их перспективными платформами для формирования на их основе супрамолекулярных [16, 17], µ-оксодимерных систем и комплексов с координированным по пероксо-типу молекулярным кислородом [15, 16, 18]. Возможность вариации аксиальных групп в экваториальных макроциклических комплексах рения делает их перспективными в сенсорике [19], катализе [13] и фотодинамической терапии, где соединения рения уже были испытаны и зарекомендовали себя [20, 21]. Замещение и присоединение различных групп по аксиальной оси высокозарядных оксопорфириновых комплексов рения существенно влияет на их основные спектральные характеристики, устойчивость по связям Re–N [22], способность образовывать π-катион радикальные формы, взаимодействовать с молекулярным кислородом [10, 15]. Дальнейшее развитие указанных областей химии рения требует изучения физической химии его соединений, в частности реакционной способности к субстратам различной природы, и механизмов сложных реакций с их участием.
Настоящая работа посвящена исследованию реакций (5,15-бис(4'-метоксифенил)-3,7,13,17-тетраметил-2,8,12,18-тетраэтилпорфинато)(оксо)(хлоро)рения(V) (O=Re(Cl)P)
с пиридином (Py). Реакция металлопорфирина (MP) с Py рассматривается как простая модель самосборки в донорно-акцепторных системах на основе металлопорфиринов и пиридильных производных таких наноформ углерода, как фуллерены [23, 24]. Порфирин-фуллереновые супрамолекулы, как уже подтверждено экспериментальными исследованиями по гашению флуоресценции и расчетами [25], проявляют свойство фотоиндуцированного переноса электрона (PET, photoinduced electron transfer) и могут “работать” в качестве активных слоев гибридных солнечных элементов.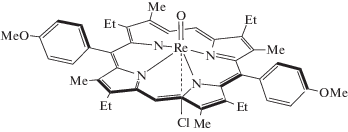
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
(5,15-бис(4'-Метоксифенил)-3,7,13,17-тетраметил-2,8,12,18-тетраэтилпорфинато)(оксо)(хлоро)рений(V), O=Re(Cl)P был получен из (5,15-бис(4'-метоксифенил)-3,7,13,17-тетраметил-2,8, 12,18-тетраэтилпорфинато)(оксо)(фенокси) рения(V) (O=Re(PhO)P), предварительно синтезированного реакцией H2ReCl6 с соответствующим порфирином согласно методике [26], пропусканием через его раствор в CH2Cl2 газообразного HCl в течение 10 мин. За это время цвет раствора менялся с зелено-желтого на винно-красный. Порфирин синтезирован [27] и любезно предоставлен для исследований профессором А.С. Семейкиным. Выход (O=Re(Cl)P) близок к 100%. Электронный спектр поглощения (ЭСП) (CH2Cl2), λмакс., нм (lg ε): 687 (3.15), 633 (3.48), 526 (3.95), 355 (4.52). 1H ЯМР спектр (CDCl3), м. д., J, Гц: 10.81 (с, 2H, CHмезо), 8.16 (д, 2H, Hо, J = 8.3); 8.08 (д, 2H, Hо, J = 8.3); 7.73 (кв, 1H, Hм, J = 8.3); 7.55 (кв, 1H, Hм, J = 7.3); 7.39 (м, 2H, Hм); 4.18 (м, 8Н, –CH2–); 2.76 (т, 12Н, CH3Et, J = 7.3); 2.20 (с, 6Н, p-OCH3); 1.96 (т, 12Н, CH3, Me, J = 7.6). Масс-спектр (MALDI TOF): m/z 891.35 [M–Cl]+. Вычислено для C46H48ReN4O3 891.12.
Синтез и свойства 1'-N-метил-2'-(пиридин-4-ил)пирролидино[3',4':1,2][60 ] фуллерена описаны ранее [25].
Py (“ч. д. а”) высушивали в течение двух суток над гранулами KOH, затем перегоняли (tкип = 115.3°С). Толуол осушали гидроксидом калия и перед использованием перегоняли (tкип = 110.6°С). Содержание воды определяли титрованием по Фишеру, оно не превышало 0.01%.
Реакцию O=Re(Cl)P с Py исследовали в дихлорметане спектрофотометрическим методом молярных отношений при 298 K. Готовили серию растворов в CH2Cl2 с постоянной концентрацией O=Re(Cl)P (2.03 × 10–5 моль/л) и различными концентрациями пиридина (8.23 × 10–5–11.17 моль/л). Константы равновесия реакций с Py (K) определяли по уравнению для трехкомпонентной равновесной системы методом наименьших квадратов (НК) с использованием программы Microsoft Excel
(1)
$\begin{gathered} K = \frac{{\left( {{{A}_{{\text{i}}}} - {{A}_{0}}} \right){\text{/}}\left( {{{A}_{\infty }} - {{A}_{0}}} \right)}}{{1 - \left( {{{A}_{{\text{i}}}} - {{A}_{0}}} \right){\text{/}}\left( {{{A}_{\infty }} - {{A}_{0}}} \right)}} \times \\ \times \;\frac{1}{{{{{\left( {{{C}_{{{\text{Py}}}}} - C_{{{\text{O}} = {\text{Re(Cl)P }}}}^{0}\frac{{{{A}_{{\text{i}}}} - {{A}_{0}}}}{{{{A}_{\infty }} - {{A}_{0}}}}} \right)}}^{n}}}}, \\ \end{gathered} $ЭСП, ИК-, флуоресцентные, 1Н ЯМР- и масс-спектры зарегистрированы, соответственно, на спектрофотометре Agilent 8453, спектрометрах VERTEX 80v, “Avantes” AvaSpec-2048, AVANCE-500 (Bruker, Германия) и Shimadzu Confidence.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Для изучения реакций O=Re(Cl)P с Py в качестве растворителя был выбран дихлорметан, так как он обладает хорошей растворяющей способностью и химической инертностью по отношению к данному комплексу. Выбор спектрофотометрии в качестве метода исследования обусловлен тем, что электронные спектры поглощения порфириновых комплексов рения(V) очень точно отражают окислительное состояние Re и чувствительны к смене аксиальных групп. Так, в работах [14, 26] для порфиринов рения(V) состава O=Re(X)P было проведено сравнение электронных спектров поглощения при смене ковалентно присоединенных X-лигандов и установлено разные соотношение интенсивностей полос в области 350–460 нм и положение полосы переноса заряда в области 440–550 нм.
На рис. 1 представлены ЭСП O=Re(Cl)P в смесях дихлорметан–Py в зависимости от концентрации Py при 298 K и соответствующие кривые титрования. Использовался широкий диапазон концентраций Py (8.23 × 10–5–11.17 моль/л). Отчетливо выделяются два семейства спектральных кривых, в которых сохраняются изобестические точки (рис. 1). Первая серия полос наблюдается при СPy = 8.23 × 10–5–2.23 × 10–2 моль/л. Здесь происходят небольшие изменения спектра – уменьшается оптическая плотность полосы переноса заряда при 526 нм и появляется новое поглощение в области 480 нм, несколько понижается интенсивность полосы Соре при 355 нм. Вторая серия полос при СPy = 2.23 × 10–2–11.17 моль/л характеризуется существенными изменениями ЭСП. Поглощение в области 480 нм растет более интенсивно с гипсохромным смещением максимума до 463 нм, в области 405 нм появляется новое поглощение, а интенсивность полосы Соре снижается с 0.7 до 0.45 единиц оптической плотности.
Рис. 1.
Изменение ЭСП O=Re(Cl)P (СO=Re(Cl)P 2.03 × 10–5 моль/л) с добавками пиридина 8.23 × 10–5–2.23 × 10–2 моль/л (а) и 2.23 × 10–2–11.17 моль/л (б). На выносках – соответствующие кривые спектрофотометрического титрования, полученные на рабочих длинах волн 526 (а) и 463 (б) нм.
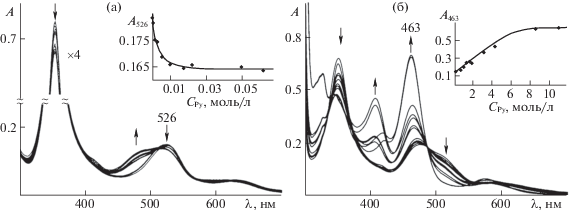
Наличие изобестических точек в обеих сериях кривых и результат спектрофотометрического титрования в пределах каждой из серий вместе с изучением тенденций изменения ЭСП при разбавлении растворов по пиридину показывают, что реакция O=Re(Cl)P с Py в дихлорметане протекает в две обратимые стадии.
Анализ зависимостей lg I–lg CPy (рис. 2) позволяет определить число молекул Py, участвующих в равновесии. Для первой стадии (концентрации Py 8.23 × 10−5–2.23 × 10−2 моль/л), равновесие в которой устанавливается сразу после смешивания растворов O=Re(Cl)P и Py, определено стехиометрическое соотношение O=Re(Cl)P : Py 1 : 1 и константа равновесия K1, равная (4.7 ± 1.1) × × 102 л/моль, что позволяет записать левую часть равновесия (2). Правая часть записывается с учетом изучения изменений ЭСП в ходе титрования и данных работ [14, 28, 29] по влиянию замещения/присоединения аксиальных лигандов в оксопорфириновых комплексах O=МV(X)P, где М – W, Mo, Re.
(2)
${\text{O}}{\kern 1pt} = {\kern 1pt} {\text{Re}}({\text{Cl}}){\text{P}} + {\text{Py}}\;\overset {{{K}_{1}}} \leftrightarrows \;{\text{O}}{\kern 1pt} = {\kern 1pt} {\text{Re}}({\text{Cl}})({\text{Py}}){\text{P}}.$Рис. 2.
Зависимости lg I от lg CPy для реакции O=Re(Cl)P с добавками пиридина 8.23 × 10–5 – 2.23 × × 10–2 моль/л (R2 = 0.97, tg α = 0.91) (1) и 2.23 × 10–2–11.17 моль/л (R2 = 0.98, tg α = 2.09) (2) при 298 K.
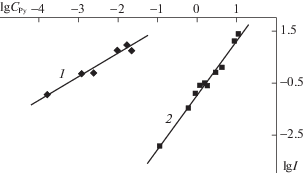
Вторая стадия (концентрации Py 2.23 × 10−2–11.17 моль/л) характеризуется временем достижения равновесия 10–15 мин, значительно с меньшей константой K2, равной (0.10 ± 0.03) л2/моль2 и стехиометрическим соотношением O=Re(Cl)P : Py 1 : 2 (рис. 1б)
(3)
${\text{O}}{\kern 1pt} = {\kern 1pt} {\text{Re}}({\text{Cl}})({\text{Py}}){\text{P}} + 2{\text{Py}}\;\overset {{{K}_{2}}} \leftrightarrows \;{{\left[ {{\text{O}}{\kern 1pt} = {\kern 1pt} {\text{Re}}{{{({\text{Py}})}}_{3}}{\text{P}}} \right]}^{ + }}{\text{C}}{{{\text{l}}}^{--}}.$Структура продукта реакции (3) подтверждена данными ИК- и 1Н ЯМР-спектроскопии, излагаемыми ниже.
Известно, что аксиальное связывание оказывает существенное влияние на колебательные частоты металлопорфириновых молекул [23]. В нашем случае, при присоединении трех молекул Py к O=Re(Cl)P наибольшие изменения, а именно низкочастотный сдвиг на 2–5 см–1, претерпевают скелетные колебания макроцикла (ν(C=N), δ(C–H) метиновых групп), что разумно связать с изменением положения атома металла в плоскости макроцикла [23]. Также на этот факт указывает высокочастотный сдвиг ν(Re–N) на 5 см–1. Колебания же периферийных заместителей макроцикла (фенильные и алкильные группы) остаются практически неизменными (табл. 1).
Таблица 1.
ИК-спектры в KBr
| ν, см–1 | Отнесение полос | ||
|---|---|---|---|
| O=Re(Cl)P | Py | [O=Re(Py)3P]+ ⋅ ⋅ Cl– | |
| 3065 3030 |
3031 |
ν(C–H)Ph | |
| 2956 2910 |
2963 2930 |
ν(С–Н)Py | |
| 2873 2856 |
2871 2854 |
ν(C–H) алкильных групп | |
| 1633 1581 |
1637 | колебания кольца Py | |
| 1608 1570 1512 |
1609 1571 1512 |
ν(C=C) | |
| 1482 | 1487 | ν(С–N)Py | |
| 1460 | 1455 | ν(C=N) | |
| 1441 | 1441 | δ(C–H) | |
| 1438 | 1398 | ν(С–N)Py | |
| 1379 | 1378 | ν(C–N) | |
| 1290 1249 |
1288 1245 |
δ(C–H) метиновых групп | |
| 1217 1147 |
1174 | ν(С–Н)Py | |
| 1104 1058 |
1104 | δ(C–H) | |
| 1069 1031 991 |
δ(С–Н)Py | ||
| 1026 982 |
981 |
ν(Cβ–С) алкильных групп | |
| 963 | 962 | ν(Re=O) | |
| 826 846 790 749 723 |
790 750 720 |
γ(C–H) | |
| 748 704 603 |
698 |
δ(С–Н)Py | |
| 542 | ν(Re–NPy) | ||
| 464 | 471 | ν(Re–N) | |
| 379 | ν(Re–Cl) | ||
В ИК-спектре продукта реакции (3) [O=Re(Py)3P]+Cl– наблюдаются новые сигналы координированного Py при 2963, 2930, 1637, 1487, 1398, 1174 и 698 см–1, отсутствующие в спектре исходного комплекса, и претерпевающие сдвиг на 4–40 см–1 по сравнению с колебаниями чистого Py (табл. 1). Новый сигнал при 542 см–1 предположительно соответствует связи Re–NPy [30]. Наличие в ИК-спектре продукта сигнала при 962 см‒1, соответствующего ν(Re=O), подтверждает химическую природу [O=Re(Py)3P]+Cl–.
O=Re(Cl)P имеет 1Н ЯМР-спектр, характерный для диамагнитных MP, с четко разделенными сигналами (Экспериментальная часть) и хорошо согласуется с литературными данными [26]. Введение Py (в концентрации, приводящей к образованию [O=Re(Py)3P]+Cl–) в раствор O=Re(Cl)P в CDCl3 сопровождается появлением трех новых сигналов протонов пиридинового кольца при 8.58, 7.62 и 7.24 м.д., которые претерпевают небольшой сильнопольный сдвиг (0.04–0.06 м.д.) по сравнению с сигналами некоординированного пиридина [31]. Аксиальное присоединение Py приводит к смещению (по сравнению со спектром O=Re(Cl)P в CDCl3) в сильное поле сигналов мезо-протонов макроцикла (на 0.06 м.д.) и сигналов орто-протонов фенильных заместителей (≈ на 0.1 м.д.), тогда как протоны –OCH3 группы смещаются в слабое поле (≈ на 0.1 м.д.). Такие сдвиги сигналов протонов макроцикла в составе порфирин-пиридиновой супрамолекулы [O=Re(Py)3P]+Cl– можно объяснить уменьшением эффекта кольцевого тока порфиринового макроцикла за счет появления дополнительного положительного заряда на атоме Re при присоединении молекул Py.
Сравнение констант равновесия K1 и K2 показывает, что присутствие в первой координационной сфере O=Re(Cl)(Py)P одной молекулы Py препятствует дальнейшему связыванию Py на второй стадии процесса. Вытеснение Cl– из координационной сферы комплекса O=Re(Cl)(Py)P и присоединение еще двух молекул Py протекает во времени.
Таким образом, в ходе реакции O=Re(Cl)P с Py из раствора могут быть выделены два пиридин-содержащих соединения рений(V)порфирина – O=Re(Cl)(Py)P и [O=Re(Py)3P]+Cl–, электронные спектры поглощения для которых различаются положением и соотношением интенсивности полос. ЭСП O=Re(Cl)(Py)P в CH2Cl2 (λмакс., нм) – 353 (I), 484 (плечо), 518 (II) (II > I), [O=Re(Py)3P]+Cl– – 409 (I), 461 (II), 545 (III) (II > I > III).
Сравнение свойств полученной донорно-акцепторной системы с ранее изученными аналогами на основе оксопорфириновых комплексов с отличающимися набором заместителей и центральным атомом металла (табл. 2) позволило сделать вывод об относительной устойчивости полученных порфирин-пиридиновых комплексов. Как видно из табл. 2, реакция оксо-координированных MP (M = Re, W, Mo) с Py представляет собой сложный процесс и заканчивается во всех случаях образованием донорно-акцепторных комплексов стехиометрического состава 1:3. Реакция O=Re(Cl)P характеризуется более низкими значениями констант равновесия по сравнению с аналогичными реакциями O=W(OH)TPP и O=Mo(OH)TPP. Последовательность изменения величин K повторяет ряд изменения ковалентного радиуса W (162 пм), Mo (154 пм) и Re (151 пм) [32]. Однако данная корреляция, возможно, случайная, так как резко изменяется природа ступенчатых реакций MP с Py. Если реакция O=Re(Cl)P с Py не затрагивает имеющуюся в составе молекулы оксо-группу, то в случае комплексов Mo и W последняя становится реакционным центром на второй и третьей стадии, соответственно, реакции с Py. Установленная зависимость между константами устойчивости порфирин-пиридиновых супрамолекул и природой металла порфириновой составляющей донорно-акцепторного комплекса может быть использована при разработке и синтезе донорно-акцепторных систем с прогнозируемой устойчивостью, что является важным фактором при оптимизации химических структур соединений для создания на их основе гибридных материалов – каталитических систем, сенсоров, фотоактивных компонентов для фотовольтаических устройств.
Таблица 2.
Количественные параметры реакций металлопорфиринов с пиридином в толуоле
| Уравнение реакции | Kn, л/моль | Источник |
|---|---|---|
| O=Re(Cl)P + Py $\overset {{{K}_{1}}} \leftrightarrows $ O=Re(Cl)(Py)P | (4.7 ± 1.1) × 102 | |
| O=Re(Cl)(Py)P + 2Py $\overset {{{K}_{2}}} \leftrightarrows $ [O=Re(Py)3P]+Cl– | (0.10 ± 0.03) л2/моль2 | |
| O=W(OH)TPP + Py $\overset {{{K}_{1}}} \leftrightarrows $ O=W(OH)(Py)TPP | (1.33 ± 0.22) × 104 | [25] |
| O=W(OH)(Py)TPP + Py $\overset {{{K}_{2}}} \leftrightarrows $ [O=W(Py)2TPP]+OH– | (8.42 ± 1.58) × 103 | |
| [O=W(Py)2TPP]+OH- + Py + H2O $\overset {{{K}_{3}}} \leftrightarrows $ [(OH)W(Py)3TPP]2+ 2OH– | 89 ± 13 | |
| O=Mo(OH)TPP + Py $\overset {{{K}_{1}}} \leftrightarrows $ [O=Mo(Py)TPP]+OH– | (9.1 ± 1.2) × 103 | [26] |
| [O=Mo(Py)TPP]+OH– + Py + H2O $\overset {{{K}_{2}}} \leftrightarrows $ [(OH)Mo(Py)2TPP]+ 2OH– | 39.3 ± 5.2 | |
| [(OH)Mo(Py)2TPP]+ 2OH– + Py $\overset {{{K}_{3}}} \leftrightarrows $ [Mo(Py)3TPP]3+ 3OH– | 1.0 ± 0.1 |
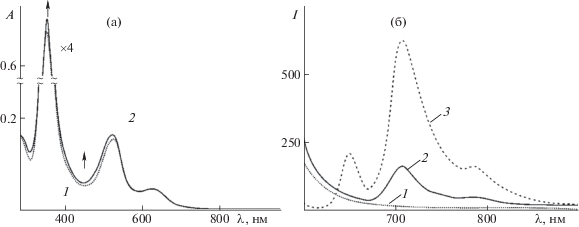
Образование устойчивого донорно-акцепторного соединения рений(V)порфирина с первой молекулой Py позволяет предложить полученные данные для использования при создании донорно-акцепторных диад металлопорфиринов с пиридильными производными наноуглерода. В работе [16] количественно изучен процесс самоорганизации в системах 5-монофенил-2,3,7,8, 12,13,17,18-октаэтилпорфинато)(оксо)(феноксо) рений(V)–2'-(пиридин-4-ил)-5'-(пиридин-2-ил)-1'-(пиридин-3-илметил)-2',4'-дигидро-1'H-пирроло[3',4':1,2][С60-Ih] [5, 6]фуллерен – дихлорметан, заканчивающийся образованием катионного донорно-акцепторного 1:1 комплекса с лигандом PhO– во внешней координационной сфере. Подобные порфирин-фуллереновые диады обладают свойством переноса электрона от донора (макроцикл) к акцептору (фуллереновый фрагмент) при фотовозбуждении (PET). Для флуоресцирующих металлопорфиринов (комплексы марганца(III), молибдена(V) и др.) свойство PET обнаруживается по тушению флуоресценции при образовании донорно-акцепторного комплекса [25, 33]. Предварительное тестирование комплекса рения O=Re(Cl)P на координирующую способность в отношении акцептора 1'-N-метил-2'-(пиридин-4-ил)пирролидино[3',4':1,2][60 ] фуллерена (PyC60)
с помощью электронной спектроскопии поглощения и флуоресценции показывает положительный результат – образование донорно-акцеторного порфирин-фуллерена в среде CH2Cl2. Несмотря на менее выраженные изменения в ЭСП O=Re(Cl)P в присутствии избытка замещенного фуллерена по сравнению с пиридином, а именно незначительные рост интенсивности полосы при 355 нм и гипсохромное смещение полосы переноса заряда (до 524 нм, рис. 3а), образование донорно-акцепторного комплекса и перераспределение заряда в нем можно зафиксировать по значительному падению интенсивности флуоресценции свободного замещенного фуллерена по сравнению со связанным металлопорфирином (рис. 3б) фуллереном.
Рис. 3.
Электронные спектры поглощения (а) и флуоресценции (б) в дихлорметане при 298 K O=Re(Cl)P (1), его комплекса с PyС60 (2) и PyС60 (3). СO=Re(Cl)P = 2.13 × 10–5 моль/л (1, 2), ${{C}_{{{\text{Py}}{{{\text{C}}}_{{60}}}}}}$ = 8.32 × 10–5 моль/л (2, 3). λexc 525 (1, 2) и 395 (3) нм.
Дополнительный аргумент в пользу расширения возможностей дизайна фотоактивных супрамолекул с использованием рений(V)порфиринов дает ссылка на результаты по модификации титанового фотоанода пленками донорно-акцептроных комплексов на основе d-металлов, в том числе рения, которые показывают увеличение конверсии световой энергии [25, 34] в присутствии модификаторов.
Работа выполнена на оборудовании Центра коллективного пользования научным оборудованием “Верхневолжский региональный центр физико-химических исследований”. Спектры флуоресценции сняты к.х.н. Губаревым Ю.А., которому авторы выражают благодарность. Синтез пирролидинил[60 ] фуллерена выполнен в рамках гранта РФФИ 16-03-00578.
Список литературы
Leeladee P., Jameson G.N.L., Siegler M.A., Kumar D. et al. // Inorg. Chem. 2013. V. 52. P. 4668.
Leung S.K.-Y., Tsui W.-M., Huang J.-S., Che C.-M. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 16629.
Abbina S., Bian S., Oian C., Du G. // ACS Catal. 2013. V. 3. P. 678.
Man W.-L., Lam W.W.Y., Yiu S.-M., Lau T.-C. et al. // J. Am. Chem. Soc. 2004. V. 126. P. 15336.
Gryca I., Machura B., Shul’pina L.S., Shul’pin G.B. // Inorg. Chim. Acta. 2017. V. 455. P. 683.
Van Bommel K.J.C., Verbom W., Hulst P., Kooijman H., et al. // Inorg. Chem. 2000. V. 39. P. 4099.
John E., Thakur M.L., De Fulvio J., McDevitt M.R., et al. // J. Nucl. Med. 1993. V. 34. P. 260.
Jürgens S., Herrmann W.A., Kühn F.E. // J. Organomet. Chem. 2014. V. 751. P. 83.
Van Bommel K.J.C., Verboom W., Kooijman H., Spek A.L. et al. // Inorg. Chem. 1998. V. 37. P. 4197.
Бичан Н.Г., Тюляева Е.Ю., Ломова Т.Н. // Журн. физ. химии. 2014. Т. 88. С. 1530.
Shi C., Jia G. // Coord. Chem. Rev. 2013. V. 257. P. 666.
Bernardo J.R., Sousa S.C.A., Florindo P.R., Wolff M., et al. // Tetrahedron. 2013. V. 69. P. 9145.
Toganoh M., Fujino K., Ikeda S., Furuta H. // Tetrahedron Lett. 2008. V. 49. P. 1488.
Bichan N.G., Tyulyaeva E.Yu., Khodov I.A., Lomova T.N. // J. Mol. Struct. 2014. V. 1061. P. 82.
Бичан Н.Г., Тюляева Е.Ю., Ломова Т.Н. // Журн. неорг. химии. 2014. Т. 59. С. 1692.
Бичан Н.Г., Тюляева Е.Ю., Ломова Т.Н., Семейкин А.С. // Журн. орган. химии. 2014. Т. 50. С. 1376.
Santosh G., Ravikanth M. // Inorg. Chim. Acta. 2005. V. 358. P. 2671.
Lomova T.N., Mozhzhukhina E.G., Tyulyaeva E.Yu., Bichan N.G. // Mend. Commun. 2012. V. 22. P. 196.
Ramdass A., Sathish V., Velayudham M., Thanasekaran P. et al. // Inorg. Chem. Com. 2013. V. 35. P. 186.
Jurisson S.S., Lydon J.D. // Chem. Rev. 1999. V. 99. P. 2205.
Jia Z., Deng H., Pu M., Luo Sh. // J. Nucl. Med. Mol. Imaging. 2008. V. 35. P. 734.
Тюляева Е.Ю., Бичан Н.Г., Ломова Т.Н. // Журн. неорган. химии. 2013. С. 1522.
Овченкова Е.Н., Клюева М.Е., Ломова Т.Н. // Журн. неорган. химии. 2017. Т. 62. С. 1490.
Ovchenkova E.N., Bichan N.G., Semeikin A.S., Lomova T.N. // Macroheterocycles. 2018. V. 11. № 1. P. 79.
Ovchenkova E.N., Bichan N.G., Kudryakova N.O. et al. // Dyes and Pigments. 2018. V. 153. P. 225.
Тюляева Е.Ю., Бичан Н.Г., Ломова Т.Н., Семейкин А.С. // Журн. неорган. химии. 2017. Т. 62. С. 1585.
Lubimova T.V., Syrbu S.A., Semeikin A.S. // Macroheterocycles. 2016. V. 9. P. 141.
Типугина М.Ю., Ломова Т.Н. // Журн. неорган. химии. 2004. Т. 49. С. 1285.
Типугина М.Ю., Ломова Т.Н., Моторина Е.В. // Координац. химия. 2005. Т. 31. С. 380.
Veryn M.G., Trujillo L.D.C., Piro O.E., Güida J.A. // J. Mol. Struct. 2014. V. 1076. P. 160.
Fulmer G.R., Miller A.J.M., Sherden N.H., Gottlieb H.E. et al. // Organometallics. 2010. V. 29. P. 2176.
Cordero B., Gómez V., Platero-Prats A.E., Revés M., et al. // Dalton Trans. 2008. V. 21. P. 2832.
Motorina E.V., Lomova T.N., Klyuev M.V. // Mendeleev Commun. 2018. V. 28. P. 426.
Bichan N.G., Ovchenkova E.N., Kudryakova N.O. et al. // Abstract X International Conference “Kinetics and Mechanism of crystallization” International Symposium “Smart Materials”. Suzdal. 1–6 July. 2018. P. 422.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии